

This paper describes extensive ADAR1-mediated RNA editing in the 3′ UTR of the Rho GTPase activating protein 26 (ARHGAP26) mRNA. This results in the disruption of binding sites for two miRNAs, miR-30b-3p and miR-573, and consequently leads to an up-regulation of the ARHGAP26 mRNA and protein levels. Wang and coauthors further connect this regulation to tumorigenesis by showing that alterations in ADAR1 expression during cancer lead to down-regulation of ARHGAP26, which behaves as a tumor suppressor.
Keywords: A-to-I RNA editing, ADARs, ARHGAP26, miR-30b-3p, miR-573, microRNA
Abstract
Rho GTPase activating protein 26 (ARHGAP26) is a negative regulator of the Rho family that converts the small G proteins RhoA and Cdc42 to their inactive GDP-bound forms. It is essential for the CLIC/GEEC endocytic pathway, cell spreading, and muscle development. The present study shows that ARHGAP26 mRNA undergoes extensive A-to-I RNA editing in the 3′ UTR that is specifically catalyzed by ADAR1. Furthermore, the mRNA and protein levels of ARHGAP26 were decreased in cells in which ADAR1 was knocked down. Conversely, ADAR1 overexpression increased the abundance of ARHGAP26 mRNA and protein. In addition, we found that both miR-30b-3p and miR-573 target the ARHGAP26 gene and that RNA editing of ARHGAP26 mediated by ADAR1 abolished the repression of its expression by miR-30b-3p or miR-573. When ADAR1 was overexpressed, the reduced abundance of ARHGAP26 protein mediated by miR-30b-3p or miR-573 was rescued. Importantly, we also found that knocking down ADAR1 elevated RhoA activity, which was consistent with the reduced level of ARHGAP26. Conversely, when ADAR1 was overexpressed, the amount of RhoA-GTP decreased. The similar expression patterns of ARHGAP26 and ADAR1 in human tissue samples further confirmed our findings. Taken together, our results suggest that ADAR1 regulates the expression of ARHGAP26 through A-to-I RNA editing by disrupting the binding of miR-30b-3p and miR-573 within the 3′ UTR of ARHGAP26. This study provides a novel insight into the mechanism by which ADAR1 and its RNA editing function regulate microRNA-mediated modulation of target genes.
INTRODUCTION
A-to-I RNA editing, the most common type of RNA editing in mammals, alters the genomic information of a gene through nucleotide base modification from adenosine (A) to inosine (I) post-transcriptionally (Bass and Weintraub 1988; Wagner et al. 1989; Gallo and Galardi 2008; Farajollahi and Maas 2010; Nishikura 2010). This type of RNA editing is catalyzed by a family of enzymes called adenosine deaminases acting on RNA (ADAR). There are four human ADARs: ADAR1, ADAR2, ADAR3, and TENR (Schumacher et al. 1995; Melcher et al. 1996a,b; Bass et al. 1997; Chen et al. 2000; Farajollahi and Maas 2010; Nishikura 2010). ADAR1 and ADAR2 are ubiquitously expressed in various tissues and have intrinsic A-to-I editing activity. However, neither the brain-specific ADAR3 nor the testis-specific TENR has editing activity (Saunders and Barber 2003; Farajollahi and Maas 2010; Nishikura 2010). ADAR1 and ADAR2 are conserved in their domain arrangement, which includes dsRNA-binding domains at the amino terminus and a highly conserved deaminase domain at the carboxyl terminus. They have distinct, but overlapping, editing site selections (Lehmann and Bass 2000; Wong et al. 2001; Kallman et al. 2003; Riedmann et al. 2008). ADAR2 prefers to specifically edit the R/G site, whereas ADAR1 edits more promiscuously adenosines without sequence specificity (Kallman et al. 2003). Both ADAR1 and ADAR2 share a similar 5′ neighbor preference (A ≈ U > C = G); however, the latter shows a 3′ neighbor preference (Lehmann and Bass 2000). Apart from the surrounding sequence, editing efficiency is strongly influenced by the base opposing the edited adenosine of the double-stranded RNAs stem. It has been shown that A:C mismatches are preferred to A:A and A:G mismatches, or A:U base pairs (Wong et al. 2001; Riedmann et al. 2008).
Inosine is recognized as guanosine by the translation machinery; thus, an A-to-I RNA editing event leads to an A-to-G change. If an edited nucleotide is located in the protein-coding region, RNA editing might lead to mutation of the protein sequence. However, the majority of the A-to-I RNA editing sites are found in the noncoding, repetitive sequences, and many of those have unknown functions. A-to-I RNA editing sites located in the noncoding sequences have been proposed to modulate the expression level of mature miRNAs by inhibiting Drosha cleavage (Yang et al. 2006) or Dicer cleavage (Kawahara et al. 2007a), to redirect them to a different set of target genes compared with the unedited conditions by modifying the miRNA seed sequence (Kawahara et al. 2007b), to affect splicing events by creating or abolishing splicing sites (Rueter et al. 1999), and to regulate gene expression through the nuclear retention of inosine-containing RNAs (Prasanth et al. 2005; Nishikura 2010). The diverse impact of the A-to-I RNA editing on gene expression and function has led to studies on how RNA editing contributes to the pathogenesis of human diseases. Dysregulation of A-to-I RNA editing has been associated with dyschromatosis symmetrica hereditaria (Miyamura et al. 2003), Aicardi-Goutières syndrome (Rice et al. 2012), amyotrophic lateral sclerosis (Kawahara et al. 2004), epilepsy (Schmauss 2005), and certain cancers (Maas et al. 2001; Ishiuchi et al. 2002; Paz et al. 2007; Cenci et al. 2008; Galeano et al. 2013).
