

Abstract
Diabetes increases the risk of bone fracture. Organic and inorganic bone extracellular matrix components determine bone strength. Previous studies indicate that in diabetes, glycation of collagen causes abnormal arrangements of collagen molecules and fragile bones. Diabetic bone fragility is additionally attributed to reduced levels of lysyl oxidase enzyme-dependent collagen cross-links. The mechanism underlying the presence of lower enzymatic collagen cross-links in diabetic bone has not been directly investigated. Here we determine in primary osteoblast cultures the regulation of lysyl oxidase protein by type I collagen and collagen modified by carboxymethylation (CML-collagen), a form of advanced glycation endproducts. Data indicate that non-glycated collagen up-regulates lysyl oxidase levels both in primary non-differentiated and in differentiating mouse and rat osteoblast cultures, while CML-collagen fails to regulate lysyl oxidase in these cells. Collagen binding to Discoidin Domain Receptor-2 (DDR2) mediates lysyl oxidase increases, determined in DDR2 shRNA knockdown studies. DDR2 binding and activation were disrupted by collagen glycation, pointing to a mechanism for the diminished levels of lysyl oxidase and consequent low lysyl oxidase-derived cross-links in diabetic bone. Our studies indicate that collagen-integrin interactions may not play a major role in up-regulating lysyl oxidase. Furthermore, non-collagenous ligands for the receptor for advanced glycation end products (RAGE) failed to alter lysyl oxidase levels. Taken together with published studies a new understanding emerges in which diabetes- and age-dependent inhibition of normal collagen-stimulated DDR2- and integrin-signaling, and independent advanced glycation-stimulated RAGE-signaling, each contributes to different aspects of diabetic osteopenia.
Keywords: Discoidin Domain Receptor 2, Lysyl Oxidase, Osteoblasts, Advanced Glycation Endproducts, Diabetes
1. Introduction
Diabetes mellitus is characterized by impaired glucose metabolism and a doubled risk of bone fracture [1]. The pathogenesis of diabetic bone fragility can be broadly divided into cellular and extracellular matrix (ECM) aspects. Cellular complications include diminished osteoblast differentiation [2], increased osteoblast apoptosis [3], and increased differentiation of mesenchymal stromal cells into adipocytes at the expense of osteoblasts [4, 5]. Together, these cellular events down-regulate bone formation in diabetes, and promote diabetic osteopenia.
In addition to cellular abnormalities, alterations in the organic extracellular matrix of bone are typical in diabetes. Collagen is the most abundant protein in bone organic matrix, and it undergoes intra- and extracellular post-translational modifications in order to help form a functional extracellular matrix. To stabilize collagen fibrils, lysyl oxidase catalyzes the oxidative deamination of lysine or hydroxylysine residues in the collagen telopeptide regions resulting in formation of peptidyl aldehydes. Lysine or hydroxylysine-derived aldehydes spontaneously react to form intra- and intermolecular cross-links between collagen molecules [6]. Lysyl oxidase-dependent collagen cross-linking is essential for bone strength [7].
Elevated levels of glucose in diabetes result in non-enzymatic modifications of extra- and intra-cellular proteins. Aldoses, ketoses and oxidized lipids react with basic residues of proteins, and initiate a complex non-enzymatic cascade of modifications resulting in heterogeneous products collectively known as advanced glycation end products (AGEs). Some AGEs interfere with the function of proteins by causing non-enzymatic cross-linking. Long-lived proteins such as type I collagen accumulate higher levels of AGEs than short-lived proteins. Non-enzymatic AGE modification and cross-linking of collagen molecules interferes with the normal structure of collagen fibrils, which reduces bone strength in diabetes [8].
Saito et al. reported that diabetic bone has lower lysyl oxidase-derived enzymatic cross-links in collagen compared to non-diabetic bone, based on studies performed in a rat model [9]. Lysyl oxidase plays a pivotal role in bone development and function, as revealed in classical studies [10, 11]. Lysyl oxidase inhibition results in weak bones and joints known as osteolathyrism [12]. Despite evidence for reduced levels of lysyl oxidase-dependent collagen cross-links in diabetic bone, it remains to be determined whether lysyl oxidase itself is regulated under diabetic conditions in bone. In this study, we investigated a role for collagen glycation in regulating lysyl oxidase expression in primary osteoblast cultures. In light of previous work, our initial hypothesis was that AGEs would actively down-regulate lysyl oxidase by an AGE-receptor mediated mechanism [13]. By contrast, we present novel data showing that non-glycated (normal) collagen up-regulates levels of lysyl oxidase; a function we ascribe to physiological (unmodified) collagen. The two major classes of collagen receptors are integrins and the discoidin domain receptors, respectively [14]. Collagen up-regulation was determined to be mediated by Discoidin Domain Receptor 2 (DDR2), with possible modulation by integrin signaling only in non-differentiating osteoblasts. Interestingly, collagen glycation was seen to attenuate its ability to both up-regulate lysyl oxidase and bind to and activate DDR2. These data have important implications regarding mechanisms which contribute to connective tissue complications of diabetes.
2. Materials and Methods
2.1. Primary mouse and rat osteoblast isolation
The Boston University Institutional Animal Care and Use Committee (IACUC) approved all animal protocols. Postnatal (1–3 days) BALB/cByJ mice (Jackson Laboratory) were euthanized, and calvaria, excluding the occipital bone, were surgically dissected. Harvested bones were sequentially digested for three cycles (20, 20, and 90 minutes) using 15 ml of a collagen-trypsin cocktail (0.1% collagenase P and 7.5% of 2.5% trypsin) in PBS, with the third digest being enriched for osteoblasts as originally described [15] and previously carried out in our laboratory [16]. For each digestion cycle, suspended bone tissues were incubated at 37° C with gentle horizontal rotation. Bone tissues were washed with PBS and re-suspended in fresh cocktail between each cycle. After the final 90 minute digestion, the mixture containing residual bones was centrifuged at 700 x g for 10 minutes at 4° C. The supernatant liquid and residual bones were carefully discarded, and the cell pellet containing primarily osteoblasts was re-suspended in growth medium. Isolated primary cells were counted and seeded at 2 × 106 cells per 100 mm diameter culture dish. After 3 or 4 days, cells were passaged and plated for each experiment as described in Results.
CD®IGS 001 rats (Charles River Laboratory) were used to isolate rat osteoblasts. The calvaria of 16–18 days old embryonic rats were dissected using a pair of miniature scissors. Rat primary osteoblasts were isolated following the same protocol as mouse cells, expect that rat cells were seeded at 5 × 105 cells per 100 mm diameter culture dish.
2.2. Cell culture
Rat and mouse primary osteoblasts were each respectively grown in growth medium (α-MEM medium supplemented with 0.1 mM nonessential amino acids, 10% fetal bovine serum, and 100 Units/ml Penicillin and 0.1 mg/ml Streptomycin) at 37° C and 5% CO2 in a fully humidified incubator. For experiments with non-differentiated osteoblasts, cells were grown to 80% visual confluence. The medium was then replaced with serum-free medium containing 0.1% bovine serum albumin for a minimum of 12 hours. Cells were then treated with either experimental or control media (see Results).
