

Reduced expression of the survival motor neuron (SMN) protein causes spinal muscular atrophy (SMA) in humans and animal models of severe SMA display defects in pre-mRNA splicing late in the disease course. Using RNA deep-sequencing (RNA-seq) of wild-type and survival motor neuron (Smn) null mutant larvae, we identified a developmental arrest in the mutants that precedes the onset of widespread disruptions in splicing. Comparison of genome-wide mRNA expression profiles of Smn mutants revealed only small changes in levels of minor intron-containing mRNAs; in contrast, we observed a prominent increase in levels of stress-signaling transcripts that may be a conserved feature of SMA.
Keywords: minor intron, splicing, snRNP, survival motor neuron, spinal muscular atrophy, RNA-sequencing, transcriptome, Drosophila insulin-like peptide 8, dILP8, stress signaling pathways
Abstract
Reduced levels of survival motor neuron (SMN) protein lead to a neuromuscular disease called spinal muscular atrophy (SMA). Animal models of SMA recapitulate many aspects of the human disease, including locomotion and viability defects, but have thus far failed to uncover the causative link between a lack of SMN protein and neuromuscular dysfunction. While SMN is known to assemble small nuclear ribonucleoproteins (snRNPs) that catalyze pre-mRNA splicing, it remains unclear whether disruptions in splicing are etiologic for SMA. To investigate this issue, we carried out RNA deep-sequencing (RNA-seq) on age-matched Drosophila Smn-null and wild-type larvae. Comparison of genome-wide mRNA expression profiles with publicly available data sets revealed the timing of a developmental arrest in the Smn mutants. Furthermore, genome-wide differences in splicing between wild-type and Smn animals did not correlate with changes in mRNA levels. Specifically, we found that mRNA levels of genes that contain minor introns vary more over developmental time than they do between wild-type and Smn mutants. An analysis of reads mapping to minor-class intron–exon junctions revealed only small changes in the splicing of minor introns in Smn larvae, within the normal fluctuations that occur throughout development. In contrast, Smn mutants displayed a prominent increase in levels of stress-responsive transcripts, indicating a systemic response to the developmental arrest induced by loss of SMN protein. These findings not only provide important mechanistic insight into the developmental arrest displayed by Smn mutants, but also argue against a minor-intron-dependent etiology for SMA.
INTRODUCTION
The survival motor neuron protein (SMN) functions as part of a large multimeric complex in the assembly of Sm-class small nuclear ribonucleoproteins (snRNPs), core components of the spliceosome (Fischer et al. 2011). Loss-of-function mutations in human SMN1 cause spinal muscular atrophy (SMA), a genetic disorder characterized by motor neuron degeneration, atrophy of proximal muscles, and symmetrical paralysis (Burghes and Beattie 2009). Although the severity of SMA clearly depends on SMN protein levels (Lorson et al. 2010), the etiologic connection between loss of SMN expression and disease pathology remains unknown.
A central issue is how loss of SMN expression manifests as a neuromuscular disorder. Although SMN may well have snRNP-independent functions (Carrel et al. 2006; Burghes and Beattie 2009), the role of SMN in snRNP assembly and splicing continues to be a central focus of much of the research into SMA etiology (Winkler et al. 2005; Gabanella et al. 2007; Zhang et al. 2008; Bäumer et al. 2009; Workman et al. 2009). Nevertheless, it remains unclear how disruption of a ubiquitously required process leads to a neuromuscular phenotype.
SMN assembles Sm-class snRNPs for both the major (U2-type) and minor (U12-type) spliceosomes. Minor-class introns are very rare and the cellular abundance of minor spliceosomes is low (Montzka and Steitz 1988; Tarn and Steitz 1996). Moreover, the splicing of U12-type introns is slower than that of U2-type introns (Patel et al. 2002). Thus, a tractable hypothesis posits the existence of a neuromuscular-specific minor-intron splicing event that is highly sensitive to disruptions in snRNP assembly. Testing this hypothesis in Drosophila models of SMA, two recent reports dispute the extent to which defects in minor intron splicing may account for SMA-like phenotypes (Lotti et al. 2012; Praveen et al. 2012). First, we showed that transgenic expression of low levels of wild-type dSMN protein could fully rescue larval motility and viability without fully rescuing snRNA levels (Praveen et al. 2012). Second, Smn-null animals showed modest (30%–50%) decreases in the levels of four minor intron-containing transcripts; however, the levels of these four mRNAs did not correlate with expression of the wild-type Smn rescue construct (Praveen et al. 2012). This study provides strong evidence uncoupling the snRNP assembly functions of dSMN from the organismal motility and viability defects.
