

Abstract
Intermittent hypoxia (IH) during sleep, such as occurs in obstructive sleep apnea (OSA), leads to degenerative changes in the hippocampus, and is associated with spatial learning deficits in adult mice. In both patients and murine models of OSA, the disease is associated with suppression of growth hormone (GH) secretion, which is actively involved in the growth, development and function of the central nervous system (CNS). Recent work showed that exogenous GH therapy attenuated neurocognitive deficits elicited by IH during sleep in rats. Here we show that administration of the Growth Hormone Releasing Hormone (GHRH) agonist JI-34 attenuates IH-induced neurocognitive deficits, anxiety, and depression in mice along with reduction in oxidative stress markers such as MDA and 8-OHDG, and increases in HIF-1α DNA binding and up-regulation of IGF-1 and erythropoietin expression. In contrast, treatment with a GHRH antagonist (MIA-602) during intermittent hypoxia did not affect any of the IH-induced deleterious effects in mice. Thus, exogenous GHRH administered as the formulation of a GHRH agonist may provide a viable therapeutic intervention to protect IH-vulnerable brain regions from OSA-associated neurocognitive dysfunction.
Keywords: Intermittent hypoxia, Morris water maze, malondialdehyde, 8-hydroxydeoxyguanosine, hippocampus, cortex
Introduction
Obstructive sleep apnea (OSA) is a highly prevalent condition characterized by repeated episodes of intermittent hypoxia (IH) and hypercapnia during sleep, sleep fragmentation (SF) and increased intrathoracic pressure swings (Dempsey et al. 2010). Among the vast spectrum of morbidities associated with OSA, neurocognitive deficits, anxiety and depression have emerged as prominent consequences of the disease (Beebe & Gozal 2002, Jackson et al. 2011). Development of rodent models mimicking components of OSA have yielded important insights into the mechanistic pathways underlying neural injury in the context of IH and SF, and have particularly pointed out the critical role of oxidative stress and inflammation-mediated neural injury in this context (Nair et al. 2011b, Nair et al. 2011a). Earlier studies from our laboratory indicated that exogenous growth hormone (GH) administration was anti-apoptotic and neuroprotective in the context of IH-induced neuronal injury, and that physical activity-associated neuroprotective effects during IH were mediated, at least in part, by endogenous increases in neuronal expression of IGF-1 (Li et al. 2011, Gozal et al. 2001, Gozal et al. 2010). However, the possibility that growth hormone-releasing hormone (GHRH) may also be neuroprotective was not explored.
GHRH is secreted by the hypothalamus and regulates the release of GH from the anterior pituitary gland. However, GHRH receptors are expressed in cortex and hippocampus and multiple other CNS sites (Muccioli et al. 1998). Furthermore, GHRH plays integral roles in the regulation of sleep, particularly deep restorative non-rapid eye movement sleep (NREM), and exerts its effects in both a systemic and localized fashion (Obal & Krueger 2004, Liao et al. 2010), and changes in GHRH regulation and expression are observed in a rodent model of OSA (Tarasiuk et al. 2011). More recently, studies have implicated a potential role for GHRH in mechanisms of declarative memory and mood regulation (Hallschmid et al. 2011, Telegdy & Schally 2012a, Telegdy & Schally 2012b, Telegdy et al. 2011), suggesting that GHRH may play a role in the cognitive, mood and behavioral consequences of sleep-disordered breathing. Furthermore, GHRH-related compounds appear to have anti-oxidant properties as well (Banks et al. 2010), such that modulation of free radicals in brain could underlie some of the beneficial effects previously attributed to GH in OSA rodent models (Li et al. 2011). We therefore undertook the present study to examine whether systemic administration of a long-acting and potent GHRH agonist (JI-34) and a new GHRH antagonist (MIA 602) would alter the effects of IH during sleep on spatial learning and depression.
Materials and Methods
Animals
Male mice C57BL/6J mice Stock No. 000664 (20–22 grams) were purchased from Jackson Laboratories (Bar Harbor, Maine), housed in a 12 hr light/dark cycle (lights on from 7:00 am to 7:00 pm) at a constant temperature (26 ±1°C). Mice were housed in groups of four in a standard clear polycarbonate cages, and were allowed access to food and water ad libitum. All behavioral experiments were performed during the light period. Mice were randomly assigned to either IH or room air (RA) exposures. The experimental protocols were approved by the Institutional Animal Use and Care Committee and are in close agreement with the National Institutes of Health Guide in the Care and Use of Animals and with the ARRIVE Guidelines (http://www.nc3rs.org.uk/page.asp?id=1357). All efforts were made to minimize animal suffering and to reducethe number of animals used.
Intermittent Hypoxia Exposures
Animals were maintained in 4 identical commercially-designed chambers (30″×20″×20″; Oxycycler model A44XO, BioSpherix, Redfield, NY) operated under a 12 hour light-dark cycle (7:00 am-7:00 pm) for 14 days prior to behavioral testing, which required an additional 7 days during which IH-exposures were continued. Oxygen concentration was continuously measured by an O2 analyzer, and was changed by a computerized system controlling gas outlets, as previously described (Gozal et al, 2001; Nair et al.,, 2011; Kaushal et al, 2012), such as to generate oxyhemoglobin nadir values (SaO2) in the 65–72% range alternating every 180 seconds with normoxia (SaO2>95%). In addition, time-matched normoxic exposures (RA) were conducted. Ambient temperature was kept at 24–26°C.
Preparation of the drugs and treatments
GHRH analog (JI-34) and GHRH antagonist (MIA 602) were synthesized in the laboratory of one of us (AVS) (Izdebski et al. 1995, Klukovits et al. 2012) and were dissolved in 0.1% DMSO in 10% aqueous propylene glycol solution (vehicle solution). Each dose of the agonist or antagonist was contained in 0.1 ml vehicle solution and stored at 4° C. As control, 0.1 ml of 0.1% DMSO in 10% aqueous propylene glycol solution was used (Veh). GHRH Analog: Mice exposed to either IH or RA received daily s.c. injections of the JI-34 or Veh at a dosage of 50 mg/kg body weight for 21days. Mice were randomly assigned to one of four experimental groups (n=12/ experimental group) consisting of: (i) RA exposure and Veh injection; (ii) RA exposure and JI-34; (iii) IH exposure and Veh; (iv) IH exposure and JI-34. All daily injections were administered at 7:00 AM. GHRH Antagonist: The GHRH antagonist MIA-602 was administered using an identical approach and dosage as with the GHRH agonist.