To induce differentiation, growth medium was replaced with differentiation medium [growth medium containing freshly prepared ascorbic acid (50 μg/ml), β-glycerophosphate (10 mM), and dexamethasone (10 nM)] at 100% visual confluence. During the course of differentiation the media were refreshed every second day. Experiments with differentiating rat and mouse osteoblasts were conducted on 7-day differentiating cells. This time point corresponds to a phase of osteoblast development in which lysyl oxidase levels and collagen deposition are both increasing ([17] and Figure S1). On day 7, cells were then treated under the experimental conditions described in Results.
2.3. Antibodies
Affinity purified rabbit anti-lysyl oxidase antibody was prepared according to a previously described report [18], and then affinity purified using a CYDTYERPRSGSRHRPG peptide affinity column. This peptide sequence is unique to lysyl oxidase and is not shared by any other lysyl oxidase isoform. This antibody was used at a concentration of 1 μg/ml for Western blots. Western blot signals were visualized with an anti-rabbit horseradish peroxidase coupled IgG (Cell Signaling cat#7074) at a 1:3000 dilution. Neutralizing anti-RAGE (cat#8230) and normal goat IgG (cat#S2028) antibodies were purchased from Santa Cruz Biotechnology. Anti-DDR2 antibody (Abcam cat#ab126773) was used at a 1:1000 dilution. Anti-phospho-FAK (Ty576/577) and anti-FAK antibodies were purchased from Cell Signaling and used at a dilution of 1:1000.
Glycated collagen and bovine serum albumin preparation
Acid soluble calf skin type I collagen (Sigma cat#3515) was chemically modified to generate N-ε-(carboxymethyl)-lysine-collagen (CML-collagen) and control collagen as we have previously described [19, 20]. Collagen was dissolved in 1 mM HCl, and was then divided into two batches: Non-glycated control-collagen, and CML-collagen, respectively. To prepare CML-collagen, dissolved collagen was incubated overnight at 37° C with 0.45 M sodium cyanoborohydride and 155 mM sodium glyoxylate in 0.2 M sodium phosphate buffer pH 7.8. The control collagen batch was incubated identically but without sodium glyoxylate, thereby making CML-modification impossible. Control- and CML-collagen were then dialyzed against fresh deionized water three times for 12 hours each time. Successful modification was indicated by slower mobility of CML-collagen on SDS-PAGE gels and by its high immunoreactivity with anti-CML antibody (R&D system cat#3247) compared to control-collagen (Figure 1A). Circular dichroism analyses of non-modified and modified collagen resulted in identical spectra (Figure 1B). The intact collagen triple helix has an unique spectrum [21]. Thus, data indicate that the chemical modification did not disrupt the triple helical structure in CML collagen (Figure 1B). Circular dichroism spectra were obtained using an AVIV-400 spectropolarimeter with thermoelectric temperature control at 37° C at a data accumulation rate of 10 sec/nm. Amino acid analysis of CML- and control collagen samples after acid hydrolysis was carried out by Ansynth Service, Roozendaal, Netherlands further revealed 1.3 residues of carboxymethyl lysine per 1000 amino acids in CML collagen which is equivalent to modification of 5% of the 26 lysine residues per 1000 residues determined in control collagen. This value is in the range of physiologic levels of lysine modification seen in aging and diabetes for long lived proteins which approaches 10% modification of lysine plus hydroxylysine [22]. Fraction V bovine serum albumin (EMD Biosciences cat#126609) was subjected to the same protocol and CML modification was confirmed by SDS PAGE and Western blot (Figure 1C).
Figure 1. Characterization of CML- and control collagen and bovine serum albumin.
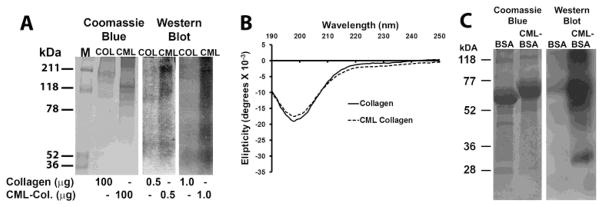
(A) Control and CML-collagen were prepared as described in Materials and Methods and subjected to 6% SDS PAGE and either stained with Coomassie Blue, or subjected to Western blotting for CML content. (B) circular dichroism of control and CML-collagen at a concentration of 0.2 mg/ml in 20 mM sodium phosphate buffer pH 7.4, 37° C. (C) SDS PAGE and Western blot for CML content of control- and CML-BSA.
2.4. Cell treatment with collagen or glycated collagen
Osteoblasts were serum-depleted using serum-free growth medium supplemented with 0.1% BSA for 18 hours. Cells were then treated with collagen, CML-collagen, BSA, CML-BSA or vehicle and cell lysates were collected in SDS PAGE sample buffer and analyzed as described in the Results section.
2.5. Lentivirus transduction
A set of 5 DDR2 shRNAs (Open Biosystems cat# RMM4534) were screened in both rat and mouse osteoblasts. TRCN0000023549 clone for mouse osteoblasts and TRCN0000023553 clone for rat osteoblasts, respectively knocked down DDR2 protein by 60 –80%, assessed by Western blot analysis. Lentiviral transduction of primary osteoblasts was executed as follows: Human 293T cells were transfected with either DDR2 shRNA plasmid or control shRNA plasmid (target sequence: CCTAAGGTTAAGTCGCCCTCG; Addgene, #1864), in the presence of the packaging plasmids (VSVG and Delta-R 8.2; Addgene), using FuGene 6 (Roche# 11914443001) [23]. A firefly luciferase shRNA plasmid was used as the negative control [24]. Media containing viral particles were collected on days 3 and 5 post-transfection. Collected media were pooled and centrifuged at 40,000 x g and the viral pellet was suspended in a small volume of PBS. DDR2 shRNA or control shRNA viral particles titration analysis in the presence of 4 μg/ml puromycin indicated an optimal dose of 19 × 108 transduction units/ml (TU/ml) for 1 × 106 primary osteoblasts [25]. In knockdown experiments, primary osteoblasts were transduced with DDR2 shRNA or control shRNA at 80–90% visual confluence for 24 hours. The medium was then refreshed to include 4 μg/ml puromycin to select for transduced cells. After two days, transduced cells were serum-depleted for 18 hours and treated with vehicle or collagen as explained in the Results for each experiment. In experiments performed with differentiating cells, the shRNA was transduced just before culturing cells in differentiation medium without loss of cell viability.
2.6. ELISA for DDR2 activation
Cells were treated with 0.2 mg/ml collagen or CML-collagen, and then lysed [1% NP-40, 20 mM Tris (pH 8.0), 137 mM NaCl, 10% glycerol, 2 mM EDTA, 1 mM activated sodium orthovanadate, 1:100 Sigma proteinase inhibitor cocktail]. Total protein was measured using the BCA assay (Pierce cat#23225), and (15 μg) aliquots were used in ELISA kits for phosphorylated DDR2 and total DDR2 (R&D system # DYC6170 and # DYC2538, respectively). The same capture anti-DDR2 antibody is used in both kits and 100 μL of 4 μg/ml in PBS was immobilized on a 96 well plate (Costar Cat # 2592) overnight at the room temperature. After 5 washes of 400 μL each with PBS containing 0.05% Tween 20, the plate was blocked with the blocking buffer (PBS containing 1% BSA) for three hours. Next, wells were washed once and cell lysates or known amounts of recombinant phosphorylated or non-phosphorylated DDR2 standards were respectively applied. The plate was incubated at room temperature for 5 hours and then washed 5 times. HRP-conjugated anti-total DRR2 detection antibody (#DYC2538, R&D Systems) or anti-phospho-tyrosine DDR2 detection antibody (#DYC6170 R&D Systems) (1:2000) were added and incubated for three hours. The plate was next washed six times before incubating with the chromogenic substrate (tetramethylbenzidine) to detect the respective levels of total and phosphorylated DDR2. Standard curves were generated and the relative amounts of phosphorylated- and total DDR2 present in each lysate were calculated.