In contrast, Lotti et al. (2012) analyzed Smn-null larvae at later stages of development and reported similar decreases in snRNA levels, along with defects in splicing of seven minor intron-containing transcripts. Among these seven genes, two of them (CG11839 and CG33108) overlapped with the four described by Praveen et al. (2012). In addition, Lotti et al. (2012) reported that two other transcripts, CG16941 and CG8408 (Stasimon), exhibited minor intron retention. The investigators further demonstrated that transgenic expression of Stasimon, either in all neurons or specifically in cholinergic neurons, rescued an electrophysiological defect at larval neuromuscular junctions in Smn mutants (Lotti et al. 2012). However, Stasimon expression failed to rescue motor rhythm, locomotion, or organismal viability (Lotti et al. 2012). These data support a role for Smn in the proper functioning of motor circuits (Imlach et al. 2012), but do not provide convincing evidence for aberrant splicing of minor introns as the molecular basis for SMA.
The situation described above is very similar to two recent studies using a mouse model of severe SMA. Using microarray analyses in late symptomatic mice, Zhang et al. (2008) reported widespread tissue-specific changes in the splicing of both minor- and major-class introns. Subsequently, Bäumer et al. (2009) showed that these changes were limited to late-symptomatic mice; both pre- and early-symptomatic SMA mice do not display such splicing defects. Thus, the observed differences in splicing are likely a downstream consequence of severe SMN loss (Bäumer et al. 2009). Because microarray (Zhang et al. 2008; Bäumer et al. 2009) and quantitative (q)RT-PCR experiments (Lotti et al. 2012; Praveen et al. 2012) have significant drawbacks, we sought a more sensitive and robust approach to address whether Smn loss causes changes in pre-mRNA splicing and mRNA abundance in Drosophila.
RESULTS AND DISCUSSION
To determine if dSMN depletion disrupts pre-mRNA splicing and mRNA levels, we performed high-throughput RNA-sequencing (RNA-seq) on poly(A)-selected RNA from age-matched Smn-null and wild-type (Oregon R, Ore-R) larvae. Two biological replicates were sequenced per genotype. Each sample consisted of RNA from 40–60 larvae isolated at an age (72–76 h post–egg laying) when they display locomotor defects but have not yet begun to die (Praveen et al. 2012). As expected, RNA-seq confirmed the absence of Smn transcripts in the Smn-null larvae (Fig. 1A).
FIGURE 1.
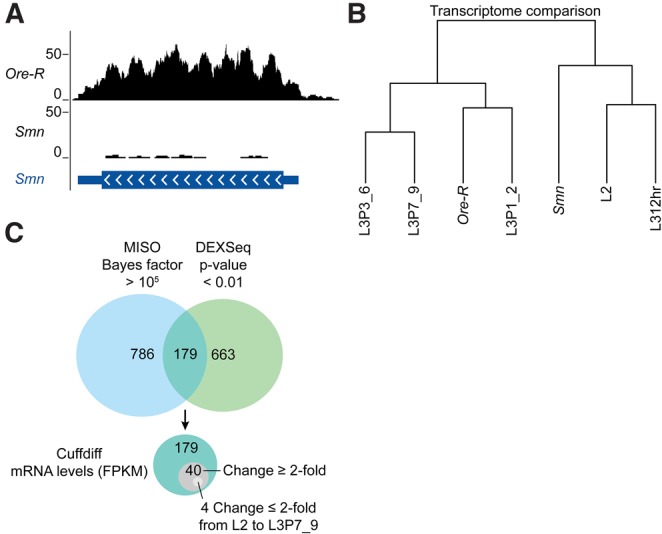
Developmental arrest phenotype of Smn mutants. (A) University of California Santa Cruz (UCSC) genome browser snapshot of mapped RNA-seq reads at the Smn locus from wild-type and Smn mutant animals. Track counts in the wild-type (Ore-R) bedGraph file were normalized using the median of track counts from the Smn mutant bedGraph files. (B) Dendrogram of wild-type (Ore-R) and Smn mutants compared with the developmental transcriptome data from second instar (L2), third instar 12 h post-molt (L312hr), third instar puffstage 1 to 2 with dark blue, bromophenol blue fed guts (L3P1_2), third instar puffstage 3 to 6 with light blue guts (L3P3_6), and the last larval puffstage with clear guts (L3P7_9) (Graveley et al. 2011). Wild-type and Smn mutant RNA-seq data are each the average of two biological samples. Dendrogram line segments correspond to Jensen-Shannon distances (Trapnell et al. 2010). (C) Venn diagrams of DEXSeq, MISO, and Cuffdiff analysis of differences between wild-type and Smn mutant RNA-seq results. For comparison, DEXSeq and MISO results were converted into a nonredundant list of genes that overlap the noted differences in exon usage and alternative splicing events, respectively.
To determine whether gene expression is changed in Smn mutants, the Tophat (Trapnell et al. 2009) and Cufflinks/Cuffdiff (Trapnell et al. 2010) analysis pipeline was used to map sequenced cDNA reads and to quantify genome-wide changes in gene expression (Trapnell et al. 2012). Measurements of mRNA levels are denoted as fragments per kilobase per million reads mapped (FPKM) (see Supplemental Table S1). Analysis of gene expression differences between Smn-null and wild-type animals uncovered several transcripts whose levels changed significantly but did not appear to be differentially spliced. A large number of genes up-regulated in the Smn-null mutants encode proteins from cuticle protein families, specifically the CPR (cuticular protein with Rebers and Riddiford consensus domain) and Tweedle families (Supplemental Table S1). These transcripts have been shown to be elevated during the early third instar developmental stage (Graveley et al. 2011), and their overabundance in Smn-null versus wild-type animals of equivalent age is consistent with a previously reported developmental delay phenotype caused by loss of dSMN (Shpargel et al. 2009).