Behavioral Testing
The Morris water maze was used to assess spatial reference learning and memory as previously described (Xu et al., 2004). The maze protocol is similar to that described by Morris (1984) with modifications for mice. The maze system included a white circular pool, 1.4m in diameter and 0.6m in height, filled to a level of 35cm with water maintained at a temperature of 21°C (Morris 1984). The transparency of the pool water was abolished by addition of 150 ml of non-toxic white tempera paint. An escape platform (10 cm in diameter) made of Plexiglas was positioned 1 cm below the water surface and located at various locations throughout the pool. Extramaze cues surrounding the maze were placed at fixed locations in the surrounding curtains, and visible to the mice while these were in the maze. Maze performance was recorded by a video camera suspended above the maze that was interfaced with a video tracking system (HVS Imaging, Hampton, UK).
Briefly, a standard place-training reference memory task was conducted on mice in the water maze following exposure to 14 days of RA or IH. One day prior to place learning, mice were habituated to the water maze by allowing them to freely swim for several minutes. Place learning was then assessed over six consecutive days using a spaced training regimen that has been demonstrated to produce optimal learning in mice (Gerlai & Clayton 1999). Each training session consisted of three trials separated by a 10 minute inter-trial interval (ITI). On a given daily session, each mouse was placed into the pool from 1 of 4 quasirandom start points (N, S, E or W) and allowed a maximum of 90 seconds to escape to the platform where the mice were allowed to stay for 15 sec. Mice that failed to escape were led to the platform. The position of the platform was kept constant during the trials. 24 h following the final training session, the platform was removed for a probe trial to obtain measures of spatial bias. To assess the performance in the water maze, mean escape latencies and swim distance were analyzed.
Forced swimming test (FST)
Briefly, mice were individually forced to swim in an open cylindrical container (diameter 14 cm, height 20 cm), with a depth of 15 cm of water at 25 ± 1 °C. The immobility time, defined as the absence of escape-oriented behaviors, was scored during 6 min, as previously described (Eckeli et al. 2000, Nair et al. 2011a, Zomkowski et al. 2005). Each mouse was judged to be immobile when it ceased struggling, and remained floating motionless in the water, making only those movements necessary to keep its head above water. The average percentage immobility was calculated by a blinded experimenter.
Lipid Peroxidation Assay
To assess lipid peroxidation in brain tissue samples, we used commercially available kits (cat# MDA-586 kits; OxisResearch, Portland OR) that allow for assessment of the relative malondialdehyde (MDA) production, a commonly used indicator of lipid peroxidation using the manufacturer’s instructions. Briefly and as previously described (Nair et al., 2011a), anesthesia with pentobarbital (50 mg/kg intraperitoneally) was administered, after which, mice were perfused with 0.9% saline buffer for 5 minutes and the cortex was dissected, snap frozen in liquid nitrogen, and stored at −80°C until assay the following day. Cortical tissues were homogenized in 20 mM phosphate buffer (pH 7.4) containing 0.5 mM butylated hydroxytoluene to prevent sample oxidation. After protein concentration measurements, equalamounts of proteins (2.0–2.5 mg protein from each sample) were used in triplicate to enable the reaction with the chromogenic reagents at 45°C in 500 μL buffer for 1 to 2 hours. The samples were then centrifuged and clear supernatants were measured at 586 nm. The level of MDA production was then calculated with the standard curve obtained from the kit.
8-hydroxydeoxyguanosine (8-OHDG) Tissue Levels
To assess levels of 8-OHDG in harvested hippocampal samples, a commercially available assay was employed (Cell Biolabs, San Diego, CA), as previously described (Nair et al., 2011a). In brief, samples or 8-OHDG standards were first added to an 8-OHDG/BSA conjugate pre-absorbed enzyme immunoassay plate. After a short incubation period, an anti–8-OHDG mAb was added to the plates, followed by an horseradish peroxidase-conjugated secondary antibody. The 8-OHDG content in the tissue samples was then determined by comparison with the 8-OHDG standard curves as directed by the manufacturer’s instructions.
HIF-1α Assay
Nuclear extracts were isolated from harvested hippocampal and cortical samples and subjected to a commercially available HIF-1α Assay (Cayman Chemical; cat #10006910) using manufacturer’s instructions. This assay is a non-radioactive, sensitive method for detecting specific transcription factor DNA binding activity in nuclear extracts and whole cell lysates. The 96-well enzyme-linked immunosorbent assay (ELISA) format employs a specific double stranded DNA (dsDNA) consensus sequence containing the HIF-1α response element that is immobilized to the wells of 96-well plates. HIF-1α contained in the tissue lysates will then bind specifically to the HIF-1α response element. The HIF-1α transcription factor complex is detected by addition of a specific primary antibody directed against HIF-1α. A secondary antibody conjugated to HRP is then added to provide a sensitive colorimetric readout at 450 nm. To standardize the assay across experiments, we used hippocampal extracts obtained from 2 mice exposed to sustained hypoxia (6.5% oxygen) for 6 hours, and all results were expressed as ratios with the mean 450 nm absorbance readings of these samples as the denominator.