2.7. Collagen-DDR2 binding assay
A previously published collagen-DDR2 binding assay was employed with modifications [14]. Control collagen or CML-collagen (100 μL at 10 μg/ml per well in PBS) was immobilized on a 96-well plate overnight at room temperature. After washing (PBS containing 0.05% Tween 20), wells were blocked with 150 μL of 1 mg/ml BSA in PBS for 1 hour at room temperature. Recombinant DDR2-(His)6 (30 pmole) (Creative Biomart cat#773M) was serially diluted in the incubation buffer (PBS with 0.5 mg/ml BSA) in a final volume of 50 μL and added to each well. The plate was incubated at room temperature for 3 hours on a gently shaking horizontal rotator. Wells were washed 6 times with the washing buffer, and incubated for 2 hours with 1:2000 diluted HRP conjugated anti-His6 antibody (Abcam #22226) in the incubation buffer in a final volume of 50 μL. The level of bound DDR2 was detected using 100 μl of chromogenic substrate (tetramethylbenzidine). The reaction was stopped with 100 μl of 2 M sulfuric acid after 20 minutes. Absorbance of samples at 450 nm was determined with a Berthold plate reader. All binding assays were carried out in six replicates and showed less than 15% variation.
Independent control analyses confirmed that the wells contained equivalent levels of bound control and CML-collagen. Briefly, control- and CML-collagen were biotinylated using an EZ-link Amine-PEG2-Biotin (Pierce cat#21346), and biotin/total collagen measured. Next, 1 μg of biotinylated control- and CML-collagen were respectively immobilized on a 96-well plate (24 wells per group) overnight. Wells were then washed five times with PBS containing 0.05% Tween 20, and were incubated with an anti-biotin HRP conjugated (Cell signaling cat#7075) for 2 hours. The plate was washed five times and the levels of HRP were detected using a chromogenic substrate (tetramethylbenzidine) kit. Biotin levels indicated that equivalent relative amounts of control and CML-collagen bound the plates as expected.
2.8. Densitometry analysis
Digital photos from the X-ray films were captured using a VersaDoc Imaging System (Biorad). Densitometry analysis was carried using the gel analyzer feature of the Image J software (http://rsbweb.nih.gov/ij/) according to the user manual (IJ version 1.46r, section 30.13). In brief, the density of lanes on a blot was calculated using Gel Analyzer feature of the Image J. This feature creates a density plot vs distance in each lane, permitting quantitative integration of peaks. Then, the areas of peaks were integrated after subtracting the background baseline. The percent area for each peak (band) was then calculated and normalized to an endogenous housekeeping protein (β-actin or tubulin). Moreover, films with non-saturated exposure were employed for densitometry. Finally, data were validated by simple visual observation to ensure accuracy.
2.9. Statistical analysis
Student’s t-test or ANOVA with post-hoc Tukey at 95% confidence was employed in our analyses. Unequal variance (heteroscedastic) assumption was set in the Student’s t-test since the standard deviations varied for groups which the null hypothesis was tested in all our experiments. Significance was declared at p<0.05.
3. Results
3.1. Collagen regulation of lysyl oxidase
To investigate the mechanism underlying the reduced levels of lysyl oxidase enzyme-derived collagen cross-links in diabetic bones, we determined the regulation of lysyl oxidase by control- and CML-collagen in primary osteoblasts. CML-collagen is an advanced glycation end product that activates RAGE signaling in osteoblasts, whereas non-glycated collagen does not [3]. Isolated primary mouse osteoblasts were cultured under non-differentiating conditions, serum-depleted and then treated with 0.2 mg/ml collagen, CML-collagen, or vehicle for 24 hours and cell lysates were subjected to Western blot analyses. Data indicate that non-glycated control collagen up-regulated mature 32 kDa lysyl oxidase protein 2-fold, while CML-collagen had no effect (Figure 2A). Glycosylated and non-glycosylated pro-lysyl oxidase (~50 kDa) [26] was similarly up-regulated by collagen (Figure 2A). These data suggest that CML-collagen effects on lysyl oxidase levels are independent of RAGE signaling.
Figure 2. Collagen up-regulates lysyl oxidase in non-differentiated primary mouse osteoblasts, while glycated collagen fails to induce lysyl oxidase. The RAGE ligand CML-BSA does not down-regulate lysyl oxidase.

(A) Non-differentiated primary mouse osteoblasts were serum depleted and then treated with control collagen (0.2 mg/ml) or CML-collagen (glycated collagen; 0.2 mg/ml). Total protein was extracted and subjected to Western blotting for 50 kDa pro-lysyl oxidase and mature 32 kDa lysyl oxidase and β-actin (loading control). The bar graph shows regulation of both 50 kDa and 32 kDa lysyl oxidase protein levels in non-differentiated mouse primary osteoblasts in response to collagen or CML-collagen (n=3). Data are from one of three independent experiments with the same outcome. Data are mean ± SD (*, p<0.05, N.S., not significant; Student’s t-test). (B) Non-differentiated primary mouse osteoblasts were serum-depleted and then treated with BSA (0.2 mg/ml) or CML-BSA (0.2 mg/ml) for 24 hours and subjected to Western blotting for pro-lysyl oxidase and mature 32 kDa lysyl oxidase and β-actin (loading control). Data are representative of two independent experiments with the same outcome. Data are mean ± SD (N.S., not significant; Student’s t-test). (C) Primary calvarial mouse osteoblasts at 80% visual confluence were serum starved for 18 hours and treated with BSA or CML-BSA for either 15 or 30 minutes. Total protein from cell layers were collected and subjected to Western blotting to measure the phosphorylation levels of ERK, an indicator of AGE-RAGE axis activation. The graph shows the results of the densitometry analysis. Data are presented as means ± SD (n=3; *, p<0.05). Data are from one of two independent experiments with the same outcomes.
3.2. Lysyl oxidase is not down-regulated by a RAGE ligand
To independently investigate the apparent lack of CML-collagen/RAGE-mediated down-regulation of lysyl oxidase, we prepared CML-modified albumin, a known non-collagenous ligand for RAGE that we and others have previously shown activates RAGE [3, 20] and that inhibits bone healing in-vivo [13]. Non-differentiated primary mouse osteoblasts were treated with CML-BSA and control non-glycated-BSA for 24 hours. Western blotting analysis showed no difference in ~50 kDa or 32 kDa lysyl oxidase protein levels in cells treated with CML-BSA compared to control-BSA (Figure 2B), confirming that AGE-initiated signaling does not regulate lysyl oxidase protein expression in non-differentiated primary mouse osteoblasts. The activation of AGE-RAGE signaling by CML-BSA was verified by observing increased levels of phosphorylated ERK1/2 levels in CML-BSA-treated cells compared to control BSA (Figure 2C) [13].