Rho GTPase activating protein 26 (ARHGAP26, also known as GRAF, GRAF1, OPHN1L) is a regulator of the Rho family that converts the small G proteins RhoA and Cdc42 to their inactive GDP-bound forms (Hildebrand et al. 1996). Through the negative regulation of small G protein RhoA, ARHGAP26 is critical for muscle development (Doherty et al. 2011b). ARHGAP26-depleted embryos in Xenopus exhibited elevated RhoA activity, progressive muscle degeneration, defective motility, and embryonic lethality (Doherty et al. 2011b). Through down-regulation of Cdc42 activity, ARHGAP26 is essential for the CLIC/GEEC endocytic pathway, which is characterized by its ability to internalize glycosylphosphatidylinositol-anchored proteins, bacterial toxins, and large amounts of extracellular fluid (Lundmark et al. 2008). ARHGAP26 activity is up-regulated in spreading cells, and uptake via CLIC is concentrated at the leading edge of migrating cells. Depletion of ARHGAP26 inhibited CLIC generation and impaired cell spreading and migration (Doherty et al. 2011a). ARHGAP26 was also thought to be a putative tumor suppressor by negatively regulating RhoA and Cdc42 in human cancers, which are known for their growth-promoting effects in oncoprotein Ras-mediated malignant transformation (Khosravi-Far et al. 1995; Bojesen et al. 2006). Previous studies have shown that ARHGAP26 was significantly down-regulated in myeloid malignancies compared with controls (Qian et al. 2010) and in lung tumor metastasis within the brain compared with primary lung adenocarcinoma (Zohrabian et al. 2007). The significant decrease in ARHGAP26 observed in X-linked α-thalassemia mental retardation syndrome suggested that it might be involved in the pathogenesis of mental retardation (Barresi et al. 2010).
Extensive A-to-I RNA editing events were predicted to occur in the ARHGAP26 transcript (Kim et al. 2004), which potentially destroy the putative binding sites of miR-30b-3p and miR-573. Our work demonstrated that ADAR1 specifically catalyzes these editing sites and regulates the expression of ARHGAP26. Moreover, both miR-30b-3p and miR-573 target the ARHGAP26 gene, and RNA editing of ARHGAP26 abolished the repression of its expression by miR-30b-3p or miR-573. In human invasive ductal breast cancer and glioblastoma specimens, we observed similar expression patterns of ARHGAP26 and ADAR1. Taken together, our results indicate that ADAR1 regulates ARHGAP26 gene expression through RNA editing by disrupting miR-30b-3p and miR-573 binding.
RESULTS
The ARHGAP26 transcript undergoes extensive A-to-I RNA editing in its 3′ UTR
The human ARHGAP26 transcript has been predicted to undergo extensive A-to-I RNA editing in its 3′ UTR (Kim et al. 2004). To verify these predicted RNA editing sites experimentally, we sequenced the matching DNA and RNA samples derived from the same specimens and compared the cDNA sequence with the corresponding genomic template in the human colon cancer cell line SW480, the human Burkitt's lymphoma cell line Raji, the human prostate carcinoma cell line DU145, the human osteosarcoma cell line U2OS, the human lung adenocarcinoma epithelial cell line A549, the estrogen receptor (ER)–positive human breast cancer cell line MCF-7, the nontransformed human mammary epithelial cell line MCF-10A, the human hepatoma cell line HepG2, and the human immortalized nontumor cell line Chang liver. The editing level was defined as the ratio of the G peak over the sum of the G and A peaks in the sequencing chromatogram. The experiments were repeated three times and a representative result is shown in Figure 1 and Supplemental Figure S1. We found that the ARHGAP26 transcript underwent extensive A-to-I RNA editing in these cell lines. The DU145 cells have the most editing sites (13 editing sites), with editing levels ranging from 25.7% to 85.5%. In contrast, the Raji cell line has the fewest editing sites (eight editing sites), with editing levels ranging from 11.0% to 34.9%. Twelve out of 13 (12/13) editing sites of ARHGAP26 pre-mRNA in DU145 were predicted to undergo A-to-I RNA editing (named U1–12), and a novel editing site N1 in its 3′-UTR sequence was identified (Fig. 1; Supplemental Fig. 1). The new editing site N1 was specific to DU145 cells, with 29.1% editing levels, and did not exist in the other cells tested in this study. Eight editing sites (except U2, U7, U8, and U11) were found to be present in all cell lines. Editing sites U2 and U11 were present in these cell lines, except Raji cells, with at least 22% or 8.7% editing levels, respectively. Editing site U7 was found in DU145, U2OS, MCF-7, MCF-10A, HepG2, and Chang liver cell lines with at least 7.5% editing levels. Compared with the U7 site, editing site U8 existed in all six cells except MCF-7, with at least 20.7% editing levels. Taken together, our results suggest that the ARHGAP26 transcript undergoes extensive A-to-I RNA editing in its 3′ UTR, although the number of the RNA editing sites varies, and the editing levels of the same editing site also vary among the cell lines tested in this study.
FIGURE 1.
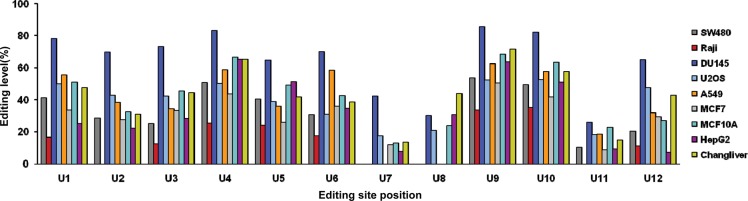
RNA editing levels of ARHGAP26 transcript in its 3′ UTR in different human cells. Twelve known A-to-I RNA editing sites of the ARHGAP26 transcript in its 3′ UTR are named U1–U12, starting from the 5′ to the 3′. They were analyzed in SW480, Raji, DU145, U2OS, A549, MCF-7, MCF-10A, HepG2, and Chang liver cells by comparing the sequences of RT-PCR products with the corresponding genomic templates. The editing level was defined to be the ratio of the G peak over the sum of the G and A peaks in the sequencing chromatogram.