To further characterize this developmental phenotype, total RNA-seq reads from Smn-null and wild-type animals were compared with the modENCODE Drosophila developmental transcriptome data (Graveley et al. 2011). This comparison clustered the Smn-null mutants with the late second and early third instar stages (Fig. 1B, L2 to L312hr). Notably, the transcript profile from our control animals more closely matched a later third instar stage (Fig. 1, L3P1_2). Genome-wide transcriptome profiling thus provides molecular evidence for a developmental arrest in the Smn-null animals. Because wild-type transcript levels vary over developmental time, it is necessary to compare RNA levels in Smn-null animals with those of younger wild-type controls. Due to the developmental arrest in the mutants, this stipulation is especially important for the later larval stages.
Although Cufflinks was useful in obtaining FPKMs to quantify mRNA abundance, its ability to unambiguously differentiate between mRNA isoforms is more limited. To address this limitation and to determine if dSMN depletion leads to changes in pre-mRNA splicing, we used two additional bioinformatics packages, DEXSeq (Anders et al. 2012) and MISO (Katz et al. 2010; see Supplemental Tables S2–S6). Put simply, DEXSeq is a statistical algorithm that tests for differential exon usage given a set of annotated mRNA exons. MISO is an algorithm that uses a mixture-of-isoforms probabilistic model to test for differences in a supplied set of annotated alternative splicing events. Using relatively relaxed significance cutoffs, both DEXSeq (P-value < 0.05) and MISO (Bayes factor > 100) uncovered more splicing changes between Smn mutants and wild type than those that occur over a similar span of development (L2 thru L3P1_2). Indeed, DEXSeq and MISO identified 2153 and 2484 differences between Smn and wild type, respectively, whereas a similar analysis of the modENCODE data found 951 and 877 splicing changes, respectively.
However, these alternative splicing events did not correlate with changes in mRNA abundance, as measured by Cuffdiff. This lack of correlation persists even when one limits the analysis to the most significant events, as determined by both methods. As shown in Figure 1C, Cuffdiff was used to measure mRNA abundance in 179 differentially spliced genes identified by both DEXSeq (P-value < 0.01) and MISO (Bayes factor > 105). Of these 179 genes, which are the least likely to be false positives, the FPKMs of fewer than one quarter of them (40) changed by more than twofold. Furthermore, only four of these genes changed by less than twofold over larval development (Fig. 1C). Thus, the levels of most of these 179 mRNAs are within the normal fluctuations that occur over developmental time, and none of them, including the final four, were significantly different as determined by Cuffdiff (q-value < 0.05, where the q-value is a P-value adjusted for false discovery rate) (Trapnell et al. 2012).
Recent reports specifically dispute the extent to which dSMN depletion affects minor intron splicing and levels of minor-intron-containing transcripts (Lotti et al. 2012; Praveen et al. 2012). Consistent with our previously published qRT-PCR results (Praveen et al. 2012), RNA-seq confirmed that levels of minor-intron-containing mRNAs are largely unaffected by loss of Smn (Fig. 2A). Of the seven mRNAs identified by Lotti et al. (2012) whose expression was reduced in Smn mutants, none were significantly reduced in our RNA-seq data set (Fig. 2A). To determine if developmental fluctuations in minor-intron-containing mRNA levels could account for observed differences between our results (Praveen et al. 2012) and those of Lotti et al. (2012), the levels of these transcripts in the Smn-null and wild-type RNA-seq data were compared with the modENCODE developmental data (Graveley et al. 2011). As shown in Figure 2B, the levels of minor-intron-containing mRNAs varied more over developmental time than they did between wild-type and Smn mutants. Thus, previously noted qRT-PCR discrepancies between Praveen et al. (2012) and Lotti et al. (2012) may reflect differences in larval staging, rather than Smn-dependent decreases.
FIGURE 2.
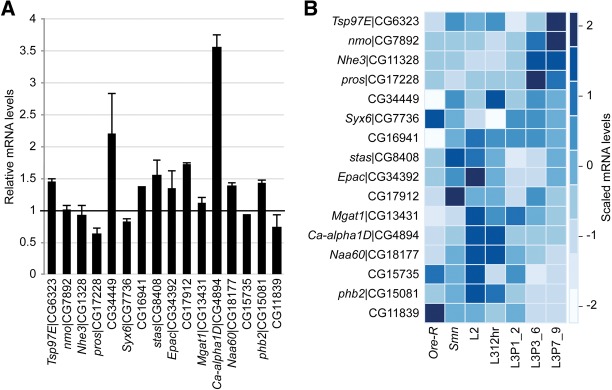
Relatively unaltered minor-intron-containing mRNA levels in Smn mutants. (A) Pairwise comparison of Cuffdiff FPKM levels between wild type and the Smn mutant. FPKM levels were normalized to the wild type, and error bars indicate standard deviation. (B) Heatmap comparison of minor-intron-containing mRNA FPKM levels over development and between wild type and the Smn mutant. Heatmap colors were rescaled for each row, and rows were manually reordered based on trends in gene expression over development. CG33108 was excluded here because Cufflinks’ FPMK analysis did not distinguish between CG33108 and LSm3.