3.3. DDR2 mediates collagen induction of lysyl oxidase
To elucidate the mechanism by which collagen up-regulates lysyl oxidase in non-differentiated primary mouse osteoblasts, we investigated roles for different collagen receptors. Discoidin Domain Receptor-2 and collagen-binding integrins (α1β1, α2β1, α10β1, and α11β1) represent two distinct collagen receptor classes in bone. We initially focused on DDR2. The ability of DDR2 to mediate collagen up-regulation of lysyl oxidase in osteoblasts was evaluated using a lentiviral shRNA against DDR2 as indicated in Experimental Procedures. DDR2 protein levels were reduced by approximately 60% with DDR2 shRNA compared to cells transduced with a control shRNA (Figure 3A). The transduced cells were next treated with 0.2 mg/ml of collagen or the vehicle for 24 hours. Western blots of cell layer extracts indicate that collagen up-regulated 32 kDa and 48 – 50 kDa pro-lysyl oxidase protein expression nearly 3-fold in control shRNA transduced cells, while lysyl oxidase levels were not increased significantly above vehicle control levels in DDR2 knocked-down osteoblasts (Figure 3B). These data indicate that collagen induction of lysyl oxidase is mediated by DDR2 in primary osteoblast cultures. Taken together, data presented above constitute the first direct determination that collagen regulates lysyl oxidase, and that this regulation is mediated by DDR2.
Figure 3. Collagen up-regulates lysyl oxidase via Discoidin Domain Receptor 2 (DDR2) in non-differentiated primary mouse osteoblasts.

Primary mouse osteoblast cultures were transduced with DDR2 shRNA lentivirus and a control shRNA lentivirus. Cells were serum-depleted and then treated with control collagen (0.2 mg/ml) for 24 hours and cell layer proteins were subjected to Western Blotting for (A) DDR2 and (B) 50 kDa prolysyl oxidase and mature 32 kDa lysyl oxidase. Data are means +/− SD, (*, p<0.05, N.S., not significant; ANOVA with post-hoc Tukey; n = 3) from one of two representative experiments with the same outcomes.
3.4. Integrin or DDR2-mediated collagen up-regulation of lysyl oxidase in non-differentiated osteoblasts
Next, we investigated a role for integrins in the up-regulation of lysyl oxidase by collagen. Cells were treated as described above with control collagen in the presence and absence of 2 mM EDTA which inhibits all integrin signaling in live cell cultures [27]. Western blot analyses of cell layer extracts indicate that collagen significantly up-regulated both 50 kDa pro-lysyl oxidase and 32 kDa mature lysyl oxidase protein levels in the presence or absence of EDTA (Figure 4A). To establish that EDTA in fact inhibited collagen-stimulated integrin signaling, activation of downstream focal adhesion kinase (FAK) was measured in the presence and absence of EDTA. FAK phosphorylation on Tyr-576/Tyr-577 was used as an index of collagen-induced integrin signaling activation [28, 29]. Western blot analysis showed that in the presence of 2 mM EDTA FAK phosphorylation was not significantly stimulated by collagen, while significant stimulation was observed in the absence of EDTA confirming that integrin signaling was mostly blocked by EDTA. (Figure 4B). Taken together, data in Figures 3 and 4 suggest that collagen up-regulation of lysyl oxidase depends primarily on DDR2, with a smaller role played by integrin signaling in these non-differentiated osteoblasts.
Figure 4. Integrin signaling partly mediates collagen up-regulation of lysyl oxidase in non-differentiated primary mouse osteoblasts.

Non-differentiated primary mouse osteoblasts were serum-depleted and then treated with control collagen (0.2 mg/ml) for 24 hours (A) or 9 hours (B) in the presence or absence of 2 mM EDTA. (A) Cell layer protein was extracted and subjected to Western blotting for 50 kDa pro-lysyl oxidase, mature 32 kDa lysyl oxidase and β-actin (loading control). Data are means +/− SD, (*, p<0.05; n = 3) from one of two representative experiments with the same outcomes. (B) Western blotting for Tyr-576/Tyr-577 phosphorylated FAK, total FAK, and β-actin (loading control). Data are means +/− SD, (*, p<0.05, N.S., not significant; ANOVA with post-hoc Tukey; n = 3).
3.5. Collagen glycation interferes with its binding to DDR2
We next reasoned that glycation of collagen could interfere with collagen-DDR2 binding and thereby block its ability to up-regulate lysyl oxidase in osteoblasts. To examine this idea, we compared recombinant DDR2 binding to collagen and CML-collagen. Control non-glycated and glycated collagens first were respectively immobilized on 96-well plates and incubated with increasing amounts of soluble rDDR2. Independent control analyses confirmed that the wells contained equivalent levels of bound control and CML-collagen (Experimental Procedures). The extent of rDDR2 binding to non-glycated control and glycated collagen was measured as explained in the Experimental Procedures. Data (Figure 5A) reveal that DDR2 binding to collagen exceeded its ability to bind CML-collagen above 10 pmoles/well. DDR2 binding to CML-collagen was only 40% of the level bound to collagen at 30 pmol DDR2 (Figure 5A). These data support that glycation of collagen interferes with DDR2-collagen binding.
Figure 5. Glycation of collagen interferes with collagen-DDR2 binding and does not activate collagen-DDR2 signaling.

(A) Collagen or CML-collagen (10 μg/ml) was coated on wells of a 96-well plate. Increasing amounts of horseradish peroxidase-conjugated recombinant DDR2 was incubated for three hours at room temperature. The plate was washed and the relative levels of bound rDDR2 were measured utilizing a chromogenic substrate (Experimental Procedures). Data show binding curves for rDDR2 to collagen (solid line) and rDDR2 to CML-collagen (dotted line) and are means +/− SD (n = 6; p<0.05; Student’s t-test). Data are from one of two experiments with the same outcomes. (B) Non-differentiated primary mouse osteoblasts were serum-depleted and then treated with collagen (0.2 mg/ml). Total protein was extracted, and the phospho-DDR2 levels and total DDR 2 were measured using a phospho- and total-DDR2 ELISA kits. Collagen increased the ratio of phospho-DDR2 to DDR2, while CML-collagen did not induce DDR2 phosphorylation. Data are from one of two independent experiments with the same outcomes (n=3; *, p<0.05, N.S., not significant; Student’s t-test).
3.6. CML-collagen does not activate collagen-DDR2 signaling
In order to verify the results of the binding assays, we next assessed the activation of the collagen-DDR2 axis by determining the relative degree of DDR2 tyrosine phosphorylation as a function of collagen or CML-collagen treatment. DDR2 phosphorylation, unlike that of other tyrosine receptor kinase receptors, occurs with slow and sustained kinetics of phosphorylation detectable 30 minutes after ligand binding and remains at a high steady-state level for at least 18 hours [30]. Non-differentiated primary mouse osteoblasts were treated with 0.2 mg/ml collagen, CML-collagen or vehicle for 5 hours following serum starvation. Total cell layer proteins were subjected to ELISA analysis for tyrosine-phosphorylated DDR2 and total DDR2 (Experimental Procedures). Data (Figure 5B) show a robust DDR2 phosphorylation following treatment with control non-glycated collagen, while no increased DDR2 phosphorylation was seen in response to CML-collagen.