ADAR1 but not ADAR2 edits ARHGAP26 transcript
ADAR1 and ADAR2 are both known to catalyze A-to-I RNA editing in mammals. The ARHGAP26 transcript is edited at multiple sites; therefore, we attempted to determine which enzyme was responsible for these editing events. To address this question, we silenced ADAR1 or ADAR2 expression using RNA interference. As shown in Figure 2, A and B, MCF-10A cells were transfected with three double-stranded ADAR1-specific siRNAs, or with a nontargeting siRNA as negative control at the concentration of 30 nM. Total RNA, gDNA, and protein were prepared 48 h post-transfection and subjected to qPCR, Western blotting, and the RNA editing assays. The mRNA levels of ADAR1 were significantly decreased in cells transfected with ADAR1 siRNA oligonucleotides (Fig. 2A, top), which was further supported by Western blotting (Fig. 2A, bottom). ADAR2 and β-actin levels were relatively unchanged in the same assay. We then sequenced the RT-PCR products and measured the RNA editing levels to determine the effect of decreased ADAR1 expression on the editing sites in the 3′ UTR of the ARHGAP26 transcript. One representative experiment is shown in Figure 2B. Upon ADAR1 knockdown, RNA editing activity of all editing sites except U8 decreased significantly in cultured MCF-10A cells. Similar experiments were performed in MCF-10A cells in which ADAR2 instead of ADAR1 was silenced. qPCR, Western blotting, and RNA editing assays were performed in cells treated with the siADAR2s at the concentration of 30 nM. As shown in Figure 2, C and D, three siRNA oligonucleotides specifically knocked down the expression of ADAR2 at both mRNA and protein levels; however, in contrast to ADAR1 knockdown, no significant changes in RNA editing levels were observed. To further determine the effect of ADAR2 knockdown, we also checked a known ADAR2 editing site in the CYFIP2 transcript (Paz et al. 2007; Tariq et al. 2013) and found that the editing levels decreased from 10.6% to zero (Supplemental Fig. 2). To exclude any nonspecific effects, we repeated the experiments in A549 cells and obtained the same result (Supplemental Fig. 3). These results indicated that all the RNA editing sites except U8 in the ARHGAP26 transcript are mediated by ADAR1.
FIGURE 2.

ADAR1, but not ADAR2 enzyme, edits the ARHGAP26 transcript. (A) Effect of siRNAs on endogenous ADAR1 mRNA and protein levels assayed using qPCR (top) and Western blotting (bottom) in human MCF-10A cells. (B) Editing levels of RNA editing sites in the 3′ UTR of the ARHGAP26 transcript after specifically knocking down ADAR1 expression. (C) Effect of siRNAs on endogenous ADAR2 mRNA and protein levels assayed using qPCR (top) and Western blotting (bottom) in human MCF-10A cells. (D) Editing levels of RNA editing sites in the 3′ UTR of the ARHGAP26 transcript after specifically knocking down ADAR2 expression. (E) Effect of double silencing of ADAR1 and ADAR2 assayed using Western blotting in MCF-10A cells. (F) Editing levels of RNA editing in the 3′ UTR of the ARHGAP26 transcript after knocking down both ADAR1 and ADAR2 expression. (G) Western blotting analysis of ADAR1 after transfecting the overexpression vector FLAG-ADAR1 or control plasmid in MCF-10A cells. (H) Editing levels of RNA editing sites in the 3′ UTR of the ARHGAP26 transcript after overexpressing ADAR1.
To examine the effect of double silencing of ADAR1 and ADAR2 on RNA editing, MCF-10A cells were cotransfected with si-ADAR1-2# and si-ADAR2-1# at a concentration of 30 nM of each siRNA (Fig. 2E,F). Forty-eight hours post-transfection, whole-cell lysates were prepared and analyzed by Western blotting. As shown in Figure 2E, the protein levels of ADAR1 and ADAR2 decreased dramatically. We sequenced the RT-PCR product and measured the RNA editing levels. A representative experiment is shown in Figure 2F. Compared with the knockdown of ADAR1, there was no further inhibition of RNA editing levels in ARHGAP26 transcript except U8 by knocking down both ADAR1 and ADAR2. These results further confirmed that ADAR1 plays a critical role in editing the ARHGAP26 transcript.
To further confirm our results, we overexpressed ADAR1 in MCF-10A cells and examined the RNA editing activity. MCF-10A cells were transiently transfected with an expression vector encoding Flag-tagged ADAR1 or empty vector as a control. Whole-cell lysates were prepared 36 h post-transfection, and the expression levels of ADAR1 were determined by Western blotting (Fig. 2G). To monitor the editing activity after ADAR1 overexpression, we collected total RNA and amplified the cDNA using the oligonucleotides ARHGAP26-F and ARHGAP26-R, followed by sequencing of the cDNA products. Overexpression of ADAR1 increased the editing activity on the ARHGAP26 transcript at all editing sites except U8 (Fig. 2H). These data suggested that the majority of editing of the ARHGAP26 transcript was mediated by ADAR1. As shown in Figure 2D (si-ADAR2-3#), Figure 2F (double siADAR1/ADAR2), and Figure 2H (up-regulation of ADAR1), it seems that editing at the U8 site of the ARHGAP26 transcript is mediated by ADAR2.
ADAR1 regulates expression of the ARHGAP26 gene
Previous studies have shown that A-to-I RNA editing in the 3′ UTR regulates gene expression by anchoring hyper-edited transcripts in nuclear para-speckles (Prasanth et al. 2005; Tang et al. 2012). To test whether the A-to-I RNA editing affects ARHGAP26 gene expression, we first determined the effect of ADAR1 knockdown on the expression of ARHGAP26 by qPCR and Western blotting. MCF-10A and A549 cells were transfected with ADAR1-specific siRNAs, or nontargeting siRNA as a control. Total RNA and protein were prepared after 48 h and subjected to qPCR and Western blotting. The reduction in endogenous ADAR1 led to a decrease in ARHGAP26 mRNA and protein abundance (Fig. 3A; data not shown). Conversely, overexpression of ADAR1 resulted in an increase of ARHGAP26 mRNA and protein expression in MCF-10A cells (Fig. 3B). These data suggest that ADAR1 regulates the abundance of ARHGAP26 mRNA and protein in cells.
FIGURE 3.

ADAR1 regulates the gene expression of ARHGAP26. (A) qPCR (top) and Western blotting (bottom) analysis of ARHGAP26 expression levels following transient silencing of ADAR1 in human MCF-10A cells. (B) qPCR (top) and Western blotting (bottom) analysis of ARHGAP26 expression levels after transfecting MCF-10A cells with the ADAR1 expression vector or the empty vector.