Having detected only low-level changes in mRNA expression in Smn mutants, it was not clear what effect these subtle changes might have on protein levels. To assess the outcome of dSMN depletion on a protein encoded by a minor-intron-containing mRNA, we performed Western blotting for Prohibitin 2 (Phb2) in larval lysates from wild type, Smn mutants, and U6atac mutants. We chose phb2 because it is the only Drosophila minor-intron-containing gene that is expressed at reasonably high levels (Fig. 3A), and its splicing is known to be disrupted in U6atac mutants (Pessa et al. 2010; Praveen et al. 2012). As shown in Figure 3B, Phb2 protein levels were unaffected in Smn mutants but decreased as expected in U6atac hypomorphs. Although Smn mutant phb2 mRNA levels were unaltered in our current RNA-seq analysis (Fig. 2A), our previous qRT-PCR experiments (Praveen et al. 2012) showed a roughly twofold decrease in phb2 transcript levels in Smn-null mutants. These data suggest that small changes (twofold or less) in minor-intron-containing mRNAs may fall within the experimental noise of both qRT-PCR and RNA-seq methodologies, and that such small changes are unlikely to significantly alter cognate protein levels.
FIGURE 3.

Prohibitin 2 protein levels unchanged in Smn mutants. (A) UCSC browser snapshot of normalized RNA-seq tracks at the Prohibitin 2 gene locus. (B) Western blot of Prohibitin 2 (Phb2). Phb2 levels were analyzed in protein lysates from age-matched wild-type (Ore-R), Smn mutant (Smn), and U6atac mutant (U6atac) larvae as well as a later-stage wandering wild-type larva (Ore-R late L3). The anti-prohibitin antibody recognizes the minor-intron-containing PHB2, upper band, and PHB1, lower band, which doesn't contain a known U12-type minor intron. The Drosophila mitochondrial ATP synthase bellwether (BLW) served as a loading control.
Irrespective of changes in mRNA or protein levels, disruptions in splicing may have other negative phenotypic consequences, such as the production of nonfunctional mRNAs or proteins. In their study, Lotti et al. (2012) highlighted a single-minor-intron-containing mRNA, Stasimon, and its role in a specific electrophysiological phenotype. In addition to a slight decrease in Stasimon mRNA levels, they noted an increase in Stasimon minor intron retention in Smn mutants (Lotti et al. 2012). To quantify the degree of minor intron retention in Stasimon and other minor-intron-containing transcripts, we designed a custom Perl script to count specific reads spanning exon–exon and intron–exon junctions (Fig. 4A; Supplemental Table S7). We found that Stasimon and three other minor-intron-containing mRNAs exhibited significant (P-value < 0.01) increases in minor intron retention in the Smn mutants (Fig. 4; Supplemental Table S7). However, intron retention for these genes is also readily detectable in the wild-type samples, including those from the modENCODE project (Fig. 4B; Supplemental Table S7; Graveley et al. 2011). Strikingly, the levels of intron retention for these genes fluctuate during larval development (Supplemental Table S7). In particular, retention of the minor intron in Stasimon was greatest at the L312hr time point in the modENCODE data set (Fig. 4B). Because Smn mutants are developmentally arrested at this particular stage, the elevated intron retention identified by our analysis, and noted by Lotti et al. (2012), appears to be a consequence of the developmental arrest and thus not a result of dSMN depletion.
FIGURE 4.

Increase in Stasimon mRNA minor intron retention in Smn mutants and early in the third instar larval stage. (A) UCSC browser snapshot of normalized RNA-seq tracks at the Stasimon gene locus and diagram of junction spanning reads used to measure intron retention. (Gray lines and numbers) Exon–intron junction reads; (black lines and numbers) exon–exon junction reads. (Gray brackets and arrows in gene model) Minor intron locations. (B) Stasimon mRNA minor intron retention levels as a percentage of exon–intron (intron) junction spanning reads over total junction spanning reads between wild type, mutant, and over the developmental time from L2 to L3P7_9. Actual junction-spanning read counts and calculations are also provided in Supplemental Table S7. For comparison with our paired-end data, we used the Celniker paired-end modENCODE developmental data for this analysis (Supplemental Table S7).
