

Abstract
The structural complexity and diversity of glycans parallel their multilateral functions in living systems. To better understand the vital roles glycans play in biological processes, it is imperative to develop analytical tools that can provide detailed glycan structural information. This was conventionally achieved by multistage tandem mass spectrometry (MSn) analysis using collision-induced dissociation (CID) as the fragmentation method. However, the MSn approach lacks the sensitivity and throughput needed to analyze complex glycan mixtures from biological sources, often available in limited quantities. We define herein the critical parameters for a recently developed fragmentation technique, electronic excitation dissociation (EED), which can yield rich structurally informative fragment ions during liquid chromatographic (LC)-MS/MS analysis of glycans. We further demonstrate that permethylation, reducing end labeling and judicious selection of the metal charge carrier can greatly facilitate spectral interpretation. With its high sensitivity, throughput, and compatibility with on-line chromatographic separation techniques, EED appears to hold great promise for large-scale glycomics studies.
Glycans play vital roles in many biological processes, including immunological response, inflammation, and cancer metastasis.1a,1b The multilateral functions of glycans derive from their structural diversity. Unlike linear biopolymers, such as oligonucleotides or proteins, whose primary structures are uniquely defined by one-dimensional sequences, glycans are built from multivalent monosaccharide units, giving rise to a large number of potential topological and linkage isomers. Thus, the detailed structural characterization of a glycan requires determination of not only its topology, but also its linkage configuration. This analytical challenge is further exacerbated by the lack of glycan amplification methods, and the presence of many closely-related structures in most naturally-occurring glycan mixtures. Among the current methodologies for biopolymer analysis, tandem mass spectrometry (MS/MS) is one of the most powerful owing to its specificity, low sample requirement, and compatibility with chromatographic separation methods. However, when used for glycan structural determinations, the conventional collision-induced dissociation (CID)-based MS/MS method often fails to produce sufficient numbers of the cross-ring fragments that are crucial for linkage determination.2 Although multistage tandem MS analysis (MSn) can ultimately provide more exhaustive linkage information, MSn approaches suffer from relatively low throughput and sensitivity, and are too time-consuming to be matched with on-line separation methods. We define herein the critical parameters for electron excitation dissociation (EED) and demonstrate how use of optimum conditions achieves high yields of well-defined, informative fragment ions during on-line HPLC-MS/MS analysis of glycans. Optimized EED thus overcomes all of the recognized limitations of CID and MSn and represents an important advance toward the goal of high-throughput detailed glycan analysis, even when the available amounts of biologically derived samples are extremely limited.
Electron-activated dissociation (ExD) methods have recently emerged as promising tools for glycan structural analysis.3a-e ExD of metal-adducted glycans can be initiated by either electron transfer from anion radical reagents, as in electron transfer dissociation (ETD) or direct interaction with free electrons. In the latter case, several ExD fragmentation pathways exist; these differ from one another in terms of the electron energy, including: electron capture dissociation (ECD) at low energies, hotECD at intermediate energies, and EED at high energies.4 As shown in Figure S1a, EED takes place at an electron energy of >9 eV, as made evident by the presence of doubly charged fragment ions, which cannot be produced by ECD from doubly charged precursor ions. EED is also different from the electron ionization dissociation (EID) process,5a,5b because a 9-eV electron does not have energy sufficient to induce fragmentation after ionization or to induce double ionization. Theoretical investigation suggested that EED is initiated by electron detachment from an oxygen atom, forming a distonic ion. The subsequent electron re-capture produces a di-radical which can undergo extensive fragmentation (Scheme S1). As a charge remote process, EED is capable of producing many more structurally informative fragments than ECD, regardless of the type of metal charge carriers (Figure S1).