Editing of the ARHGAP26 transcript leads to destruction of miR-30b-3p and miR-573 target sites
To explore how the RNA editing at specific sites affects the expression of ARHGAP26, we performed in silico analysis of these RNA editing sites in its 3′ UTR. MicroRNAs (miRNAs) are endogenous regulatory molecules that modulate gene expression or mRNA stability by pairing to the 3′ UTRs. Target recognition by an miRNA is mainly achieved by pairing to the miRNA seed sequence. A single-nucleotide mutation in the miRNA seed sequence could abolish miRNA binding and repression (Lewis et al. 2003; Brennecke et al. 2005). Recently, it was reported that A-to-I RNA editing may block miRNA:target recognition (Liang and Landweber 2007). We scanned the 3′-UTR sequence of ARHGAP26 using miRanda (Miranda et al. 2006) to identify putative miRNA binding sites and calculate minimum free energy (MFE) changes induced by the unedited (adenosine at the editing sites) or edited transcripts (guanine at the editing sites) of ARHGAP26. We observed that editing sites U3 and U4 were located in the putative target site of miR-30b-3p and editing site U12 resides in the putative target sites of miR-573. RNA editing increased the MFE of the corresponding RNA duplex and led to weaker miR-30b-3p:mRNA or miR-573:mRNA binding. This mechanism may also lead to the destruction of target sites presented in the ARHGAP26 transcript.
To investigate whether miR-30b-3p and miR-573 regulate ARHGAP26 gene expression, we performed transient transfection with miR-30b-3p or miR-573 mimics and inhibitors. MCF-10A cells were transfected with miR-30b-3p mimics or miR-573 mimics at a concentration of 80 nM, and whole-cell lysates were prepared 48 h post-transfection and subjected to Western blotting. As expected, ARHGAP26 protein levels were decreased in cells treated with the miR-30b-3p or miR-573 mimics, while no detectable reduction of the β-actin level was observed. In contrast, transfection with the miR-30b-3p or miR-573 inhibitors increased the protein levels of ARHGAP26 (Fig. 4A,B). These data suggest that miR-30b-3p and miR-573 repress the expression of ARHGAP26.
FIGURE 4.

Effects of A-to-I RNA editing at the U3 and U4 or U12 sites on the regulation of miR-30b-3p or miR-573. (A) Western blotting analysis of ARHGAP26 in MCF-10A cells transfected with miRNA negative control (NS-m) or miR-30b-3p mimics, miRNA inhibitor negative control (NS-i), or miR-30b-3p inhibitor. (B) Western blotting analysis of ARHGAP26 in MCF-10A cells transfected with miRNA negative control (NS-m) or miR-573 mimics, miRNA inhibitor negative control (NS-i), or miR-573 inhibitor, respectively. (C) Schematic representation of luciferase reporter constructs containing unedited (adenosine at the U3 and U4 editing sites) or edited (A → G at the U3 and U4 editing sites) form within the miR-30b-3p binding site. (D) Schematic representation of luciferase reporter constructs containing unedited (adenosine at the U12 editing sites) or edited (A → G at the U12 editing sites) formed within the miR-573 binding site. (E) The miR-30b-3p:target interaction was shown with the unedited or edited sites highlighted (upper panel). The effect of the unedited (A) or edited (G) form on the miR-30b-3p:target interaction by analysis of luciferase activity (lower panel). Relative luciferase activities for pGL3-26UTR-unedit or pGL3-26UTR-edit1 cotransfected with the predicted interacting miR-30b-3p and the negative control (=1); the results are presented as mean ± SD of three independent experiments performed in a total of six replicates. (**) Significant differences between unedited and edited targets (P < 0.01). (F) The miR-573:target interaction is shown with the unedited or edited sites highlighted (upper panel). The effect of the unedited (A) or edited (G) form on the miR-573:target interaction by analysis of luciferase activity (lower panel). Relative luciferase activities for pGL3-26UTR-unedit or pGL3-26UTR-edit2 cotransfected with the predicted interacting miR-573 and the negative control (=1); the results are presented as the mean ± SD of three independent experiments performed in a total of six replicates. (**) Significant differences between the unedited and edited targets (P < 0.01). (G) Immunoblotting of ARHGAP26 in MCF-10A cells transfected with control-miRNA, miR-30b-3p, or cotransfected with miR-30b-3p and ADAR1 expression vectors. (H) Immunoblotting of ARHGAP26 in MCF-10A cells transfected with control-miRNA, miR-573, or cotransfected with miR-573 and ADAR1 expression vectors. (I) qPCR analysis of miR-30b-3p in MCF-10A, MCF-7, Chang liver, and HepG2 cells. (J) qPCR analysis of miR-573 in MCF-10A, MCF-7, Chang liver, and HepG2 cells.
To further determine whether the RNA editing sites U3, U4, and U12 affect the binding of miR-30b-3p or miR-573 to the target 3′ UTR of ARHGAP26, we performed luciferase reporter assays. We constructed two pGL3-26UTR vectors carrying either the unedited type (adenosine at the editing sites) or the edited type (A → G at the U3, U4, and U12 editing sites) of the 3′-UTR transcript (Fig. 4C,D). HEK 293T cells were cotransfected with miR-30b-3p, miR-573 mimics, or the scrambled control. As shown in Figure 4, E and F, miR-30b-3p and miR-573 reduced the luciferase activity from the unedited plasmid by 30% and 20%, respectively; however, no suppression of the luciferase activity from the reporter containing edited-type sequence was observed. Thus, RNA editing inhibited the suppression of ARHGAP26 expression mediated by miR-30b-3p and miR-573, indicating that RNA editing of ARHGAP26 by ADAR1 may destroy the miR-30b-3p and miR-573 target sites in the 3′ UTR of ARHGAP26.
To investigate whether ADAR1 could rescue ARHGAP26 expression that was down-regulated by miR-30b-3p or miR-573 transfection, we cotransfected miR-30b-3p or miR-573 mimics with the ADAR1 expression vector into MCF-10A cells. Western blotting confirmed that the reduction in the amount of ARHGAP26 protein mediated by miR-30b-3p or miR-573 was rescued by ADAR1 overexpression (Fig. 4G,H). In addition, we examined the endogenous levels of miR-30b-3p or miR573 in MCF-10A cells, and in MCF-7, HepG2, and Chang liver cells. As shown in Figure 4, I and J, miR-30b-3p has a similar expression in all cell lines, whereas lower levels of miR-573 were observed in MCF-7 and HepG2 cells compared with the noncancerous control MCF-10A or Chang liver cells. Taken together, these results suggested that editing of ARHGAP26 transcript by ADAR1 leads to destruction of miR-30b-3p and miR-573 target sites.