3.7. DDR2 mediates collagen induction of lysyl oxidase in differentiating primary rat osteoblasts
In order to confirm the finding that DDR2 mediates collagen induction of lysyl oxidase, we employed differentiating rat osteoblasts which undergo typical osteoblast differentiation (Figure S1) with knock-down of LOX using an independent shRNA sequence. This experiment controls for any unanticipated off-target effects of the shRNA employed and tests the generality of DDR2 mediation of LOX regulation by collagen by investigating effects of DDR2 knockdown in a different species. Non-differentiated cells were first transduced with lentiviral particles containing DDR2 shRNA or control shRNA and transduced cells were selected with puromycin. The target sequence of DDR2 shRNA used for this experiment was different from the sequence used for non-differentiated primary mouse osteoblasts. DDR2 knockdown was 70% compared to controls as determined by Western blotting (Figure 6A). After seven days of differentiation of confluent cultures, cells were treated with collagen and CML-collagen for 24 hours and then extracted for Western blot analyses. Data show that 24-hour collagen induction of lysyl oxidase in DDR2 knock-down differentiating primary rat osteoblasts was largely suppressed (Figure 6B). Essentially identical data were obtained in differentiating primary mouse osteoblasts in which DDR2 was knocked down with its respective shRNA employed in Figures 2 and 3 (data not shown).
Figure 6. Collagen up-regulation of lysyl oxidase in differentiating primary rat osteoblasts is mediated by DDR2.

DDR2 was knocked down in non-differentiating primary rat osteoblasts using shRNA lentivirus technology with a different shRNA from what was used in experiments with mouse osteoblasts (see Methods). Transduced cells were differentiating for seven days, serum starved and then treated with control collagen (0.2 mg/ml) for 24 hours. Cell layer protein was extracted and subjected to Western blotting for 50 kDa pro-lysyl oxidase, mature 32 kDa lysyl oxidase and β-actin (loading control). (A) Western blot showing 70% knockdown of DDR2 on day 7 of differentiation in knockdown cells; (B) Western blots for pro- and mature lysyl oxidase protein and quantitation. Data (means +/− SD) are from one of two experiments each performed in triplicate with the same outcome (n = 3; *, p<0.05, N.S., not significant; ANOVA with Tukey post-hoc).
Taken together, data demonstrate that collagen glycation interferes with normal collagen/DDR2-dependent up-regulation of lysyl oxidase in osteoblasts, potentially providing insights into a novel mechanism of diminished collagen cross-linking in both aging and diabetic bone that contributes to osteopenia.
4. Discussion
Cellular and extracellular matrix complications contribute to diabetic bone fragility. Specifically, cellular complications disturb bone-remodeling homeostasis through diminished bone formation, leading to weak bones in diabetes. Furthermore, accumulation of AGE adducts on type I collagen, the prime constituent of the organic extracellular matrix of bone, interferes with bone structure and thereby reduces bone strength in diabetes. Evidence for reduced levels of lysyl oxidase-dependent collagen cross-links in rat diabetic bones has also been reported [9]. However, lysyl oxidase regulation in osteoblasts as a function of diabetic conditions has not, to our knowledge, been directly studied. Here we provide evidence for a novel mechanism, which could contribute to diabetic bone fragility. Our studies reveal that lysyl oxidase levels are increased through the interactions of osteoblasts with collagen. Collagen that has been non-enzymatically glycated, as is typical in diabetes, fails to promote lysyl oxidase production by osteoblasts.
This is the first report showing that collagen regulates lysyl oxidase in osteoblasts. In order to understand the mechanism of collagen up-regulation of lysyl oxidase, we investigated roles for the major classes of collagen receptors: integrins and Discoidin Domain Receptors. We demonstrate that DDR2, a receptor expressed on a variety of mesenchymal cells [31], functions to mediate collagen regulation of lysyl oxidase. Moreover, we show that glycated collagen made by chemical modification has a diminished ability to bind and DDR2. In future studies it will be of interest to study the ability of collagens derived from diabetic and non-diabetic bones to stimulate lysyl oxidase levels and activate DDR2 in osteoblasts to further assess the physiological importance of the studies presented here.
Our studies investigating a role for integrins in collagen induction of lysyl oxidase may also suggest a cooperative function for integrins in DDR2-mediated up-regulation of lysyl oxidase by collagen. It is interesting to note, however, that the level of 32 kDa lysyl oxidase in the presence of EDTA was partially reduced compared to its level in the no EDTA controls treated with collagen (Figure 4A). Pro-lysyl oxidase conversion to mature lysyl oxidase is catalyzed by procollagen C-proteinases which are metalloenzymes [32, 33]. We suspect that the effect of EDTA on mature 32 kDa lysyl oxidase levels may be partially due to inhibition of pro-lysyl oxidase processing by EDTA by contrast to inhibiting integrin signaling.
The DDR2-mediated lysyl oxidase induction in osteoblasts reported here is a novel function for this receptor. Various functions have been reported for DDR2. In an in-vitro study, Zhang and colleagues showed that DDR2 activation leads to Runx2 phosphorylation, which regulates osteoblast differentiation in pre-osteoblasts [34]. Mice in which DDR2 has been knocked out show abnormal post-natal bone growth and development [35]. Diminished chondrocyte proliferation was shown as a mechanism explaining this bone phenotype. Craniofacial deformities associated with DDR2 −/− mice [36] indicate other potential possibilities as well, since craniofacial skeletal growth and development is mostly based on intramembranous osteogenesis, which lacks the transient cartilage formation phase. Furthermore, DDR2 mutated mice (Slie/Slie), similar to DDR2 null mice, present with post-natal growth abnormalities in craniofacial and long bones. This spontaneous autosomal-recessive mutation was mapped to an approximately 150 kbp deletion that extended into the DDR2 gene [37]. The mutated mice also presented with gonadal dysfunction. The authors ruled out a central mechanism, namely hypopituitarism, for these complications and suggested local bone tissue-specific pathogenesis and only concentrated on determining a mechanism for the gonadal dysfunction. Our findings that DDR2 mediates collagen regulation of lysyl oxidase provide a new potential mechanism for skeletal complications, which occur in both DDR2 deficient and mutant mice. Our findings combined with studies of DDR2 mutant mice, suggest that DDR2 regulates various cellular and extracellular events in bone growth and development, some of which may depend on lysyl oxidase.
Mice null for the structurally related DDR1 receptor are small in stature [38], and develop osteoarthritis specifically in the temporomandibular joint and not in other joints [39]. Females have mammary gland defects, and exhibit decreased fertility. A role for DDR1 in atherosclerosis development and arterial calcification is known and may be related to its role in regulating the proliferation and differentiation of a variety of cell types including vascular smooth muscle cells and macrophages [40]. A possible role for type I collagen/DDR1 signaling in promoting megakaryocyte development has been reported [41], and contributions to fibrosis in lungs and kidney are known [42, 43]. To our knowledge, possible functions of DDR1 in extracellular matrix production by osteoblasts or in other mineralized extracellular matrix abnormalities have not been explored.