Despite its promise, the application of EED has so far been limited by its low fragmentation efficiency.5a Scheme S2 shows a generic setup for ExD experiments. The electron current measured at the hexapole reflects the electron flux passing through the ion cyclotron resonance (ICR) cell. To perform EED, it is necessary to increase the potential bias of the electron emitter to obtain high-energy electrons. However, the larger negative cathode bias also produces a higher electron emission current due to the stronger Schottky effect (Table S1). The resultant higher electron density inside the ICR cell can significantly perturb the quadrupolar electrostatic trapping potential. This leads to efficient coupling of the electron axial motion to its high-frequency cyclotron rotation, which is rapidly cooled by radiative decay, producing low-energy electrons.6 Consequently, even at the higher negative cathode bias, the low-energy ECD process still dominates (Figure S2b). Thus, to attain efficient EED, it is important to reduce the electron flux into the ICR cell, which can be achieved either by decreasing the cathode heating current (Figures S2c and S2e) or by reducing the electric field at the emitter surface with application of an extraction lens potential that is close to the cathode bias voltage (Figures S2d and S2f). Note that these parameters were found to be critical for electron detachment dissociation (EDD), for a very different reason. In EDD, it is important to control the electron density to minimize the radial repulsive field that pushes anionic analytes away from the electrons, further reducing the EDD efficiency.7a,7b Although EED involves interaction between two mutually attractive oppositely charged particles, we demonstrate here that it is equally important to control the electron density to minimize competition from the more efficient ECD process. As shown in Figure 1, the relative abundance of the ECD diagnostic ion, [M + Na]+, decreased as the electron emission current decreased. Meanwhile, the EED efficiency, represented here by the abundance of an EED fragment ion, [1,5X2 + Na]+, raised gradually as the extraction lens voltage decreased, and reached its maximum with a moderate electron emission current, before diminishing again when the extraction lens voltage was negative enough to block the majority of electrons. Under the optimized conditions, 200 to 500 ms irradiation time was sufficient for efficient EED (Figure S2d).
Figure 1.
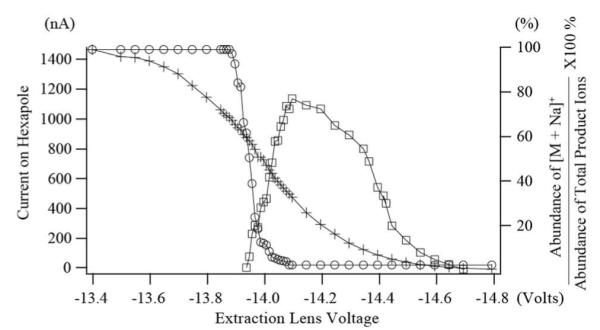
Plots of the relative abundance of the ECD diagnostic ion, [M + Na]+ (m/z 1497.7295, ○), absolute peak intensity (arbitrary units) of an EED diagnostic ion, [1,5X2+ Na]+ (m/z 491.2099, □), and the electron emission current measured at the hexapole (+) as a function of the extraction lens voltage. The cathode bias was −14.0 V and the cathode heating current was 1.52 A.
The utility of EED was recently demonstrated in the structural characterization of simple linear and branched glycan standards.3d Here, with improved efficiency, the performance of EED was evaluated on glycans having more complex structures. Two tumor antigens which play important roles in cell-cell recognition, Sialyl Lewis A (SLeA) and Sialyl Lewis X (SLeX), were selected as models, because they are linkage isomers containing analytically challenging residues: sialic acid (N-acetyl neuraminic acid, Neu5Ac) and fucose (Fuc).2 Permethylation was performed to stabilize these labile groups and to block unpredictable fragmentation pathways such as Fuc migration. Permethylation also facilitates de novo topology analysis as each glycosidic cleavage leaves a free hydroxyl “scar”, enabling differentiation of terminal and internal fragment ions and assignment of the initial branch point. However, ambiguities arising from the potential for isomeric non-reducing end and reducing end fragments may still exist for permethylated glycans. Although isomer-based ambiguity is more common for glycans with symmetrical structures, it can also occur in highly asymmetrical glycans. For example, for permethylated SLeX, the 0,2A2α and 2,5X1α fragments both have a neutral mass of 523.2629 u. Such ambiguity can be removed by labeling the reducing end hemiacetal oxygen with 18O prior to permethylation.