ADAR1 regulates the activity of RhoA
ARHGAP26 is known to be a GAP for RhoA and Cdc42 (Hildebrand et al. 1996). Therefore, based on our finding that ADAR1 regulates the abundance of ARHGAP26, we hypothesized that ADAR1 might inhibit RhoA activity by regulating ARHGAP26 expression. To test the RhoA activity, we used a Rho binding domain of Rhotekin fused to GST (GST-RBD) pull-down assay, which only captures the active GTP-bound form of RhoA. As shown in Figure 5, A and B, knocking down ADAR1 caused an increase in the amount of RhoA-GTP, but when Flag-tagged ADAR1 was overexpressed, the amount of RhoA-GTP decreased; the total amount of RhoA was not affected. These results indicated that ADAR1 modulates RhoA activity probably through ARHGAP26.
FIGURE 5.

ADAR1 regulates the activity of RhoA. (A) Knockdown of ADAR1 increased the activity of RhoA in MCF-10A cells. MCF-10A cells were transfected with control siRNA or ADAR1 siRNA as indicated. GTP-RhoA was assayed using GST-RBD. (B) Overexpression of ADAR1 decreased the activity of RhoA in MCF-10A cells. MCF-10A cells were transfected with control vector or ectopic ADAR1 plasmid, as indicated. GTP-RhoA was assayed using GST-RBD.
ADAR1 and ARHGAP26 expression in human tissues
To test whether there is a correlation between ADAR1 and ARHGAP26 expressions in human tissues, we analyzed the expressions of ARHGAP26, ADAR1, ADAR2, miR-30b-3p, and miR-573 in nine human invasive ductal breast cancer samples versus the matched adjacent normal tissues using qPCR. We found that ADAR1 and ARHGAP26 showed a similar expression pattern in the nine pairs of human samples (Fig. 6A,B). In addition, an inverse expression pattern between ARHGAP26 and miR-573 was observed in six out of nine paired samples. However, no correlation was observed between ADAR2 and ARHGAP26 (Supplemental Fig. 4). MiR-30b-3p levels could not be detected in most of these samples (Fig. 6C,D).
FIGURE 6.

ADAR1 and ARHGAP26 expression in human invasive ductal breast cancers. qPCR analysis of (A) ADAR1, (B) ARHGAP26, (C) miR-573, and (D) miR-30b-3p in nine paired human invasive ductal breast cancers.
To determine whether there is a correlation between ADAR1 and ARHGAP26 expressions in other human cancers, we collected glioblastoma (GBM) specimens in which ADAR1 might be down-regulated (Paz et al. 2007), which would correlate with ARHGAP26's role as a tumor suppressor gene. Three pairs of human adult glioblastoma tissues versus the adjacent noncancerous tissue were used for qPCR, Western blotting, and RNA editing analyses. As expected, the mRNA and protein levels of ARHGAP26 and ADAR1 were decreased in the glioblastoma samples, while ADAR2 expression showed no change in two out of three glioblastoma samples (Fig. 7A–F). A significant decrease in the amount of RNA editing in the ARHGAP26 transcript was observed in human glioblastoma compared with the adjacent noncancerous tissues (Supplemental Fig. 5). Editing at sites U3, U4, and U12 was predicted to destroy miRNA binding sites (Fig. 4), indicating that decreased editing levels at these three sites are required for the repression of ARHGAP26 mediated by miR-30b-3p or miR-573. To exclude the possibility that decreased levels of ARHGAP26 were caused by overexpression of miR-30b-3p and miR-573, we used qPCR to examine the endogenous levels of miR-30b-3p or miR573 in the glioblastoma tissues. No significant increases in miR-30b-3p and miR-573 levels were found in most of the samples (Fig. 7G,H). These data suggested that the changes in the mRNA and protein levels of ARHGAP26 in the human tissues were associated with altered ADAR1 expression.
FIGURE 7.

Reduced expression of ARHGAP26 and ADAR1 in three human adult glioblastoma tissues. (A,C,E) Western blotting analysis of ARHGAP26, ADAR1, and ADAR2 expression. (B,D,F) qPCR analysis of ARHGAP26, ADAR1, and ADAR2 expression. (G) qPCR analysis of miR-30b-3p expression. (H) qPCR analysis of miR-573 expression.
DISCUSSION
In this study, we demonstrate that ADAR1 regulates the expression of ARHGAP26 by RNA editing, which disrupts the miR-30b-3p and miR-573 binding sites. A-to-I RNA editing in 3′ UTR of genes provides a layer of regulation in addition to microRNA modulation of target genes. The levels of A-to-I RNA editing determine the degree of disruption of miRNA-mediated repression. RNA editing increases the diversity of miRNA regulation of the target genes (Blow et al. 2006; Yang et al. 2006). Our data support the hypothesis that A-to-I RNA editing has the potential to block the miRNA:target recognition revealed by computational modeling (Liang and Landweber 2007). This is the first study to provide experimental evidence supporting this hypothesis. The prediction showed that more than 100 editing positions reside in miRNA seed sequences, although computational simulation suggests that RNA editing tends to avoid miRNA target sites in general (Liang and Landweber 2007). Further investigation is needed to verify the miRNA target sites and evaluate the functional consequences of specific A-to-I RNA editing. In contrast, A-to-I editing events have been demonstrated to have the capability to create functional miRNA target sites (Borchert et al. 2009). Destruction or creation of functional miRNA target sites are important features of A-to-I RNA editing for cross talk with miRNA:target recognition.