We have shown that the glycation of collagen hinders collagen-DDR2 binding. McCarthy et al. previously showed reduced binding of rat osteosarcoma-derived cells (UMR106) to glycated collagen. The authors speculated that glycation of collagen attenuates collagen binding to collagen-specific integrins on these cells. They showed, however, that glycated collagen does not compete with RGD and DGEA peptides (specific sequence for α1β1 and α2β1 integrins), and proposed that glycation of collagen interferes with different collagen-integrin binding sites [44]. Based on our current study, we suggest that loss of collagen interaction with DDR2 under diabetic conditions could potentially also contribute to the observed loss of cell adhesion. It is interesting that although carboxymethylation of lysine residues in our collagen preparations was about 5%, that a strong inhibition of collagen stimulation of lysyl oxidase was found. DDR2 binding to collagens occurs at a consensus sequence of GVMGFO where O is hydroxyproline [45]. We speculate that regions of collagens in the vicinity of this sequence are likely to be highly susceptible to chemical modifications of lysine residues, suggesting that they may be more exposed to solvent conditions than other regions of collagen, or that other regions of collagen may also bind and activate DDRs [46]. If true, this notion could have important physiological implications with respect to mechanisms of AGE-dependent extracellular matrix complications, including effects on lysyl oxidase regulation and collagen homeostasis in diabetes: disproportionately high AGE modification of regions of collagen which consist of DDR2 ligands would result in a strong down regulation of lysyl oxidase levels.
Global inhibition of integrin signaling with EDTA here suggests that integrin signaling potentially cooperates with DDR2 in collagen induction of lysyl oxidase. This notion is supported by a recent study that shows DDR2 cooperation with specific integrins in cell adhesion to collagen [47]. It is of interest that collagen-specific α1 integrin-ablations in mice manifested as no overt phenotype or anatomical anomalies [48] although bone fracture healing in these mice was impaired [49]. Homozygous mice deficient for collagen-specific α10β1 integrin showed after birth growth plate defects with no morphological skeletal deformities [50]. Integrin α2 deletion in mice revealed no striking anatomical phenotype [51, 52]. In a recent study, mice ablated in α2β1-, α11β1-, or both α2β1-α11β1-integrins were born with no bone phenotype; however, long bone post-natal growth were reduced in mice lacking α11β1- and α2β1-α11β1-integrins. The authors demonstrated that a proportional dwarfism in these mice is a consequence of insufficient systemic IGF-1 levels, and neither osteoblast dysfunction nor growth plate alterations contributed to this phenotype [51]. Thus, collagen-integrin axis activation can be considered to indirectly regulate bone growth and development [51], whereas collagen-activation of DDR2 appears to regulate activities of bone cells which directly synthesize bone extracellular matrix.
We have previously shown that the receptor for advanced glycation end products (RAGE) is expressed by osteoblasts in-vitro and in-vivo and that a RAGE ligand interferes with calvarial bone healing [53]. Combined with the current study, an understanding emerges that AGE-modification of collagen inhibits binding to integrins [44] and DDR2, while independent AGE-activation of RAGE signaling each contributes to bone complications seen in diabetes.
In summary, we have shown that collagen stimulates osteoblasts to increase lysyl oxidase levels, thereby maintaining its steady state normal physiological level. Elevated glycation of collagen, which occurs in aging and in diabetes interferes with DDR2 binding and activation, hence failing to maintain lysyl oxidase levels made by osteoblasts. Previous studies indicate that activation of AGE/RAGE signaling by glycated proteins including collagen leads to increased pro-inflammatory cytokine production, which independently diminishes osteoblast productivity [3]. This new appreciation of multiple mechanisms by which glycation can affect osteoblasts may provide novel opportunities for addressing diabetic osteopenia.
Highlights.
Lysyl oxidase-dependent biosynthetic cross-linking in bone occurs at abnormally low levels in diabetes, contributing to diabetic osteopenia.
Lysyl oxidase was here unexpectedly found to be up-regulated by collagen in primary osteoblast cultures.
Collagen glycation prevented its ability to collagen up-regulate lysyl oxidase.
Collagen up-regulation of lysyl oxidase was found to be primarily mediated by the Discoidin Domain 2 receptor.
The inability of glycated collagen to up-regulate collagen is here proposed to be a potential contributing mechanism for diabetic osteopenia.
Acknowledgments
This work was supported by R01DE14066 to PCT. We are grateful for assistance with circular dichroism studies from Associate Professor Olga Gursky, Department of Physiology and Biophysics, Boston University School of Medicine.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Roozbeh Khosravi, Email: Roozbeh.kh@gmail.com.
Katharine L. Sodek, Email: katharinelise@gmail.com.
Michael Faibish, Email: mfaibish@gmail.com.
Philip C. Trackman, Email: Trackman@bu.edu.
References
- 1.Bouillon R. Diabetic bone disease. Calcif Tissue Int. 1991;49:155– 160. doi: 10.1007/BF02556109. [DOI] [PubMed] [Google Scholar]
- 2.Bouillon R, Bex E, Van Herk E, Laureys J, Dooms L, Lesaffre E, Ravussin E. Influence of age, sex, and insulin on osteoblast dysfunction in diabetes mellitus. J Clin Endocrinol Metab. 1995;80:1194– 1202. doi: 10.1210/jcem.80.4.7714089. [DOI] [PubMed] [Google Scholar]
- 3.Alikhani M, Alikhani Z, Boyd C, Maclellan CM, Raptis M, Liu R, Pischon N, Trackman PC, Gerstenfeld L, Graves DT. Advanced glycation end products stimulate osteoblast apoptosis via the MAP kinase and cytosolic apoptotic pathways. Bone. 2007;40:345–53. doi: 10.1016/j.bone.2006.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Botolin S, Faugere MC, Malluche H, Orth M, Meyer R, McCabe LR. Increased bone adiposity and peroxisomal proliferator-activated receptor-gamma2 expression in type I diabetic mice. Endocrinology. 2005;146:3622–31. doi: 10.1210/en.2004-1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Botolin S, McCabe LR. Bone loss and increased bone adiposity in spontaneous and pharmacologically induced diabetic mice. Endocrinology. 2007;148:198–205. doi: 10.1210/en.2006-1006. [DOI] [PubMed] [Google Scholar]