Quantitative PCR
Since both IGF-1 and erythropoietin (EPO) exert neuroprotective effects in the context of IH (Li et al. 2011, Dayyat et al. 2012), we examined the effect of treatment with GHRH-related compounds on the expression of these 2 genes of interest. Total RNA was prepared from hippocampal tissue samples using TRIzol reagent (Invitrogene) following the manufacturer’s instructions. Isolated total RNA was quantified spectrophotometrically. Aliquots of total RNA (1 μg) were reverse transcribed using Superscript II-Reverse Transcriptase (Invitrogene, Carlsbad, CA) according to the manufacturer’s protocol. cDNA equivalent to 20 ng of total RNA were subjected to real-time PCR analysis (MX4000, Stratagene, La Jolla, CA) following the manufacturer’s protocol. PCR Primers and Taqman probes for IGF-1 (5′-AGAAGGTGCTGTTGAGTAA and 3′-AACTAGAAGTCAGAAGTGTCA) and EPO (5′-GGTACTGGGAGCTCAGAAGG and 3′-TGGGGAAACCCCCATGAGATC) were purchased from ABI (Applied Biosystems). Each reaction (25μl) contained 2.5 μl reaction buffer (10×), 6 mM MgCl2, 0.2 μM dNTP, 0.6 μM each primer, 0.25 μl SureStar Taq DNA Polymerase and 2 μl cDNA dilutions. The cycling condition consisted of 1 cycle at 95°C for 10 min and 40 three-segment cycles (95°C for 30 s, 55°C for 60 s and 72°C for 30 s). Standard curves for gene of interest and housekeeping gene (β-actin) were included in each reaction. Real-time PCR results were analyzed using MX4000 software (Stratagene, La Jolla, CA).
Data Analysis
To elucidate the nature of interactions between IH and RA conditions and treatment effects, either two-way repeated measures ANOVA or MANOVA were employed to analyze each trial block, and then followed by post-hoc Tukey tests. Similar statistical approaches were used to compare probe trials, reference memory tests, and the FST. In all the experimental conditions, the data were partitioned into 6 blocks (containing 3 trials/day). We used a multivariate MANOVA model (SPSS software 17; Chicago) that included latency, pathlength and swim speed and two between factors: (1) Groups (four levels): RA Veh, IH Veh, RA J1-34, and IH J1-34 compound (2) Condition (two levels): RA or IH. All F statistics are reported using Pillai’s Trace. The interaction of three different factors, i.e., time, condition and group were determined using this mixed model repeated measures MANOVA. Similar statistics were conducted on all groups for GHRH antagonist MIA 602 studies. For all comparisons, a p value <0.05 was considered to achieve statistical significance.
Results
Spatial Learning Performance
GHRH Analog – JI-34
On the standard place discrimination task, wild type mice exposed to 21 days of IH (IH-Veh) exhibited longer latencies and path lengths to locate the hidden platform when compared to either controls (RA-JI-34, RA-Veh ) or to IH-JI-34 mice exposed to 21 days IH ( n=12 per experimental condition; Figures 1A and B). Overall latency analysis for the entire trial blocks revealed significant changes between the different treatment groups, [F= 26.744; p<0.001] and pathlength, [F=25.413; p<0.001] indicating that IH adversely affected task performance. Significant differences in latencies were observed during blocks 1 [F=30.819; p<0.001], 2 [F= 6.204; p<0.003], 3 [F= 10.575; p<0.001], 4 [F=3.124; p<0.045], 5 [F=7.720; p<0.001] and 6 [F=14.898; p<0.001] (Figure 1A). Repeated measures ANOVA revealed significant differences in path lengths during blocks 1 [F=9.310; p<0.001], 2 [F=52.397; p<0.001], 3 [F=13.264; p<0.001], 4 [F=9.649; p<0.001], 5 [F=6.885; p<0.002] and 6 [F=23.318; p<0.001] (Figure 1B). In the probe-trial test, one-way ANOVA revealed a significant effect of treatment [IH vs. RA: F=14.833; p<0.001]. The magnitude of impairment was present only in IH Vehicle treated mice (Figure 2A).
Figure 1.

Treatment with the GHRH agonist JI-34 prevents IH-induced adverse effects on acquisition of a hippocampal spatial task in the water maze in mice. Administration of the GHRH antagonist MIA-602 adversely affects spatial task learning in normoxic mice but is not additively deleterious to IH. (A, B) Mean pathlengths (cm) and latencies (seconds) to locate the target platform during spatial task acquisition during treatment with either J1-34 or vehicle, in mice exposed to either RA or intermittent hypoxia (IH). Data are mean ± SEM. (*IH-VEH vs. RA-VEH, p <0.05; One-way ANOVA). (C, D) Mean pathlengths (cm) and latencies (seconds) to locate the target platform during spatial task acquisition treatment with either MIA-602 or vehicle in mice exposed to either normoxia or intermittent hypoxia. Data are mean ± SEM. (*IH-VEH and IH-MIA 602 vs. RA-VEH, p <0.05; One-way ANOVA).
Figure 2.

Treatment with the GHRH agonist JI-34 prevents IH-induced adverse effects on retention of a hippocampal spatial task in the water maze and the deleterious effects of IH on forced swim test in mice. Administration of the GHRH antagonist MIA-602 adversely affects spatial task retention and forced swim performance in normoxic mice but is not additively deleterious to IH. Mean percentage time in the target quadrant during probe trials after completion of water maze testing in mice treated with either J1-34 (A) and Mice exposed to intermittent hypoxia and treatment with J1-34 (B) Data are mean ± SEM. (*IH-VEH vs. RA-VEH, p <0.05; One-way ANOVA).
Mean percentage time in the target quadrant during probe trials after completion of water maze testing in mice treated MIA-602 (C) during intermittent hypoxia and mice exposed to intermittent hypoxia and treatment with MIA 602 (D) during the forced swim test. Data are mean ± SEM. (*IH-VEH and IH-MIA 602 vs. RA-VEH, p <0.05; One-way ANOVA).
Repeated measures MANOVA with latency, groups and conditions [F(56,12) = 4.812; p< 0.0001]; revealed that IH-JI-34, RA-JI-34 and RA-Veh mice required significantly less time than IH-Veh to find the hidden platform in a Morris water maze (Figure 1A); Repeated measures MANOVA with pathlength, groups and conditions [F(56,12) = 14.771; p< 0.0001]; indicated that as the training progressed the IH-JI-34, RA-JI-34 and RA-Veh mice could reach the hidden platform and covered the shortest distance when compared to the distance covered by IH-Veh treated mice (Figure 1B). Thus, JI-34 treatment in the context of IH exposures was associated with preservation of spatial learning performance while vehicle treatment during IH demonstrated the anticipated deficits in the acquisition and retention of the spatial task.