To identify genes whose differences in expression level could not be accounted for by developmental fluctuations, we searched within our Cuffdiff list of significantly altered transcripts (Supplemental Table S1) for differences that were inconsistent with and/or exceeded normal developmental changes. Using Gene Ontology (GO) enrichment analysis, we noted a large number of stress responsive transcripts that were up-regulated in the Smn mutants (Fig. 5A; Supplemental Table S1). Unlike the CPR and Tweedle gene families mentioned above, the expression of these stress response genes was inconsistent with the timing of the Smn mutant developmental arrest and exceeded normal developmental fluctuations. Thus, changes in levels of these mRNAs appear to be a consequence of dSMN depletion. The Toll, Imd (Immune deficiency), JNK (c-Jun N-terminal kinase), and JAK-STAT (Janus kinase–signal transducer and activator of transcription) signaling pathways regulate expression of these transcripts in response to infectious and noninfectious stresses (Leclerc and Reichhart 2004; Biteau et al. 2011; Teixeira 2012). For example, the mRNAs for AttacinA (AttA) and Drosocin (Dro) encode antimicrobial peptides that are normally increased in response to septic injury, but their expression has also been linked to noninfectious stressors (Sano et al. 2005; Tsuzuki et al. 2012).
FIGURE 5.
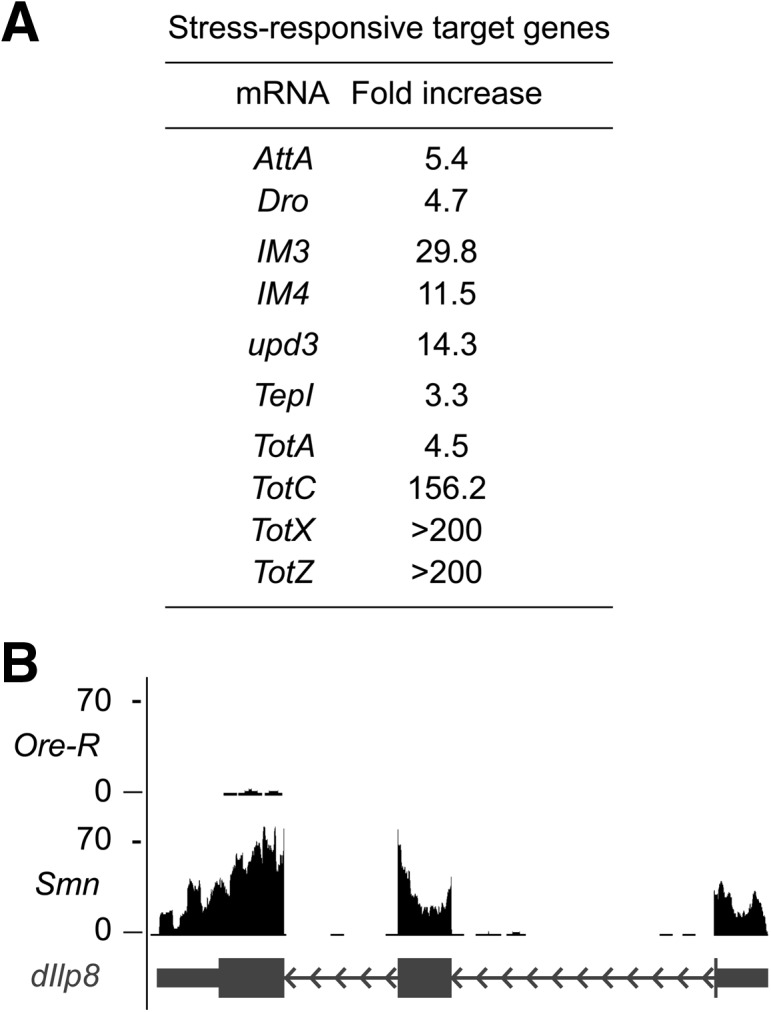
Increased stress-responsive target genes in Smn mutants. (A) Stress-responsive transcripts that were significantly increased in the Smn mutants expressed here as fold increase above wild-type levels (from average Cufdiff FPKM values) (Supplemental Table S1). (B) UCSC browser snapshot of normalized RNA-seq tracks at the dILP8 gene locus.
At least two additional signaling pathways have been linked to the noninfectious induction of antimicrobial peptides, the IIS (insulin/insulin-like growth factor) (Becker et al. 2010; Karpac et al. 2011), and the GBP (growth blocking peptide) growth-factor signaling pathways (Tsuzuki et al. 2012). Transcripts for traditional cytokines associated with these pathways, such as dILP1-5 (Insulin-like peptides 1–5), dILP7 and GBP (CG15917) are largely unaltered in Smn mutants, whereas dIILP6 showed a nearly threefold decrease (Supplemental Table S1). Perhaps most notable was the dramatic increase in expression of dILP8 (Insulin-like peptide 8), a recently discovered cytokine (Fig. 5B). Secretion of dILP8 by damaged or slow-growing imaginal discs was shown to coordinate organismal growth with developmental progression (Colombani et al. 2012; Garelli et al. 2012). In particular, dILP8 was shown to block the transition from larval to pupal stages (Colombani et al. 2012; Garelli et al. 2012). Interestingly, we find that Smn-null mutant larvae have either very small or nonexistent imaginal discs (data not shown). Therefore, we submit that up-regulation of dILP8 contributes to the pupariation failure observed in Smn-null mutants.