- 6.Knott L, Bailey A. Collagen cross-links in mineralizing tissues: a review of their chemistry, function, and clinical relevance. Bone. 1998;22:181–187. doi: 10.1016/s8756-3282(97)00279-2. [DOI] [PubMed] [Google Scholar]
- 7.Saito M, Marumo K. Collagen cross-links as a determinant of bone quality: a possible explanation for bone fragility in aging, osteoporosis, and diabetes mellitus. Osteoporos Int. 2010;21:195–214. doi: 10.1007/s00198-009-1066-z. [DOI] [PubMed] [Google Scholar]
- 8.Paul RG, Bailey AJ. Glycation of collagen: the basis of its central role in the late complications of ageing and diabetes. Int J Biochem Cell Biol. 1996;28:1297–310. doi: 10.1016/s1357-2725(96)00079-9. [DOI] [PubMed] [Google Scholar]
- 9.Saito M, Fujii K, Mori Y, Marumo K. Role of collagen enzymatic and glycation induced cross-links as a determinant of bone quality in spontaneously diabetic WBN/Kob rats. Osteoporos Int. 2006;17:1514–23. doi: 10.1007/s00198-006-0155-5. [DOI] [PubMed] [Google Scholar]
- 10.Pinnell SA, Martin GR. The cross-linking of collagen and elastin: enzymatic conversion of lysine in peptide linkage to alpha-amino adipic-delta-semialdehyde (allysine) by an extract from bone. Proc of the Natl Acad Sci, USA. 1968;61:708– 716. doi: 10.1073/pnas.61.2.708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Selye H. Lathyrism. Revue Canadienne de Biologie. 1957;16:3– 82. [PubMed] [Google Scholar]
- 12.Geiger BJ, Steenbock H, Parsons HT. Lathyrism in the rat. J Nutr. 1933;6:427–42. doi: 10.1111/j.1753-4887.1976.tb05778.x. [DOI] [PubMed] [Google Scholar]
- 13.Santana RB, Xu L, Chase HB, Amar S, Graves DT, Trackman PC. A role for advanced glycation end products in diminished bone healing in type 1 diabetes. Diabetes. 2003;52:1502–10. doi: 10.2337/diabetes.52.6.1502. [DOI] [PubMed] [Google Scholar]
- 14.Leitinger B. Molecular analysis of collagen binding by the human discoidin domain receptors, DDR1 and DDR2. Identification of collagen binding sites in DDR2. J Biol Chem. 2003;278:16761–9. doi: 10.1074/jbc.M301370200. [DOI] [PubMed] [Google Scholar]
- 15.Bellows CG, Aubin JE, JNMH, Antosz ME. Mineralized bone nodules formed in vitro from enzymatically released rat calvaria cell populations. Calcif Tissue Int. 1986;38:143–154. doi: 10.1007/BF02556874. [DOI] [PubMed] [Google Scholar]
- 16.Vora SR, Palamakumbura AH, Mitsi M, Guo Y, Pischon N, Nugent MA, Trackman PC. Lysyl oxidase propeptide inhibits FGF-2-induced signaling and proliferation of osteoblasts. J Biol Chem. 2010;285:7384–93. doi: 10.1074/jbc.M109.033597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hong HH, Pischon N, Santana RB, Palamakumbura AH, Chase HB, Gantz D, Guo Y, Uzel MI, Ma D, Trackman PC. A role for lysyl oxidase regulation in the control of normal collagen deposition in differentiating osteoblast cultures. J Cell Physiol. 2004;200:53–62. doi: 10.1002/jcp.10476. [DOI] [PubMed] [Google Scholar]
- 18.Jeay S, Pianetti S, Kagan HM, Sonenshein GE. Lysyl oxidase inhibits ras-mediated transformation by preventing activation of NF-kappa B. Mol Cell Biol. 2003;23:2251–63. doi: 10.1128/MCB.23.7.2251-2263.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kadler KE, Hojima Y, Prockop DJ. Assembly of collagen fibrils de novo by cleavage of the type I pC-collagen with procollagen C-proteinase. Assay of critical concentration demonstrates that collagen self-assembly is a classical example of an entropy-driven process. J Biol Chem. 1987;262:15696–701. [PubMed] [Google Scholar]
- 20.Kislinger T, Fu C, Huber B, Qu W, Taguchi A, Du Yan S, Hofmann M, Yan SF, Pischetsrieder M, Stern D, Schmidt AM. N(epsilon)-(carboxymethyl)lysine adducts of proteins are ligands for receptor for advanced glycation end products that activate cell signaling pathways and modulate gene expression. J Biol Chem. 1999;274:31740–9. doi: 10.1074/jbc.274.44.31740. [DOI] [PubMed] [Google Scholar]
- 21.Greenfield NJ. Using circular dichroism spectra to estimate protein secondary structure. Nat Protoc. 2006;1:2876–90. doi: 10.1038/nprot.2006.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Verzijl N, DeGroot J, Oldehinkel E, Bank RA, Thorpe SR, Baynes JW, Bayliss MT, Bijlsma JW, Lafeber FP, Tekoppele JM. Age-related accumulation of Maillard reaction products in human articular cartilage collagen. Biochem J. 2000;350(Pt 2):381–7. [PMC free article] [PubMed] [Google Scholar]
- 23.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 24.Guo W, Keckesova Z, Donaher JL, Shibue T, Tischler V, Reinhardt F, Itzkovitz S, Noske A, Zurrer-Hardi U, Bell G, Tam WL, Mani SA, van Oudenaarden A, Weinberg RA. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell. 2012;148:1015–28. doi: 10.1016/j.cell.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Geraerts M, Willems S, Baekelandt V, Debyser Z, Gijsbers R. Comparison of lentiviral vector titration methods. BMC Biotechnol. 2006;6:34. doi: 10.1186/1472-6750-6-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trackman PC, Tang J, Bedell-Hogan D, Kagan HM. Posttranslational glycosylation and proteolytic processing of a lysyl oxidase precursor. J Biol Chem. 1992;267:8666– 8671. [PubMed] [Google Scholar]
- 27.Knight CG, Morton LF, Peachey AR, Tuckwell DS, Farndale RW, Barnes MJ. The collagen-binding A-domains of integrins alpha(1)beta(1) and alpha(2)beta(1) recognize the same specific amino acid sequence, GFOGER, in native (triple-helical) collagens. J Biol Chem. 2000;275:35–40. doi: 10.1074/jbc.275.1.35. [DOI] [PubMed] [Google Scholar]
- 28.Calalb MB, Polte TR, Hanks SK. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Mol Cell Biol. 1995;15:954–63. doi: 10.1128/mcb.15.2.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ohkawa Y, Miyazaki S, Hamamura K, Kambe M, Miyata M, Tajima O, Ohmi Y, Yamauchi Y, Furukawa K. Ganglioside GD3 enhances adhesion signals and augments malignant properties of melanoma cells by recruiting integrins to glycolipid-enriched microdomains. J Biol Chem. 2010;285:27213–23. doi: 10.1074/jbc.M109.087791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vogel W. Discoidin domain receptors: structural relations and functional implications. FASEB J. 1999;13 (Suppl):S77–82. doi: 10.1096/fasebj.13.9001.s77. [DOI] [PubMed] [Google Scholar]
- 31.Alves F, Vogel W, Mossie K, Millauer B, Hofler H, Ullrich A. Distinct structural characteristics of discoidin I subfamily receptor tyrosine kinases and complementary expression in human cancer. Oncogene. 1995;10:609–18. [PubMed] [Google Scholar]