GHRH Antagonist – MIA 602
Treatment with MIA 602 showed a significant difference in latency on block 2 [F=6.00; p<0.002] and block 3 [F=6.0652; p<0.001] (Fig. 1C). However, only block 3 [F=3.213; p<0.035] showed a significant difference in pathlength (Fig 1D). In the probe-trial test, one-way ANOVA revealed a significant effect of treatment [IH vs. RA: F=12.0508; p<0.001]. The magnitude of impairment was present in all IH-exposed groups of mice (Figure 2C). Thus, MIA602 adversely affected spatial task acquisition and retention in normoxic conditions but did not exhibit a deleterious effect that was incremental to that induced by IH exposures. Furthermore, 2-way ANOVA followed by post-hoc tests revealed that the improvements associated with JI-34 treatment in IH-exposed mice markedly differed from the effects of MIA-602 in similarly treated mice (F=11.354; p<0.001).
Forced Swim Test
IH-Veh mice had significantly higher immobility durations during the last 4 min of the FST [F=7.614; p<0.004] when compared to all other treatment groups, including IH-J1-34 (Figure 2B). Similarly, IH-Veh mice in the antagonist group also had significantly higher immobility durations during the last 4 min of the FST [F=4.765; p<0.016] when compared to all other treatment groups. Further, IH-MIA 602 treatment also showed significantly higher immobility (Figure 2D).
Lipid Peroxidation
After the behavioral experiments, cortical tissues and hippocampus were harvested and processed for assessment of lipid peroxidation as indicated by MDA levels. Figure 3 shows MDA concentrations in cortical and hippocampal tissue homogenates from all treatment groups (Fig. 3 A and C). A significant increase in MDA levels was observed in IH-Veh and IH-MIA602 mice both in the cortex [F=15.787; p<0.001] and hippocampus [F=22.979; p<0.001], when compared to RA-Veh. In contrast, no increases in MDA levels occurred in IH-JI-34 treated mice (Figure 3).
Figure 3.

Lipid peroxidation and oxidative DNA damage levels in cortex and hippocampus of mice exposed to IH and treated with either J1-34 or MIA-602. Data are mean ± SEM. (*IH-VEH and IH-MIA 602 vs. RA-VEH, p <0.05; One-way ANOVA).
8-OHDG Levels
Figure 3 shows 8-OHdG concentrations in cortical and hippocampal tissue homogenates from all treatment groups (Fig. 3B and D). A significant increase in 8-OHdG levels was observed in IH-Veh and IH-MIA602 mice both in the cortex [F=15.361; p<0.001] and hippocampus [F=14.674; p<0.001], when compared to RA-Veh, but such increases were absent in IH-JI-34 treated mice (Figure 3).
HIF-1α Assay
HIF-1α binding activity was similar between RA-Veh and IH-Veh mice (Figure 4). However, significant increases in HIF-1α binding activity occurred in RA-JI-34 exposed mice and IH-JI34 mice both in the cortex [n=6/group; p<0.01] and hippocampus [n=5/group; p<0.001], when compared to RA-Veh or IH-Veh. In contrast, no increases in HIF-1α binding activity levels occurred in MIA602-treated mice (Figure 4).
Figure 4.
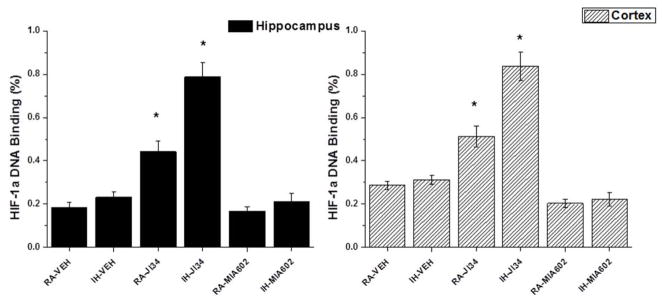
Effects of exogenous administration of J1-34 and MIA-602 on HIF-1α DNA binding. Data are mean ± SEM. (*IH-JI34 or RA-JI34 vs. IH-VEH, RA-MIA602, and IH-MIA602, n=5–6/group; p <0.01; One-way ANOVA). All results are expressed as ratios of hippocampal (left panel) or cortical (right panel) samples obtained from 2 mice exposed to sustained hypoxia for 6 hours.
IGF-1 and EPO mRNA Expression
Long-term IH exposures induced significant decreases in EPO and IGF-1 mRNA expression (p <0.05 vs. RA). However, GHRH analog J1-34 treatment significantly increased both the expression of EPO [F=10.883; p<0.005] (Fig 5 A) and mRNA for IGF-1 [F=5.625; p<0.030] (Fig. 4B). In contrast, treatment with GHRH antagonist MIA 602 was not associated with any significant differences compared to vehicle treated mice (Figure 5).
Figure 5.
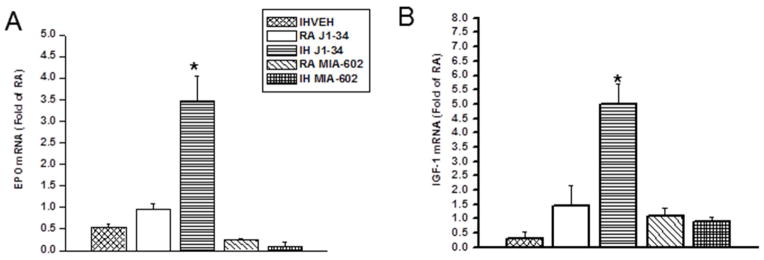
Effects of exogenous administration J1-34 and MIA-602 on EPO (A) and IGF-1 (B) mRNA expression. EPO and IGF-1 mRNA expression was assessed by quantitative real-time RT-PCR. J1-34 administration induced significant increases in IGF-1 mRNA and EPO expression during exposures to IH. Data are mean ± SEM. (*IH-VEH and IH-JI34 vs. RA-VEH, p <0.01; One-way ANOVA).