Figures S3, S4, and 2, respectively, show the CID, ECD, and EED spectra and cleavage maps of the [M + 2Li]2+ ions from the 18O-labeled and permethylated SLeX. As an ergodic fragmentation method, CID produced mostly glycosidic cleavages with the most abundant product ions resulting from the facile loss of a sialic acid (Figure S3). A few more cross-ring cleavages were observed in the ECD spectrum (Figure S4), but none occurred within the reducing end N-acetyl glucosaminyl (GlcNAc) residue. The new fragments did not contribute toward differentiation of SLeA and SLeX. Sialic acid loss was still the predominant pathway, presumably because the ECD process was initiated by electron capture by the metal charge carrier, and the sialic acid residue was the preferred metal binding site. By contrast, EED produced extensive glycosidic and cross-ring cleavages throughout the molecule (Figure 2). Similar ExD fragmentation patterns were observed for SLeA (Figures S5 and S6).
Figure 2.
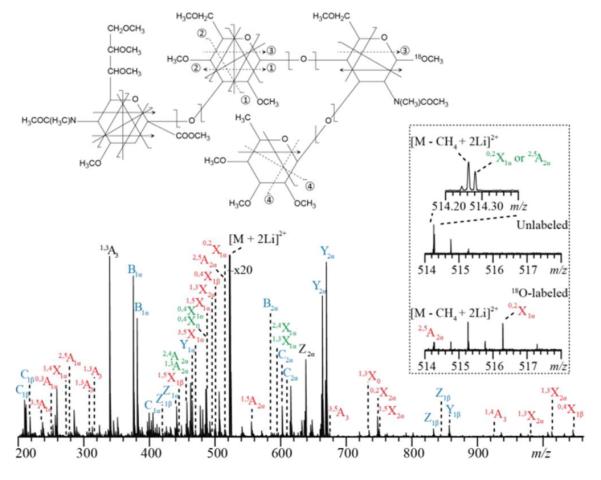
The EED spectrum and cleavage map of the permethylated and 18O-labeled SLeX precursor ion [M + 2Li]2+ m/z 523.2830. The detailed list of fragments can be found in Table S4.
The inset in Figure 2 shows a zoomed-in view of the region containing the 2,5A2α and 0,2X1α ions in the EED spectra of two isotopic forms of SLeX with (bottom) and without (top) the reducing-end 18O-labeling. Both 2,5A2α and 0,2X1α ions (m/z 514.2834 for the unlabeled SLeX) are keys to determination of the Neu5Ac2→3galactose (Gal) linkage, but they are isomeric and cannot be assigned definitively in the spectrum of the unlabeled glycan. When this spectrum is compared to the spectrum of the 18O-labeled glycan, the 0,2X1α ion is offset by 2.0043 u whereas the 2,5A2α ion is unaffected. This result confirms their assignments and allows confident linkage determination.
The number of building blocks with unique masses is smaller for glycans than for peptides, and each monosaccharide residue can potentially be cleaved into 22 different glycosidic and cross-ring fragments. Consequently, the presence of isobaric fragment ions is quite common in complex glycan EED spectra. A 36-mDa peak splitting due to the mass difference between a CH4 group and an O atom is frequently observed; their definition requires a mass resolving power of >50,000 at m/z 1,000 for baseline separation. Peaks separated by an even smaller interval may also be present. For example, in the EED spectrum of permethylated SLeX, the 0,2X1α and 2,5A2α ions (both at m/z 514.2834) and the doubly charged [M − CH4 + 2Li]2+ ion (m/z 514.2658) are separated by only 0.018 u (Figure 2 inset). The superior mass resolving power afforded by a high-field FTICR mass analyzer is essential for confident peak assignments, and for delineation of the Neu5Ac2→3Gal linkage in SLeX.
ExD experiments were also carried out on milk oligosaccharide linkage isomers, lacto-N-tetraose (LNT) and lacto-N-neotetraose (LNnT). Again, EED produced the most extensive fragmentation, including cross-ring cleavages for unambiguous determination of the non-reducing end Gal–GlcNAc linkage for isomer differentiation (Figures S7 and S8).