- 32.Panchenko MV, Stetler-Stevenson WG, Trubetskoy OV, Gacheru SN, Kagan HM. Metalloproteinase activity secreted by fibrogenic cells in the processing of pro-lysyl oxidase. J Biol Chem. 1996;271:7113– 7119. doi: 10.1074/jbc.271.12.7113. [DOI] [PubMed] [Google Scholar]
- 33.Uzel MI, Scott IC, Babakhanlou-Chase H, Palamakumbura AH, Pappano WN, Hong HH, Greenspan DS, Trackman PC. Multiple bone morphogenetic protein 1-related mammalian metalloproteinases process pro-lysyl oxidase at the correct physiological site and control lysyl oxidase activation in mouse embryo fibroblast cultures. J Biol Chem. 2001;276:22537–43. doi: 10.1074/jbc.M102352200. [DOI] [PubMed] [Google Scholar]
- 34.Zhang Y, Su J, Yu J, Bu X, Ren T, Liu X, Yao L. An essential role of discoidin domain receptor 2 (DDR2) in osteoblast differentiation and chondrocyte maturation via modulation of Runx2 activation. J Bone Miner Res. 2011;26:604–17. doi: 10.1002/jbmr.225. [DOI] [PubMed] [Google Scholar]
- 35.Labrador JP, Azcoitia V, Tuckermann J, Lin C, Olaso E, Manes S, Bruckner K, Goergen JL, Lemke G, Yancopoulos G, Angel P, Martinez C, Klein R. The collagen receptor DDR2 regulates proliferation and its elimination leads to dwarfism. EMBO Rep. 2001;2:446–52. doi: 10.1093/embo-reports/kve094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dullin C, Missbach-Guentner J, Vogel WF, Grabbe E, Alves F. Semi-automatic classification of skeletal morphology in genetically altered mice using flat-panel volume computed tomography. PLoS Genet. 2007;3:e118. doi: 10.1371/journal.pgen.0030118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kano K, Marin de Evsikova C, Young J, Wnek C, Maddatu TP, Nishina PM, Naggert JK. A novel dwarfism with gonadal dysfunction due to loss-of-function allele of the collagen receptor gene, Ddr2, in the mouse. Mol Endocrinol. 2008;22:1866–80. doi: 10.1210/me.2007-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vogel WF, Aszodi A, Alves F, Pawson T. Discoidin domain receptor 1 tyrosine kinase has an essential role in mammary gland development. Mol Cell Biol. 2001;21:2906–17. doi: 10.1128/MCB.21.8.2906-2917.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schminke B, Muhammad H, Bode C, Sadowski B, Gerter R, Gersdorff N, Burgers R, Monsonego-Ornan E, Rosen V, Miosge N. A discoidin domain receptor 1 knock-out mouse as a novel model for osteoarthritis of the temporomandibular joint. Cell Mol Life Sci. 2013 doi: 10.1007/s00018-013-1436-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ahmad PJ, Trcka D, Xue S, Franco C, Speer MY, Giachelli CM, Bendeck MP. Discoidin domain receptor-1 deficiency attenuates atherosclerotic calcification and smooth muscle cell-mediated mineralization. Am J Pathol. 2009;175:2686–96. doi: 10.2353/ajpath.2009.080734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abbonante V, Gruppi C, Rubel D, Gross O, Moratti R, Balduini A. Discoidin domain receptor 1 protein is a novel modulator of megakaryocyte-collagen interactions. J Biol Chem. 2013;288:16738–46. doi: 10.1074/jbc.M112.431528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gross O, Girgert R, Beirowski B, Kretzler M, Kang HG, Kruegel J, Miosge N, Busse AC, Segerer S, Vogel WF, Muller GA, Weber M. Loss of collagen-receptor DDR1 delays renal fibrosis in hereditary type IV collagen disease. Matrix Biol. 2010;29:346–56. doi: 10.1016/j.matbio.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 43.Avivi-Green C, Singal M, Vogel WF. Discoidin domain receptor 1-deficient mice are resistant to bleomycin-induced lung fibrosis. Am J Respir Crit Care Med. 2006;174:420–7. doi: 10.1164/rccm.200603-333OC. [DOI] [PubMed] [Google Scholar]
- 44.McCarthy AD, Uemura T, Etcheverry SB, Cortizo AM. Advanced glycation endproducts interefere with integrin-mediated osteoblastic attachment to a type-I collagen matrix. Int J Biochem Cell Biol. 2004;36:840–8. doi: 10.1016/j.biocel.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 45.Konitsiotis AD, Raynal N, Bihan D, Hohenester E, Farndale RW, Leitinger B. Characterization of high affinity binding motifs for the discoidin domain receptor DDR2 in collagen. J Biol Chem. 2008;283:6861–8. doi: 10.1074/jbc.M709290200. [DOI] [PubMed] [Google Scholar]
- 46.Fu HL, Valiathan RR, Arkwright R, Sohail A, Mihai C, Kumarasiri M, Mahasenan KV, Mobashery S, Huang P, Agarwal G, Fridman R. Discoidin domain receptors: unique receptor tyrosine kinases in collagen-mediated signaling. J Biol Chem. 2013;288:7430–7. doi: 10.1074/jbc.R112.444158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu H, Bihan D, Chang F, Huang PH, Farndale RW, Leitinger B. Discoidin domain receptors promote alpha1beta1- and alpha2beta1-integrin mediated cell adhesion to collagen by enhancing integrin activation. PLoS One. 2012;7:e52209. doi: 10.1371/journal.pone.0052209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gardner H, Kreidberg J, Koteliansky V, Jaenisch R. Deletion of integrin alpha 1 by homologous recombination permits normal murine development but gives rise to a specific deficit in cell adhesion. Dev Biol. 1996;175:301–13. doi: 10.1006/dbio.1996.0116. [DOI] [PubMed] [Google Scholar]
- 49.Ekholm E, Hankenson KD, Uusitalo H, Hiltunen A, Gardner H, Heino J, Penttinen R. Diminished callus size and cartilage synthesis in alpha 1 beta 1 integrin-deficient mice during bone fracture healing. Am J Pathol. 2002;160:1779–85. doi: 10.1016/s0002-9440(10)61124-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bengtsson T, Aszodi A, Nicolae C, Hunziker EB, Lundgren-Akerlund E, Fassler R. Loss of alpha10beta1 integrin expression leads to moderate dysfunction of growth plate chondrocytes. J Cell Sci. 2005;118:929–36. doi: 10.1242/jcs.01678. [DOI] [PubMed] [Google Scholar]
- 51.Blumbach K, Niehoff A, Belgardt BF, Ehlen HW, Schmitz M, Hallinger R, Schulz JN, Bruning JC, Krieg T, Schubert M, Gullberg D, Eckes B. Dwarfism in mice lacking collagen-binding integrins alpha2beta1 and alpha11beta1 is caused by severely diminished IGF-1 levels. J Biol Chem. 2012;287:6431–40. doi: 10.1074/jbc.M111.283119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Holtkotter O, Nieswandt B, Smyth N, Muller W, Hafner M, Schulte V, Krieg T, Eckes B. Integrin alpha 2-deficient mice develop normally, are fertile, but display partially defective platelet interaction with collagen. J Biol Chem. 2002;277:10789–94. doi: 10.1074/jbc.M112307200. [DOI] [PubMed] [Google Scholar]
- 53.Santana RB, Trackman PC. Controlled release of fibroblast growth factor 2 stimulates bone healing in an animal model of diabetes mellitus. Int J Oral Maxillofac Implants. 2006;21:711–8. [PubMed] [Google Scholar]