Discussion
In this study, treatment with a GHRH analog (JI-34) during IH exposures mimicking OSA was accompanied by significant attenuation of neurocognitive deficits and depression, the latter being assessed by the forced swim test. Conversely, we found no evidence for further deterioration in any of the outcome measures when the GHRH antagonist MIA602 was administered during IH exposures, even if adverse effects were detected in normoxic mice treated with MIA602. Furthermore, JI-34 administration led to decreases in oxidative stress markers, such as MDA and 8-OHDG, as well as increased gene expression of 2 recognized endogenous cellular survival pathways, namely IGF-1 and EPO, suggesting that exogenous GHRH analog administration may serve as an effective therapeutic intervention aiming to mitigate the extent and severity of OSA-associated cognitive dysfunction. These results suggest that peripheral GHRH administration stimulated either directly through GHRH receptor binding or by alternatively inducing a downstream response to increase cellular GH and downstream pathways in the brain that may ameliorate the hypoxia-induced injury of OSA.
Before we address the potential significance of our findings, some technical and methodological issues deserve comment. First, IH exposures do not constitute the full complement of OSA-associated physiological disturbances, but rather serve as a corollary to one of its major characteristics, namely the recurring nature of hypoxia and re-oxygenation cycles that are characteristically present in the vast majority of patients. In addition, IH exposures do induce some degree of sleep perturbation, particularly centered around fragmentation of sleep (Polotsky et al. 2006, Kaushal et al. 2012), even though the pattern of such sleep disruption is not as prominent as in OSA itself, most likely due to the absence of concomitant hypercapnia and chest wall afferent inputs induced by increased respiratory efforts during airway occlusion (Szollosi et al. 2004, Calero et al. 2006, Buchanan & Richerson 2010, Wilcox et al. 1990). It is likely that the interactions between IH alone and IH-associated sleep perturbations may lead to reduced harnessing of GHRH synthesis and bioavailability in regions such as the hippocampus and cortex, thereby increasing their susceptibility to injury. In summary, our results show that exogenous administration of the GHRH analog J1-34 during the course of IH exposures attenuates behavioral impairments associated with IH and decreases oxidative stress markers (i.e., MDA and 8-OHDG), indicating that the GHRH analog J1-34 confers neuronal protection against IH-induced oxidative stress. Indeed, GHRH appears to play a significant role in the regulation of sleep, particularly NREM sleep (Obal & Krueger 2004, Szentirmai et al. 2007, Liao et al. 2010), such that the alterations in sleep architecture induced by IH could be mediated, at least in part, by IH-induced reductions in GHRH gene transcription and bioavailability. The mechanisms underlying the putative effects on GHRH and potentially other downstream related peptides (i.e., GH and IGF-1) remain unknown. Preliminary evidence using GHRH antagonists would suggest the presence of reciprocal interactions between hypoxia-inducible factor-1α (HIF-1α) and GHRH (Munoz-Moreno et al. 2012). Since HIF-1α is an important transcriptional regulator of both IGF-1 and EPO in CNS (Acker & Acker 2004, Freeman & Barone 2005), it is possible that the relatively reduced recruitment of HIF-1α during chronic IH exposures may result in a disproportionately lower expression of neuroprotective elements, such as IGF-1, and EPO (Dayyat et al. 2012), and that exogenous administration of GHRH agonists may restore the desirable protective effects of these genes. Furthermore, our data also show that the GHRH antagonist MIA602 reduces cognitive performance in room air-exposed control animals. It is possible that GHRH-related pathways may play an intrinsic role in maintenance of specific aspects of cognition, and the present observation should prompt further study in this direction. However, when IH exposures were added, MIA602 did not induce any further discernible deleterious effects on performance in the water maze. One thing that needs to be taken into account is that the treatment for IH was for 21 days, a period during which most of the adverse effects of IH have already taken place. Evidence supporting such possibility derives from previous studies in aging, whereby class II G protein-coupled receptors, which play major roles in regulating the function and plasticity of neuronal circuits in CNS, can be activated by GHRH, and increase the resistance of neuronal cells to oxidative, metabolic, and excitotoxic injury (Thornton et al. 2000, Martin et al. 2005). To further confirm the potential beneficial effect of a GHRH agonist in IH-induced cognitive impairments, we sought to explore whether JI-34 modified any of the previously identified markers of either injury or protection. We found that animals treated daily with the GHRH agonist while undergoing IH exposures exhibited not only attenuated increases in oxidative stress markers, but also displayed increased IGF-1 mRNA and EPO mRNA expression in the hippocampus, suggesting that the somatotropic axis in the brain was effectively activated by exogenous subcutaneous administration of an agonist of GHRH. Some of these findings appear to be in contradiction with previously published work on IH and HIF-1α expression, which most likely represent differences in HIF-1α regulation in different tissues or when using different IH profiles (Belaidi et al., 2009; Nanduri et al, 2008, Yuan et al, 2008, 2011). Indeed, our previous study (Dayyat et al, 2012) and the study by Aviles-Reyes et al (2010) clearly showed that long-term exposures to IH are associated with initial increases in HIF-1α transcriptional activity, but are followed by progressive reductions of such activity to baseline normoxic levels despite ongoing IH exposures. We now show that administration of the GHRH analog JI-34, but not the GHRH antagonist MIA602, elicited increases in HIF-1α DNA binding activity in hippocampus and cortex, in addition to increased expression of the HIF-1α target genes EPO and IGF-1. Secondly, our behavioral phenotype reporter assays explored only selective aspects of cognition, i.e., spatial task acquisition and retention using the Morris water maze, and of mood, i.e., forced swim test. It is possible that different aspects of cognition and behavioral functioning may be differentially modulated by OSA-associated perturbations, and that GHRH-related cognitive and behavioral effects may differ under normal conditions and conditions such as IH during sleep (Telegdy & Schally 2012a, Telegdy et al. 2011, Telegdy & Schally 2012b). Indeed, the dual beneficial cognitive and mood effects of both GHRH antagonists (Telegdy et al. 2011, Klukovits et al. 2012), and of agonists (Vitiello et al. 2006) may reflect the divergent interactions of these compounds on the endogenous regulation of the somatotropic axis and IH-effects. The GHRH analogs can cross the blood brain barrier (BBB) (Jaeger et al. 2005) and exert their effects in CNS, or whether such effects are due to modulation of alternative peripheral mechanisms. Peptides such as GHRH can cross the BBB (Dogrukol-Ak et al. 2004), and previously reported effects of GHRH-related compounds on several neurotransmitters would suggest that the beneficial effects of JI-34 were likely centrally mediated.