The ARHGAP26 protein is essential to the CLIC/GEEC endocytic pathway and muscle development via its capacity to regulate the GTPase activity of RhoA and Cdc42 proteins. Via the CLIC/GEEC endocytic pathway, ARHGAP26 is concentrated at the leading edge of spreading cells and facilitates cell morphological changes, which is important during the transformation of a normal cell into a cancer phenotype. Knocking down of ARHGAP26 decreased CLIC generation and impaired cell spreading and migration (Doherty et al. 2011a). Therefore, based on our findings that ADAR1 modulates ARHGAP26 protein abundance and negatively regulates the activity of RhoA, we hypothesize that ADAR1 affects the functions of the ARHGAP26 protein. In addition, ARHGAP26 was also thought to be a putative tumor suppressor, by negatively regulating RhoA and Cdc42 in human cancers, which have growth-promoting effects in oncoprotein Ras-mediated malignant transformation (Khosravi-Far et al. 1995; Bojesen et al. 2006). Thus, we selected brain cancer tissues in which ADAR1 was reported to be decreased (Glioma cell line U87 cells) (Paz et al. 2007) to determine if its expression correlated with the tumor suppressor function of ARHGAP26. A loss of ADAR1 and ARHGAP26 expression was observed in the three human adult glioblastomas studied here. However, ADAR1 expression in GBM is a controversial point: Two other studies showed either no difference in ADAR1 expression (Maas et al. 2001) or ADAR1 overexpression in pediatric GBMs (Cenci et al. 2008). Until now, there has been no report of ARHGAP26's involvement in brain cancer. Considering the limited samples in our study, the role of ADAR1 and ARHGAP26 in human brain cancer requires further investigation.
Thirteen A-to-I RNA editing sites showing a high level of editing were found in the 3′ UTR of human ARHGAP26 pre-mRNA (Fig. 1; Supplemental Fig. 1). RNA editing sites U3, U4, and U12 disrupt the miR-30b-3p and miR-573 target sites; however, the function of the other editing sites remains unknown. The 13 editing sites studied here are embedded in Alu elements, which are conserved ∼300-nucleotide (nt)–long repeat sequences in the human genome. Recent studies have suggested several potential roles of A-to-I RNA editing within Alu elements in gene expression (Chen and Carmichael 2008). Alu editing located in the coding region may change the amino acid sequence and thus potentially affect the protein function, while Alu editing within introns might influence splicing and cross talk with the microRNA pathway and regulate gene expression via nuclear retention (Chen and Carmichael 2008). Therefore, it remains to be determined whether the editing events identified in the present study would result in the nuclear retention of ARHGAP26. According to a previous study (Chen and Carmichael 2008), unedited-type ARHGAP26 protein is preferentially exported to the cytoplasm, whereas the highly edited RNA isoforms are selectively retained in the nucleus. However, ARHGAP26 was found to be distributed in the cytoplasm of HeLa and NIH 3T3 cells (Lundmark et al. 2008). Further study is warranted to explore the correlation between ARHGAP26 mRNA editing and protein cellular localization at high resolution.
In summary, we identified extensive A-to-I RNA editing sites in the 3′ UTR of the ARHGAP26 transcript that were specifically catalyzed by ADAR1. We found that ADAR1 regulated the expression of the ARHGAP26 gene through A-to-I RNA editing by destroying the miR-30b-3p and miR-573 binding sites. Furthermore, ARHGAP26 expression in the human tissues was associated with altered ADAR1 expression. Moreover, the regulation of ARHGAP26 expression by ADAR1 leads to changes in RhoA activity. Our results provide a novel insight into the mechanism by which ADAR1 and its RNA editing function regulate the microRNA modulation of target genes.
MATERIALS AND METHODS
Human tumor and control tissues
Three paired glioblastoma and adjacent noncancerous tissues were obtained from the 301 Hospital of PLA. Nine paired human invasive ductal breast cancer and the adjacent noncancerous tissues were obtained from the 307 Hospital of PLA and Peking Union Medical College Hospital. The ethics committee of the Beijing Institute of Biotechnology approved all the experiments using the tissues.
Cell culture and transfection
Ten cell lines were used in this study. The human prostate carcinoma cell line DU145, the human colon cancer cell line SW480, the human lung adenocarcinoma epithelial cell line A549, and the human Burkitt's lymphoma cell line Raji were originally purchased from ATCC and maintained in RPMI 1640 media supplemented with 10% fetal bovine serum, 100 units/mL penicillin, and 0.1 mg/mL streptomycin. The human hepatoma cell line HepG2, the human estrogen receptor (ER)–positive breast cancer cell line MCF-7, the human immortalized nontumor cell line Chang liver, the human osteosarcoma cell line U2OS, and the human renal epithelial cell line HEK-293T cell lines were grown in Dulbecco's modified Eagle's medium (DMEM) with 10% FBS, 100 units/mL penicillin, and 0.1 mg/mL streptomycin. The nontransformed human mammary epithelial cell line MCF-10A was maintained in DMEM/F12 (1:1) supplemented with 5% (v/v) heat-inactivated horse serum, 10 µg/mL insulin, 20 ng/mL EGF, 0.5 µg/mL hydrocortisone, 100 ng/mL Cholera toxin, and penicillin/streptomycin antibiotic. All cells were cultured at 37°C in a 5% CO2 incubator with humidified air. Plasmids were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instruction.
RNA extraction, RT-PCR, and real-time PCR
Total RNA was isolated with the TRIzol reagent (Invitrogen), and genomic DNA was extracted from the tissues and cultured cells according to the manufacturer's instructions of the Wizard genomic DNA purification kit (Promega). To remove genomic DNA contamination, RNA samples were treated with DNase I (Takara). First-strand cDNA was synthesized from total RNA with M-MLV (Takara) using oligo(dT) for the editing analysis and real-time PCR analysis.
For the editing analysis, PCR was performed according to standard procedures with 30 pM each primer and 2.5 units of rTaq DNA polymerase (Takara) to amplify the transcripts and the genomic DNA using the cDNA and gDNA as templates, respectively. The cycling conditions for amplification were as follows: initial denaturation at 95°C for 5 min; then 30 cycles at 95°C for 30 sec, 60°C for 30 sec, and 72°C for 50 sec; followed by a final extension at 72°C for 10 min. Control experiments were conducted without the reverse transcriptase enzyme (no RT-control) to verify that the amplified products were from the reverse-transcribed mRNA and not from contaminating genomic DNA. Specific products were gel-purified and directly sequenced. The following primers were used to amplify the 3′ UTR of ARHGAP26 containing the 13 editing sites: ARHGAP26-F (5′-GGAGGGGTATAGATTGTGCCGT-3′) and ARHGAP26-R (5′-TGTCTACAGGGATGCTTCGTGAG-3′). The following primers were used to amplify the CYFIP2 sequence: CYFIP2-F (5′-CTACCTAATGGATGGAAATGTCAGTAAC-3′) and CYFIP2-R (5′-CCCGGATCTGAACCATCTG-3′).