In addition to providing more structural information in general, EED offers a further advantage over ECD and ETD in that it does not involve charge reduction. Thus, EED is applicable to singly charged ions, making it an ideal choice for analysis of MALDI-generated ions as well as small glycans that cannot be efficiently multiply charged. As shown in Figures S9 and S10, EED of the [M + Li]+ peaks in the mass spectra of SLeX/SLeA produced fragmentation coverage similar to their [M + 2Li]2+ counterparts (Figures 2 and S6). Charge retention benefits the analysis of multiply charged precursor ions as well. First, fragment ions in a higher charge state can be more efficiently detected because the image current generated by an ion scales linearly with its charge. Second, because the mass resolving power of the ICR analyzer is inversely proportional to the ion mass-to-charge ratio, it is easier to resolve isobaric fragment ions when they are present in higher charge states and thus their signals appear at lower m/z values. Finally, charge retention increases the chance of observing both members of each set of complementary fragment ions, leading to more confident spectral interpretation. Figures S11 and S12 show the EED spectra of the [M + Li]+ and [M + 2Li]2+ peaks from permethylated Man5GlcNAc2 (Man = mannose). Whereas the 0,4X3α ion that defines the terminal, Man1→6Man linkage was present in both spectra, its complementary 0,4A2α ion was only observed in the EED spectrum of the [M + 2Li]2+ species. The general lack of non-reducing end fragments in EED of the [M + Li]+ species likely resulted from the preference for Li+-binding within the chitobiose core at the reducing end.
Unlike the protonated species commonly encountered in peptide tandem mass spectra, in which the number of charge carriers (protons) in a fragment ion is always the same as its charge state, there is no such simple correlation in ExD fragments of metal-adducted glycans, and this presents a challenge for determination of the fragment neutral mass. In addition, most metals used in ECD and ETD (lithium, magnesium and transition metals) have multiple stable isotopes, further complicating the data analysis. Some alkali metal cations, such as Na+ and Cs+, are monoisotopic, but they do not induce efficient ECD fragmentation, and favor metal loss, as a result of their weaker interaction with glycans and lower recombination energy upon electron capture.3d EED does not suffer from such restriction, as it is a charge remote process that results from a much higher energy input. EED of the [M + Cs]+ ions from SLeX and SLeA (Figures S13 and S14) provided structural information comparable to that of their [M + Li]+ species. Cesium adduction offers an additional benefit owing to its large negative mass defect (mCs = 132.9055 Da). For permethylated polyhexose with a repeating unit of C9H16O5 (204.0998 Da), each substitution of a proton with a Cs+ introduces an effective mass defect shift of −0.16 u at a given nominal mass. This mass difference is significantly higher than the variation in mass defects due to the difference in the fragment elemental composition. As illustrated in Figure 3, EED fragment ions of the [M + 2Cs]2+ of SLeX were clustered around three trend lines in the mass defect vs. mass plot, corresponding to 0, 1, or 2 Cs+-adduction, respectively. This information can be utilized for automatic metal counting and accurate determination of the fragment ion neutral mass.
Figure 3.
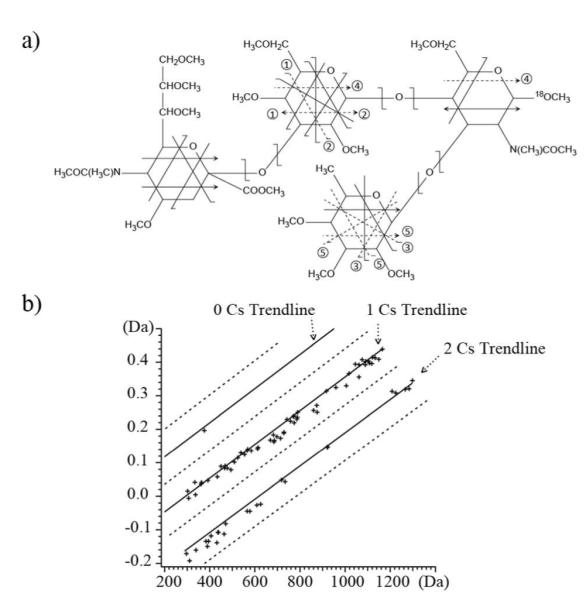
a) The EED cleavage map of the SLeX [M + Cs]+ m/z 1165.4400. b) Fragment ions from SLeX [M + 2Cs]2+ m/z 649.1724 can be clustered into different trend lines in the mass defect vs. mass plot based on their Cs+ content.