In summary, Smn-null larvae (harvested at 72–76 h post–egg laying) display significant locomotor deficits (Shpargel et al. 2009; Praveen et al. 2012), but do not show appreciable genome-wide changes in mRNA levels. Based on morphological criteria, we previously concluded that Smn-null mutants are developmentally delayed (Shpargel et al. 2009). Here, we show by transcriptome profiling that Smn mutants display a molecular signature indicating a profound developmental arrest during the early third larval instar. Consistent with this notion, Smn-null larvae never display the wandering behavior that is characteristic of the late third instar, despite the fact that a small fraction of these animals live for several weeks (Praveen et al. 2012). Proper staging of experimental and control animals is critical to any meaningful comparison. Because Lotti et al. (2012) used RNA isolated from wandering third instar larvae (108 ± 12 h post–egg laying) as controls, their qRT-PCR data may appear to show that Smn depletion negatively affects certain transcripts, when in actuality it is due to a combination of both developmental fluctuation and normal biological noise.
Transcriptome profiling reveals that Smn mutants display an increase in stress-responsive transcripts that may be a direct or indirect consequence of SMN loss. However, the role of these transcripts in the etiology of SMA-like phenotypes is not clear. The developmental arrest may be a downstream consequence of the inability of Smn-null larvae to feed properly. With this caveat in mind, the RNA-seq data provide evidence for the activation of stress signaling pathways that could represent an important systemic response to dSMN depletion. The activation of cellular stress pathways was previously observed in a mouse embryonic stem (ES) cell model of SMA and in spinal cord tissues from which these ES cells were derived (Wu et al. 2011). Thus, activation of stress-response pathways may be a conserved feature of SMA. Neuronal overexpression of certain antimicrobial peptides, which are known stress signaling targets, was recently shown to cause neurodegeneration in Drosophila (Cao et al. 2013). Taken together, these observations suggest that the activation of cellular stress signaling pathways in Smn mutants is an important pathophysiological consequence of SMN depletion that is relevant for modeling SMA.
Overall, the observed developmental arrest complicates determination of cause and effect in the pathophysiology of Smn mutants. Both fruit fly and mouse models of severe SMA display neuromuscular deficits that are detectable early in the disease course, whereas splicing defects are only apparent later on (Zhang et al. 2008; Bäumer et al. 2009; Lotti et al. 2012; Praveen et al. 2012). Our RNA-seq data reveal several differences in pre-mRNA splicing between wild-type and Smn animals. However, these splicing differences do not correlate with changes in mRNA levels. The impact, if any, of these small changes on the organism's neurophysiology remains to be elucidated. Results presented here cannot exclude the important possibility that tissue- and cell-specific splicing events below the current level of detection might be causative for SMA. Future studies of mRNA splicing and abundance in isolated cells and tissues may uncover a more direct link between SMN depletion, splicing, and SMA-like phenotypes. Nevertheless, the results presented here raise serious doubt as to the validity of conclusions reached by others regarding the connection between splicing deficiencies and motor circuit dysfunction (Roselli and Caroni 2012). Clearly, additional studies will be needed in order to fully elucidate the molecular etiology of SMA.
MATERIALS AND METHODS
Drosophila stocks
Fly stocks were cultured as in Praveen et al. (2012). Briefly, larvae were cultured on molasses and agar at room temperature (24°C ± 1°C). The wild-type allele was Oregon-R. Smn-null animals were trans-heterozygous for an SmnX7 microdeletion allele (Chang et al. 2008) and an isogenized SmnD (f01109) transposon allele from the Exelixis collection at the Harvard Medical School (Rajendra et al. 2007).
RNA-seq and bioinformatic analysis
RNA was isolated from 74 ± 2-h-old wild-type and Smn mutant larvae by homogenization in TRIzol (Invitrogen), according to the manufacturer's protocol. RNA was DNased with Amplification Grade DNase I (Invitrogen). A TruSeq RNA Sample Preparation Kit v2 (Illumina) was used for: poly(A) enrichment, barcoding for multiplexing, and cDNA library preparation. Paired end (2 × 50) sequencing was performed on an Illumina HiSeq 2000 platform, raw FastQ file output:
wild-type, EG003_ACAGTG_L005_R1_001.fastq EG003_ACAGTG_L005_R2_001.fastq and EG004_GCCAAT_L005_R1_001.fastq EG004_GCCAAT_L005_R2_001.fastq;
Smn mutant, EG007_ACTGAT_L005_R1_001.fastq EG007_ACTGAT_L005_R2_001.fastq and EG008_ATTCCT_L005_R1_001.fastq EG008_ATTCCT_L005_R2_001.fastq.