To analyze the mRNA expression of ADAR1, ADAR2, and ARHGAP26, quantitative real-time PCR was performed in triplicate on a MiniOpticon Real-Time PCR System (Bio-Rad) using SYBR Green Master Mix (Takara). The mRNA levels were normalized to the expression of the housekeeping gene GAPDH. The primers used were as follows: for GAPDH, forward (5′-CACCATCTTCCAGGAGCGAG-3′) and reverse (5′-GCAAATGAGCCCCAGCCT-3′); for ADAR1, forward (5′-GCTTGGGAACAGGGAATCG-3′) and reverse (5′-CTGTAGAGAAACCTGATGAAGCC-3′); for ADAR2, forward (5′-CGCAGGTTTTAGCTGACGC-3′) and reverse (5′-GCATCTTTAACATCTGTGCCTGT-3′); for ARHGAP26, forward (5′-TAAGAATGCTTCCAGGACCACTC-3′) and reverse (5′-GCTGTAACATCTGCCGATTTTTC-3′).
Analysis of microRNA expression by qRT-PCR
qRT-PCR assays were performed to measure the expression levels of microRNAs. Total RNA, containing microRNAs, was extracted from cells or tissues using the TRIzol reagent (Invitrogen). Hairpin-it miRNAs Real-Time PCR Quantitation Kit (GenePharma, China) containing a stem–loop-like RT primer, miRNA-specific PCR primer, and the molecular beacon probe was used to detected the miRNAs according to the protocol provided by the manufacturer. RNA was reversed-transcribed to cDNA with an miRNA-specific stem–loop-like RT primer, and U6 was used as the internal control. The expression of each miRNA relative to that of U6 was determined using the 2−ΔΔCT method.
Plasmid constructions
FLAG-ADAR1 was kindly provided by Dr. Carmo-Fonseca (Desterro et al. 2003). To assess the A-to-I RNA editing on the potential miR-30b-3p and miR-573 targets of ARHGAP26, pGL3-26UTR-unedit, pGL3-26UTR-edit1, and pGL3-26UTR-edit2 were constructed. The partial 3′ UTR of human ARHGAP26, which contains both potential miR-30b-3p and miR-573 targets, was amplified using primers 26UTR-F (5′-TAAAGATCTGCCTCAGGGGATGTGC-3′) and 26UTR-R (5′-ATAGGTACCCAGGGATGCTTCGTGAG-3′). The PCR products were cloned into pGL3-CM, in which the multiple cloning site of the pGL3-control vector (Promega) was removed and placed downstream from the luciferase gene, as previously described (Sun et al. 2008), to generate the luciferase reporter vector pGL3-26UTR-unedit. To mimic A-to-I RNA editing, we generated a luciferase reporter vector, pGL3-26UTR-edit1, which contains two A-to-G mutants in the potential miR-30b-3p targets, and pGL3-26UTR-edit2, which contains an A-to-G mutant in the potential miR-573 targets. The primers for site-directed mutagenesis were as follows: for pGL3-26UTR-edit1, forward (5′-GAACCCGGGAGGCGGAGGTT-3′) and reverse (5′-TCCGCCTCCCGGGTTCAAGT-3′); for pGL3-26UTR-edit2, forward (5′-TTGGGGGTCCGAAGTGGGC-3′) and reverse (5′-CTTCGGACCCCCAAAGTGCTG-3′). The PCR products of the mutated 3′ UTR of ARHGAP26 were cloned into pGL3-CM to generate pGL3-26UTR-edit1 or pGL3-26UTR-edit2.
Luciferase assay
To assess the effect of A-to-I RNA editing in the miRNA target sites, HEK293T cells were cotransfected in 24-well plates with pGL3 (luciferase reporter vectors), pRL-TK (Renilla; Promega), miRNA mimics, or scrambled negative control (Genepharma, Shanghai), using Lipofectamine 2000 (Invitrogen). Twenty-four hours after transfection, cells were lysed in 100 µL of passive lysis buffer according to the Dual-Luciferase reporter assay protocol (Promega), and luciferase activity was measured by using the Dual-Luciferase reporter assay system (Promega) on a TD-20/20 Luminometer (Turner Designs). All experiments were repeated at least three times.
RNA interference
To knock down ADAR1 or ADAR2 in A549 or MCF-10A cells, duplex siRNAs for target sequence and nontarget siRNA control (Negative control) were synthesized by Genepharma, Shanghai and transfected, respectively, into A549 or MCF-10A cells at a concentration of 30 nM using Lipofectamine RNAiMAX (Invitrogen), following the manufacturer's instructions. siRNAs targeting ADAR1 were as follows: si-ADAR1-1# (sense, GCGACUAUCUCUUCAAUGUTT; antisense, ACAUUGAAGAGAUAGUCGCTT); si-ADAR1-2# (sense, GGCCCGAGAUAUAAAUGCUTT; antisense, AGCAUUUAUAUCUCGGGCCTT); si-ADAR1-3# (sense, GCCCAUUUAUCUCAAAUCUTT; antisense, AGAUUUGAGAUAAAUGGGCTT). siRNAs targeting ADAR2 were as follows: si-ADAR2-1# (sense, GCGCCUUUGUUUGUCAUGUTT; antisense, ACAUGACAAACAAAGGCGCTT); si-ADAR2-2# (sense, CAGGCACAGAUGUUAAAGATT; antisense, UCUUUAACAUCUGUGCCUGTT); si-ADAR2-3# (sense, CCGCUAUUGAGGUCAUCAATT; antisense, UUGAUGACCUCAAUAGCGGTT). Controls included a nontargeting siRNA (NC). The extent and specificity of the silencing of ADARs were assessed by qPCR and Western blotting.