Because glycans often occur as complex mixtures, including many isomers, liquid chromatographic (LC) separation is usually needed before tandem MS analysis. Figure 4 shows the result of an on-line LC-MS/MS analysis of maltodextrin, (Glc-α1→4)nGlc, n = 2 to 6. All glycans were reduced and permethylated, and the singly sodiated form of each species was isolated for EED fragmentation. The total ion chromatogram for MS/MS analysis is shown in Figure 4a, with a single-scan EED spectrum of maltotetraose shown in Figure 4b. The insets show the zoomed-in view of selected regions. The 36-mDa splitting of the Z3 and C3 – 2H ions can be easily differentiated, and the measured masses are both within 2 ppm of the theoretical value with external calibration. The low-abundance cross-ring fragment ions, 3,5A3 and 0,2X2, are both easily detected with S/N ratios of 225 and 48, respectively. This result demonstrates the applicability of EED as the fragmentation method for on-line LC-MS/MS analysis of glycan mixtures. EED has a further advantage for analysis of glycans that have been labeled with a reducing-end chromophore, a modification frequently used to facilitate chromatographic separation, UV detection and quantification. However, the chromophore-labeled reducing end often serves as the preferred metal binding site, and is an electron scavenger. Consequently, reducing-end labeled glycans fragment poorly in ECD, and much of the fragment ion is generated by elimination of small neutrals containing the tag, as illustrated in Figure 5a. In sharp contrast, EED is not affected by the presence of the chromophore (Figure 5b), and this feature provides an additional advantage to its use for LC-MS analysis of reducing-end labeled glycans.
Figure 4.
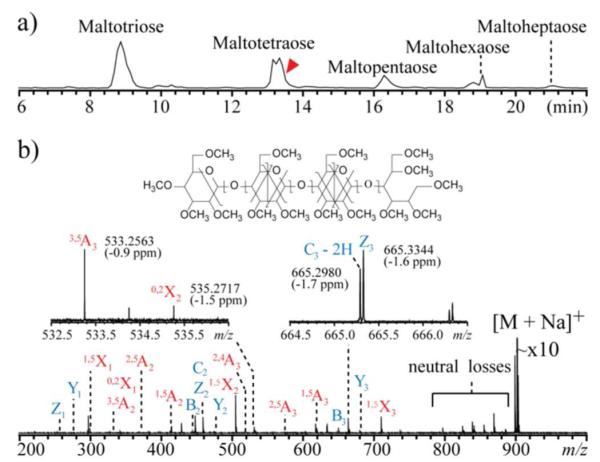
a) Total MS/MS ion chromatogram of an LC-MS/MS run of a reduced and permethylated maltodextrin mixture; b) Single-scan EED spectrum of the [M + Na]+ of maltotetraose, m/z 675.3798, acquired at the elution time indicated by the arrow.
Figure 5.