Of note, IH and SF are both associated with increased oxidative stress, the magnitude of which is a critical determinant of end-organ injury in general, and CNS dysfunction in particular (Row et al. 2003, Shan et al. 2007, Xu et al. 2004, Row et al. 2007, Douglas et al. 2010, Nair et al. 2011a). GHRH has been postulated to modulate oxidative stress in specific cancer lines, as well as other settings (Barabutis & Schally 2008, Banks et al. 2010, Wang et al. 2012), such that the beneficial effects of JI-34 in our IH-exposure murine model could be ascribable to the attenuation of free radical-induced neuronal injury. A substantial component of the neuroprotective effect conferred by the administration of the GHRH analog JI-34 may be due to the downstream effects of this compound on GH-IGF-1 dependent cellular pro-survival pathways. This possibility was suggested by the increased expression of IGF-1 after treatment with JI-34, and further confirmation of this possibility will require further study, particularly considering the previously reported favorable effects of exogenous GH treatment in rodent OSA models (Li et al. 2011). Notably, the beneficial effects of GHRH agonists on hypoxia-induced phenomena are in agreement with their effects on pancreatic β-islet survival (Ludwig et al. 2010, Ludwig et al. 2012). A recent study from our lab further supports the concept that exogenous administration of recombinant human EPO will attenuate IH-induced NADPH oxidase mediated hippocampal oxidative stress injury and cognitive and behavioral deficits (Dayyat et al. 2012), indicating the protective effects of GHRH analog JI-34 may be via the EPO pathway.
In summary, treatment with a selective GHRH agonist reduced markers of oxidative stress in the cortex and hippocampus, promoted enhanced expression of the neuroprotective genes IGF-1 and EPO, and markedly attenuated IH-induced cognitive and behavioral deficits.
Acknowledgments
This study was supported by National Institutes of Health grant HL-086662. The authors have no conflicts of interest to declare in relation with this manuscript.
Footnotes
Author contributions: DG designed research; DN, VR, and RCL performed research; AVS contributed new reagents; DN and VR analyzed data; and DG and AVS wrote the paper.
References
- Acker T, Acker H. Cellular oxygen sensing need in CNS function: physiological and pathological implications. J Exp Biol. 2004;207:3171–3188. doi: 10.1242/jeb.01075. [DOI] [PubMed] [Google Scholar]
- Aviles-Reyes RX, Angelo MF, Villarreal A, Rios H, Lazarowski A, Ramos AJ. Intermittent hypoxia during sleep induces reactive gliosis and limited neuronal death in rats: implications for sleep apnea. J Neurochem. 2010;112:854–69. doi: 10.1111/j.1471-4159.2009.06535.x. [DOI] [PubMed] [Google Scholar]
- Banks WA, Morley JE, Farr SA, Price TO, Ercal N, Vidaurre I, Schally AV. Effects of a growth hormone-releasing hormone antagonist on telomerase activity, oxidative stress, longevity, and aging in mice. Proc Natl Acad Sci U S A. 2010;107:22272–22277. doi: 10.1073/pnas.1016369107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabutis N, Schally AV. Antioxidant activity of growth hormone-releasing hormone antagonists in LNCaP human prostate cancer line. Proc Natl Acad Sci U S A. 2008;105:20470–20475. doi: 10.1073/pnas.0811209106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beebe DW, Gozal D. Obstructive sleep apnea and the prefrontal cortex: towards a comprehensive model linking nocturnal upper airway obstruction to daytime cognitive and behavioral deficits. J Sleep Res. 2002;11:1–16. doi: 10.1046/j.1365-2869.2002.00289.x. [DOI] [PubMed] [Google Scholar]
- Belaidi E, Joyeux-Faure M, Ribuot C, Launois SH, Levy P, Godin-Ribuot D. Major role for hypoxia inducible factor-1 and the endothelin system in promoting myocardial infarction and hypertension in an animal model of obstructive sleep apnea. J Am Coll Cardiol. 2009;53:1309–17. doi: 10.1016/j.jacc.2008.12.050. [DOI] [PubMed] [Google Scholar]
- Buchanan GF, Richerson GB. Central serotonin neurons are required for arousal to CO2. Proc Natl Acad Sci U S A. 2010;107:16354–16359. doi: 10.1073/pnas.1004587107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calero G, Farre R, Ballester E, Hernandez L, Daniel N, Montserrat Canal JM. Physiological consequences of prolonged periods of flow limitation in patients with sleep apnea hypopnea syndrome. Respir Med. 2006;100:813–817. doi: 10.1016/j.rmed.2005.09.016. [DOI] [PubMed] [Google Scholar]
- Dayyat EA, Zhang SX, Wang Y, Cheng ZJ, Gozal D. Exogenous erythropoietin administration attenuates intermittent hypoxia-induced cognitive deficits in a murine model of sleep apnea. BMC Neurosci. 2012;13:77. doi: 10.1186/1471-2202-13-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempsey JA, Veasey SC, Morgan BJ, O’Donnell CP. Pathophysiology of sleep apnea. Physiol Rev. 2010;90:47–112. doi: 10.1152/physrev.00043.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogrukol-Ak D, Tore F, Tuncel N. Passage of VIP/PACAP/secretin family across the blood-brain barrier: therapeutic effects. Curr Pharm Des. 2004;10:1325–1340. doi: 10.2174/1381612043384934. [DOI] [PubMed] [Google Scholar]