TopHat and Cufflinks were used to quantify and compare mRNA FPKM levels, according to the bioinformatic pipeline from Trapnell et al. (2012). We used RStudio (http://www.rstudio.org/) to interface with the R statistical suite (http://www.r-project.org), and the R-based cummeRbund package (http://compbio.mit.edu/cummeRbund/) was used as a graphical interface with the Cufflink's Cuffdiff output. DEXSeq (Anders et al. 2012) and MISO (Katz et al. 2010) were used to measure changes in exon usage and annotated alternative splicing events, respectively. For developmental comparison, all publicly available RNA-seq data from Graveley et al. (2011) (http://www.modencode.org/) from L2 to L3 puff stage 7–9 were downloaded from the NCBI Sequence Read Archive (SRA) and converted to fastq format with the SRA toolkit. The heatmap was generated with the Heatplus R package from bioconductor (http://www.bioconductor.org). Reads mapping to exon–intron junctions of U12 introns were extracted from SAM files generated by Tophat and quantified using a custom Perl script. P-values were generated using the Fisher's exact test.
Western blotting
Western analysis was performed using standard protocols, according to the methods of Praveen et al. (2012). Briefly, larval protein lysates were prepared by crushing the animals in lysis buffer (50 mM Tris-HCl at pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% NP-40) with 1× protease inhibitor cocktail (Invitrogen) and clearing the lysate by centrifugation. The anti-prohibitin 2 antibody (H-80; Santa Cruz Biotechnology) was used at a 1:100 dilution. The anti-mitochondrial ATP synthase antibody, ATP5A [15H4C4] (Abcam)—which recognizes Drosophila bellwether (BLW), was used as a loading control at a 1:1000 dilution.
DATA DEPOSITION
The data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus (Edgar et al. 2002) and are accessible through GEO Series accession number GSE49587 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE49587).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We gratefully acknowledge C. Jones and P. Mieczkowski at the University of North Carolina High Throughput Sequencing Facility (HTSF). C. Jones and P. Mieczkowski were instrumental in discussions on proper high-throughput sequencing platform, sequencing strategy, sample preparation, and sample submission. P. Mieczkowski performed the sequencing, and A. Brandt from the HTSF prepared TruSeq cDNA libraries. This work was supported by a grant (to A.G.M.) from the National Institute of Neurological Disorders and Stroke (R01 NS041617). E.L.G. was supported by a National Cancer Institute postdoctoral fellowship (T32 CA009156), administered by J. Pagano and the Lineberger Comprehensive Cancer Center. M.P.M. was supported by a pre-doctoral fellowship (T32 GM007092) from the National Institute of General Medical Sciences.
REFERENCES
- Anders S, Reyes A, Huber W 2012. Detecting differential usage of exons from RNA-seq data. Genome Res 22: 2008–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäumer D, Lee S, Nicholson G, Davies JL, Parkinson NJ, Murray LM, Gillingwater TH, Ansorge O, Davies KE, Talbot K 2009. Alternative splicing events are a late feature of pathology in a mouse model of spinal muscular atrophy. PLoS Genet 5: e1000773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker T, Loch G, Beyer M, Zinke I, Aschenbrenner AC, Carrera P, Inhester T, Schultze JL, Hoch M 2010. FOXO-dependent regulation of innate immune homeostasis. Nature 463: 369–373 [DOI] [PubMed] [Google Scholar]
- Biteau B, Karpac J, Hwangbo D, Jasper H 2011. Regulation of Drosophila lifespan by JNK signaling. Exp Gerontol 46: 349–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burghes AH, Beattie CE 2009. Spinal muscular atrophy: Why do low levels of survival motor neuron protein make motor neurons sick? Nat Rev Neurosci 10: 597–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Chtarbanova S, Petersen AJ, Ganetzky B 2013. Dnr1 mutations cause neurodegeneration in Drosophila by activating the innate immune response in the brain. Proc Natl Acad Sci 110: E1752–E1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrel TL, McWhorter ML, Workman E, Zhang H, Wolstencroft EC, Lorson C, Bassell GJ, Burghes AH, Beattie CE 2006. Survival motor neuron function in motor axons is independent of functions required for small nuclear ribonucleoprotein biogenesis. J Neurosci 26: 11014–11022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HC, Dimlich DN, Yokokura T, Mukherjee A, Kankel MW, Sen A, Sridhar V, Fulga TA, Hart AC, Van Vactor D, et al. 2008. Modeling spinal muscular atrophy in Drosophila. PLoS One 3: e3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombani J, Andersen DS, Leopold P 2012. Secreted peptide Dilp8 coordinates Drosophila tissue growth with developmental timing. Science 336: 582–585 [DOI] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE 2002. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30: 207–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer U, Englbrecht C, Chari A 2011. Biogenesis of spliceosomal small nuclear ribonucleoproteins. Wiley Interdiscip Rev RNA 2: 718–731 [DOI] [PubMed] [Google Scholar]