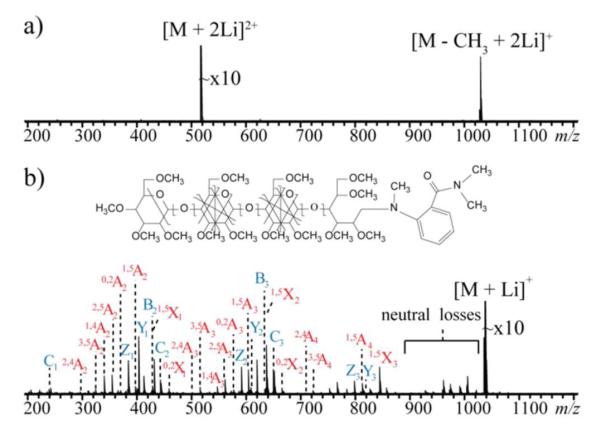
a) ECD of the [M + 2Li]2+ m/z 519.2938 and b) EED of the [M + Li]+ m/z 1031.5721 of 2-AB labeled, permethylated maltotetraose.
In conclusion, EED can provide rich structural information for a wide variety of glycans. Permethylation, reducing end labeling and high mass resolving power are helpful for accurate interpretation of the complex EED spectra. EED is amenable to analysis of singly charged precursor ions, although analysis of ions in higher charge states offers better detection efficiency, higher mass resolution and complementary fragment information. The chargeremote nature of the EED process allows a wider selection of metal charge carriers, among which Cs+ is preferred, as it is monoisotopic and has a large mass defect that enables reliable metal counting. With its high sensitivity, throughput, and compatibility with HPLC analysis of reducing-end labeled glycans, EED appears to hold great promise for large-scale glycomics studies.
Supplementary Material
ACKNOWLEDGMENT
The authors would like to thank Dr. Qi Wang for her help with sample preparation. This research was supported by NIH grants P41 RR010888/GM104603 and S10 RR025082.
Footnotes
Supporting Information This material is available free of charge via the Internet at http://pubs.acs.org.
Notes The authors declare no competing financial interest.
REFERENCES
- (1)(a).Dwek RA. Chem. Rev. 1996;96:683. doi: 10.1021/cr940283b. [DOI] [PubMed] [Google Scholar]; (b) Varki A. Essentials of Glycobiology. 2nd ed. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, N.Y.: 2009. [PubMed] [Google Scholar]
- (2).Zaia J. Mass Spectrom. Rev. 2004;23:161. doi: 10.1002/mas.10073. [DOI] [PubMed] [Google Scholar]
- (3)(a).Wolff JJ, Amster IJ, Chi LL, Linhardt RJ. J. Am. Soc. Mass Spectrom. 2007;18:234. doi: 10.1016/j.jasms.2006.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Adamson JT, Håkansson K. Anal. Chem. 2007;79:2901. doi: 10.1021/ac0621423. [DOI] [PubMed] [Google Scholar]; (c) Zhao C, Xie B, Chan SY, Costello CE, O’Connor PB. J. Am. Soc. Mass Spectrom. 2008;19:138. doi: 10.1016/j.jasms.2007.10.022. [DOI] [PubMed] [Google Scholar]; (d) Han L, Costello CE. J. Am. Soc. Mass Spectrom. 2011;22:997. doi: 10.1007/s13361-011-0117-9. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Yu X, Huang Y, Lin C, Costello CE. Anal. Chem. 2012;84:7487. doi: 10.1021/ac301589z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Nielsen ML, Budnik BA, Haselmann KF, Olsen JV, Zubarev RA. Chem. Phys. Lett. 2000;330:558. [Google Scholar]
- (5)(a).Zubarev RA. Mass Spectrom. Rev. 2003;22:57. doi: 10.1002/mas.10042. [DOI] [PubMed] [Google Scholar]; (b) Fung YME, Adams CM, Zubarev RA. J. Am. Chem. Soc. 2009;131:9977. doi: 10.1021/ja8087407. [DOI] [PubMed] [Google Scholar]
- (6).Li GZ, Guan S, Marshall AG. J. Am. Soc. Mass Spectrom. 1997;8:793. [Google Scholar]
- (7)(a).Leach FE, Wolff JJ, Laremore TN, Linhardt RJ, Amster IJ. Int. J. Am. Soc. Mass Spectrom. 2008;276:110. doi: 10.1016/j.ijms.2008.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yang J, Håkansson K. Int. J. Am. Soc. Mass Spectrom. 2008;276:144. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.