- Douglas RM, Ryu J, Kanaan A, Del Carmen Rivero M, Dugan LL, Haddad GG, Ali SS. Neuronal death during combined intermittent hypoxia/hypercapnia is due to mitochondrial dysfunction. Am J Physiol Cell Physiol. 2010;298:C1594–1602. doi: 10.1152/ajpcell.00298.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckeli AL, Dach F, Rodrigues AL. Acute treatments with GMP produce antidepressant-like effects in mice. Neuroreport. 2000;11:1839–1843. doi: 10.1097/00001756-200006260-00008. [DOI] [PubMed] [Google Scholar]
- Freeman RS, Barone MC. Targeting hypoxia-inducible factor (HIF) as a therapeutic strategy for CNS disorders. Curr Drug Targets CNS Neurol Disord. 2005;4:85–92. doi: 10.2174/1568007053005154. [DOI] [PubMed] [Google Scholar]
- Gerlai R, Clayton NS. Analysing hippocampal function in transgenic mice: an ethological perspective. Trends Neurosci. 1999;22:47–51. doi: 10.1016/s0166-2236(98)01346-0. [DOI] [PubMed] [Google Scholar]
- Gozal D, Daniel JM, Dohanich GP. Behavioral and anatomical correlates of chronic episodic hypoxia during sleep in the rat. J Neurosci. 2001;21:2442–2450. doi: 10.1523/JNEUROSCI.21-07-02442.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozal D, Nair D, Goldbart AD. Physical activity attenuates intermittent hypoxia-induced spatial learning deficits and oxidative stress. Am J Respir Crit Care Med. 2010;182:104–112. doi: 10.1164/rccm.201001-0108OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallschmid M, Wilhelm I, Michel C, Perras B, Born J. A role for central nervous growth hormone-releasing hormone signaling in the consolidation of declarative memories. PLoS One. 2011;6:e23435. doi: 10.1371/journal.pone.0023435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izdebski J, Pinski J, Horvath JE, Halmos G, Groot K, Schally AV. Synthesis and biological evaluation of superactive agonists of growth hormone-releasing hormone. Proc Natl Acad Sci U S A. 1995;92:4872–4876. doi: 10.1073/pnas.92.11.4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson ML, Howard ME, Barnes M. Cognition and daytime functioning in sleep-related breathing disorders. Prog Brain Res. 2011;190:53–68. doi: 10.1016/B978-0-444-53817-8.00003-7. [DOI] [PubMed] [Google Scholar]
- Jaeger LB, Banks WA, Varga JL, Schally AV. Antagonists of growth hormone-releasing hormone cross the blood-brain barrier: a potential applicability to treatment of brain tumors. Proc Natl Acad Sci U S A. 2005;102:12495–12500. doi: 10.1073/pnas.0504163102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushal N, Ramesh V, Gozal D. Human apolipoprotein E4 targeted replacement in mice reveals increased susceptibility to sleep disruption and intermittent hypoxia. Am J Physiol Regul Integr Comp Physiol. 2012;303:R19–29. doi: 10.1152/ajpregu.00025.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klukovits A, Schally AV, Szalontay L, Vidaurre I, Papadia A, Zarandi M, Varga JL, Block NL, Halmos G. Novel antagonists of growth hormone-releasing hormone inhibit growth and vascularization of human experimental ovarian cancers. Cancer. 2012;118:670–680. doi: 10.1002/cncr.26291. [DOI] [PubMed] [Google Scholar]
- Li RC, Guo SZ, Raccurt M, Moudilou E, Morel G, Brittian KR, Gozal D. Exogenous growth hormone attenuates cognitive deficits induced by intermittent hypoxia in rats. Neuroscience. 2011;196:237–250. doi: 10.1016/j.neuroscience.2011.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao F, Taishi P, Churchill L, Urza MJ, Krueger JM. Localized suppression of cortical growth hormone-releasing hormone receptors state-specifically attenuates electroencephalographic delta waves. J Neurosci. 2010;30:4151–4159. doi: 10.1523/JNEUROSCI.6047-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig B, Rotem A, Schmid J, et al. Improvement of islet function in a bioartificial pancreas by enhanced oxygen supply and growth hormone releasing hormone agonist. Proc Natl Acad Sci U S A. 2012;109:5022–5027. doi: 10.1073/pnas.1201868109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig B, Ziegler CG, Schally AV, et al. Agonist of growth hormone-releasing hormone as a potential effector for survival and proliferation of pancreatic islets. Proc Natl Acad Sci U S A. 2010;107:12623–12628. doi: 10.1073/pnas.1005098107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin B, Lopez de Maturana R, Brenneman R, Walent T, Mattson MP, Maudsley S. Class II G protein-coupled receptors and their ligands in neuronal function and protection. Neuromolecular Med. 2005;7:3–36. doi: 10.1385/nmm:7:1-2:003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- Muccioli G, Ghe C, Ghigo MC, et al. Specific receptors for synthetic GH secretagogues in the human brain and pituitary gland. J Endocrinol. 1998;157:99–106. doi: 10.1677/joe.0.1570099. [DOI] [PubMed] [Google Scholar]
- Munoz-Moreno L, Arenas MI, Schally AV, et al. Inhibitory effects of antagonists of growth hormone-releasing hormone on growth and invasiveness of PC3 human prostate cancer. Int J Cancer. 2012 doi: 10.1002/ijc.27716. [DOI] [PubMed] [Google Scholar]