- Gabanella F, Butchbach ME, Saieva L, Carissimi C, Burghes AH, Pellizzoni L 2007. Ribonucleoprotein assembly defects correlate with spinal muscular atrophy severity and preferentially affect a subset of spliceosomal snRNPs. PLoS One 2: e921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garelli A, Gontijo AM, Miguela V, Caparros E, Dominguez M 2012. Imaginal discs secrete insulin-like peptide 8 to mediate plasticity of growth and maturation. Science 336: 579–582 [DOI] [PubMed] [Google Scholar]
- Graveley BR, Brooks AN, Carlson JW, Duff MO, Landolin JM, Yang L, Artieri CG, van Baren MJ, Boley N, Booth BW, et al. 2011. The developmental transcriptome of Drosophila melanogaster. Nature 471: 473–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imlach WL, Beck ES, Choi BJ, Lotti F, Pellizzoni L, McCabe BD 2012. SMN is required for sensory-motor circuit function in Drosophila. Cell 151: 427–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpac J, Younger A, Jasper H 2011. Dynamic coordination of innate immune signaling and insulin signaling regulates systemic responses to localized DNA damage. Dev Cell 20: 841–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz Y, Wang ET, Airoldi EM, Burge CB 2010. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat Methods 7: 1009–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leclerc V, Reichhart JM 2004. The immune response of Drosophila melanogaster. Immunol Rev 198: 59–71 [DOI] [PubMed] [Google Scholar]
- Lorson CL, Rindt H, Shababi M 2010. Spinal muscular atrophy: Mechanisms and therapeutic strategies. Hum Mol Genet 19: R111–R118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotti F, Imlach WL, Saieva L, Beck ES, Hao le T, Li DK, Jiao W, Mentis GZ, Beattie CE, McCabe BD, et al. 2012. An SMN-dependent U12 splicing event essential for motor circuit function. Cell 151: 440–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montzka KA, Steitz JA 1988. Additional low-abundance human small nuclear ribonucleoproteins: U11, U12, etc. Proc Natl Acad Sci 85: 8885–8889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AA, McCarthy M, Steitz JA 2002. The splicing of U12-type introns can be a rate-limiting step in gene expression. EMBO J 21: 3804–3815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessa HK, Greco D, Kvist J, Wahlstrom G, Heino TI, Auvinen P, Frilander MJ 2010. Gene expression profiling of U12-type spliceosome mutant Drosophila reveals widespread changes in metabolic pathways. PLoS ONE 5: e13215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praveen K, Wen Y, Matera AG 2012. A Drosophila model of spinal muscular atrophy uncouples snRNP biogenesis functions of survival motor neuron from locomotion and viability defects. Cell Rep 1: 624–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendra TK, Gonsalvez GB, Walker MP, Shpargel KB, Salz HK, Matera AG 2007. A Drosophila melanogaster model of spinal muscular atrophy reveals a function for SMN in striated muscle. J Cell Biol 176: 831–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roselli F, Caroni P 2012. A circuit mechanism for neurodegeneration. Cell 151: 250–252 [DOI] [PubMed] [Google Scholar]
- Sano Y, Akimaru H, Okamura T, Nagao T, Okada M, Ishii S 2005. Drosophila activating transcription factor-2 is involved in stress response via activation by p38, but not c-Jun NH2-terminal kinase. Mol Biol Cell 16: 2934–2946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shpargel KB, Praveen K, Rajendra TK, Matera AG 2009. Gemin3 is an essential gene required for larval motor function and pupation in Drosophila. Mol Biol Cell 20: 90–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarn WY, Steitz JA 1996. Highly diverged U4 and U6 small nuclear RNAs required for splicing rare AT-AC introns. Science 273: 1824–1832 [DOI] [PubMed] [Google Scholar]
- Teixeira L 2012. Whole-genome expression profile analysis of Drosophila melanogaster immune responses. Brief Funct Genomics 11: 375–386 [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL 2009. TopHat: Discovering splice junctions with RNA-seq. Bioinformatics 25: 1105–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L 2010. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7: 562–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuzuki S, Ochiai M, Matsumoto H, Kurata S, Ohnishi A, Hayakawa Y 2012. Drosophila growth-blocking peptide-like factor mediates acute immune reactions during infectious and non-infectious stress. Sci Rep 2: 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler C, Eggert C, Gradl D, Meister G, Giegerich M, Wedlich D, Laggerbauer B, Fischer U 2005. Reduced U snRNP assembly causes motor axon degeneration in an animal model for spinal muscular atrophy. Genes Dev 19: 2320–2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman E, Saieva L, Carrel TL, Crawford TO, Liu D, Lutz C, Beattie CE, Pellizzoni L, Burghes AH 2009. A SMN missense mutation complements SMN2 restoring snRNPs and rescuing SMA mice. Hum Mol Genet 18: 2215–2229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CY, Whye D, Glazewski L, Choe L, Kerr D, Lee KH, Mason RW, Wang W 2011. Proteomic assessment of a cell model of spinal muscular atrophy. BMC Neurosci 12: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Lotti F, Dittmar K, Younis I, Wan L, Kasim M, Dreyfuss G 2008. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell 133: 585–600 [DOI] [PMC free article] [PubMed] [Google Scholar]