- Nair D, Dayyat EA, Zhang SX, Wang Y, Gozal D. Intermittent hypoxia-induced cognitive deficits are mediated by NADPH oxidase activity in a murine model of sleep apnea. PLoS One. 2011a;6:e19847. doi: 10.1371/journal.pone.0019847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair D, Zhang SX, Ramesh V, Hakim F, Kaushal N, Wang Y, Gozal D. Sleep fragmentation induces cognitive deficits via nicotinamide adenine dinucleotide phosphate oxidase-dependent pathways in mouse. Am J Respir Crit Care Med. 2011b;184:1305–1312. doi: 10.1164/rccm.201107-1173OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanduri J, Yuan G, Kumar GK, Semenza GL, Prabhakar NR. Transcriptional responses to intermittent hypoxia. Respir Physiol Neurobiol. 2008;164:277–81. doi: 10.1016/j.resp.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obal F, Jr, Krueger JM. GHRH and sleep. Sleep Med Rev. 2004;8:367–377. doi: 10.1016/j.smrv.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Polotsky VY, Rubin AE, Balbir A, Dean T, Smith PL, Schwartz AR, O’Donnell CP. Intermittent hypoxia causes REM sleep deficits and decreases EEG delta power in NREM sleep in the C57BL/6J mouse. Sleep Med. 2006;7:7–16. doi: 10.1016/j.sleep.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Row BW, Kheirandish L, Cheng Y, Rowell PP, Gozal D. Impaired spatial working memory and altered choline acetyltransferase (CHAT) immunoreactivity and nicotinic receptor binding in rats exposed to intermittent hypoxia during sleep. Behav Brain Res. 2007;177:308–314. doi: 10.1016/j.bbr.2006.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Row BW, Liu R, Xu W, Kheirandish L, Gozal D. Intermittent hypoxia is associated with oxidative stress and spatial learning deficits in the rat. Am J Respir Crit Care Med. 2003;167:1548–1553. doi: 10.1164/rccm.200209-1050OC. [DOI] [PubMed] [Google Scholar]
- Shan X, Chi L, Ke Y, Luo C, Qian S, Gozal D, Liu R. Manganese superoxide dismutase protects mouse cortical neurons from chronic intermittent hypoxia-mediated oxidative damage. Neurobiol Dis. 2007;28:206–215. doi: 10.1016/j.nbd.2007.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szentirmai E, Yasuda T, Taishi P, et al. Growth hormone-releasing hormone: cerebral cortical sleep-related EEG actions and expression. Am J Physiol Regul Integr Comp Physiol. 2007;293:R922–930. doi: 10.1152/ajpregu.00237.2007. [DOI] [PubMed] [Google Scholar]
- Szollosi I, Jones M, Morrell MJ, Helfet K, Coats AJ, Simonds AK. Effect of CO2 inhalation on central sleep apnea and arousals from sleep. Respiration. 2004;71:493–498. doi: 10.1159/000080634. [DOI] [PubMed] [Google Scholar]
- Tarasiuk A, Berdugo-Boura N, Troib A, Segev Y. Role of growth hormone-releasing hormone in sleep and growth impairments induced by upper airway obstruction in rats. Eur Respir J. 2011;38:870–877. doi: 10.1183/09031936.00197610. [DOI] [PubMed] [Google Scholar]
- Telegdy G, Schally AV. Involvement of neurotransmitters in the action of growth hormone-releasing hormone antagonist on passive avoidance learning. Behav Brain Res. 2012a;233:326–330. doi: 10.1016/j.bbr.2012.05.030. [DOI] [PubMed] [Google Scholar]
- Telegdy G, Schally AV. Neurotransmitter-mediated action of an antagonist of growth hormone-releasing hormone on anxiolysis in mice. Behav Brain Res. 2012b;233:232–236. doi: 10.1016/j.bbr.2012.04.011. [DOI] [PubMed] [Google Scholar]
- Telegdy G, Tanaka M, Schally AV. Effects of the growth hormone-releasing hormone (GH-RH) antagonist on brain functions in mice. Behav Brain Res. 2011;224:155–158. doi: 10.1016/j.bbr.2011.05.036. [DOI] [PubMed] [Google Scholar]
- Thornton PL, Ingram RL, Sonntag WE. Chronic [D-Ala2]-growth hormone-releasing hormone administration attenuates age-related deficits in spatial memory. J Gerontol A Biol Sci Med Sci. 2000;55:B106–112. doi: 10.1093/gerona/55.2.b106. [DOI] [PubMed] [Google Scholar]
- Vitiello MV, Moe KE, Merriam GR, Mazzoni G, Buchner DH, Schwartz RS. Growth hormone releasing hormone improves the cognition of healthy older adults. Neurobiol Aging. 2006;27:318–323. doi: 10.1016/j.neurobiolaging.2005.01.010. [DOI] [PubMed] [Google Scholar]
- Wang T, Hai J, Chen X, Peng H, Zhang H, Li L, Zhang Q. Inhibition of GHRH aggravated acetaminophen-induced acute mice liver injury through GH/IGF-I axis. Endocr J. 2012;59:579–587. doi: 10.1507/endocrj.ej11-0356. [DOI] [PubMed] [Google Scholar]
- Wilcox PG, Pare PD, Road JD, Fleetham JA. Respiratory muscle function during obstructive sleep apnea. Am Rev Respir Dis. 1990;142:533–539. doi: 10.1164/ajrccm/142.3.533. [DOI] [PubMed] [Google Scholar]
- Yuan G, Nanduri J, Khan S, Semenza GL, Prabhakar NR. Induction of HIF-1alpha expression by intermittent hypoxia: involvement of NADPH oxidase, Ca2+ signaling, prolyl hydroxylases, and mTOR. J Cell Physiol. 2008;217:674–85. doi: 10.1002/jcp.21537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan G, Khan SA, Luo W, Nanduri J, Semenza GL, Prabhakar NR. Hypoxia-inducible factor 1 mediates increased expression of NADPH oxidase-2 in response to intermittent hypoxia. J Cell Physiol. 2011;226:2925–33. doi: 10.1002/jcp.22640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Chi L, Row BW, et al. Increased oxidative stress is associated with chronic intermittent hypoxia-mediated brain cortical neuronal cell apoptosis in a mouse model of sleep apnea. Neuroscience. 2004;126:313–323. doi: 10.1016/j.neuroscience.2004.03.055. [DOI] [PubMed] [Google Scholar]
- Zomkowski AD, Santos AR, Rodrigues AL. Evidence for the involvement of the opioid system in the agmatine antidepressant-like effect in the forced swimming test. Neurosci Lett. 2005;381:279–283. doi: 10.1016/j.neulet.2005.02.026. [DOI] [PubMed] [Google Scholar]