Transient miRNA mimics/inhibitors transfection
To determine the effects of miR-30b-3p or miR-573 on the expression of ARHGAP26 in MCF-10A cells, we performed transient miRNA mimics/inhibitors transfection and Western blotting. MiR-30b-3p mimics, miR-573 mimics and negative control miRNA mimics (NC mimics), miR-30b-3p inhibitors, and miR-573 inhibitors and negative controls were synthesized by Genepharma and transfected into MCF-10A cells at a concentration of 80 nM using Lipofectamine RNAiMAX (Invitrogen) following the manufacturer's instructions. Cotransfection of ADAR1 expression constructs (1 µg of DNA) and miRNA mimics (80 nM per well of six-well plates) was done with Lipofectamine 2000 (Invitrogen).
Western blotting
Total protein extracts of transfected cells or human tissues were prepared for Western blotting in lysis buffer consisting of 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, and 50 mM Tris-HCl (pH 8.0) supplemented with a mixture of protease inhibitors (Roche). For immunoblotting, protein exacts were separated by 10% SDS-PAGE and transferred to PVDF membranes. The antibodies for ADAR1, ADAR2, and RhoA were from Santa Cruz Biotechnology; the antibody for ARHGAP26 was from Abcam; the antibody for Flag was from Sigma-Aldrich, and the antibody for β-actin was purchased from Cell Signaling Technology.
GTPase activity pull-down assay
The RhoA activity assay was performed using the Rho binding domain of Rhotekin fused to GST (GST-RBD). Briefly, after washing with PBS, cells were lysed immediately with RBD lysis buffer (50 mM Tris-HCl at pH 7.2, 500 mM NaCl, 5 mM MgCl2, 1% Triton X-100, 1 mM dithiothreitol, and Cocktail). Cell lysates were clarified by centrifugation at 16,000g for 10 min at 4°C. Equal volumes of the lysates were incubated with GST-RBD fusion proteins on glutathione-S-transferase beads, to pull down active RhoA proteins. GST proteins were used as a negative control. After incubation for 2 h at 4°C, the beads were washed three times with cold RBD lysis buffer. The RhoA proteins were eluted with sample buffer and subjected to SDS–polyacrylamide gel electrophoresis. Western blotting was performed using anti-RhoA antibodies.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Dr. Chenguang Wang for critical reading and editing. This work was supported partially by the National Natural Science Foundation of China (30800644 and 31100940).
REFERENCES
- Barresi V, Ragusa A, Fichera M, Musso N, Castiglia L, Rappazzo G, Travali S, Mattina T, Romano C, Cocchi G, et al. 2010. Decreased expression of GRAF1/OPHN-1-L in the X-linked α thalassemia mental retardation syndrome. BMC Med Genomics 3: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass BL, Weintraub H 1988. An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell 55: 1089–1098 [DOI] [PubMed] [Google Scholar]
- Bass BL, Nishikura K, Keller W, Seeburg PH, Emeson RB, O'Connell MA, Samuel CE, Herbert A 1997. A standardized nomenclature for adenosine deaminases that act on RNA. RNA 3: 947–949 [PMC free article] [PubMed] [Google Scholar]
- Blow MJ, Grocock RJ, van Dongen S, Enright AJ, Dicks E, Futreal PA, Wooster R, Stratton MR 2006. RNA editing of human microRNAs. Genome Biol 7: R27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojesen SE, Ammerpohl O, Weinhausl A, Haas OA, Mettal H, Bohle RM, Borkhardt A, Fuchs U 2006. Characterisation of the GRAF gene promoter and its methylation in patients with acute myeloid leukaemia and myelodysplastic syndrome. Br J Cancer 94: 323–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchert GM, Gilmore BL, Spengler RM, Xing Y, Lanier W, Bhattacharya D, Davidson BL 2009. Adenosine deamination in human transcripts generates novel microRNA binding sites. Hum Mol Genet 18: 4801–4807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennecke J, Stark A, Russell RB, Cohen SM 2005. Principles of microRNA–target recognition. PLoS Biol 3: e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenci C, Barzotti R, Galeano F, Corbelli S, Rota R, Massimi L, Di Rocco C, O'Connell MA, Gallo A 2008. Down-regulation of RNA editing in pediatric astrocytomas: ADAR2 editing activity inhibits cell migration and proliferation. J Biol Chem 283: 7251–7260 [DOI] [PubMed] [Google Scholar]
- Chen LL, Carmichael GG 2008. Gene regulation by SINES and inosines: Biological consequences of A-to-I editing of Alu element inverted repeats. Cell Cycle 7: 3294–3301 [DOI] [PubMed] [Google Scholar]
- Chen CX, Cho DS, Wang Q, Lai F, Carter KC, Nishikura K 2000. A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single- and double-stranded RNA binding domains. RNA 6: 755–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desterro JM, Keegan LP, Lafarga M, Berciano MT, O'Connell M, Carmo-Fonseca M 2003. Dynamic association of RNA-editing enzymes with the nucleolus. J Cell Sci 116: 1805–1818 [DOI] [PubMed] [Google Scholar]
- Doherty GJ, Ahlund MK, Howes MT, Moren B, Parton RG, McMahon HT, Lundmark R 2011a. The endocytic protein GRAF1 is directed to cell-matrix adhesion sites and regulates cell spreading. Mol Biol Cell 22: 4380–4389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty JT, Lenhart KC, Cameron MV, Mack CP, Conlon FL, Taylor JM 2011b. Skeletal muscle differentiation and fusion are regulated by the BAR-containing Rho-GTPase-activating protein (Rho-GAP), GRAF1. J Biol Chem 286: 25903–25921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farajollahi S, Maas S 2010. Molecular diversity through RNA editing: A balancing act. Trends Genet 26: 221–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galeano F, Rossetti C, Tomaselli S, Cifaldi L, Lezzerini M, Pezzullo M, Boldrini R, Massimi L, Di Rocco CM, Locatelli F, et al. 2013. ADAR2-editing activity inhibits glioblastoma growth through the modulation of the CDC14B/Skp2/p21/p27 axis. Oncogene 32: 998–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo A, Galardi S 2008. A-to-I RNA editing and cancer: From pathology to basic science. RNA Biol 5: 135–139 [DOI] [PubMed] [Google Scholar]
- Hildebrand JD, Taylor JM, Parsons JT 1996. An SH3 domain-containing GTPase-activating protein for Rho and Cdc42 associates with focal adhesion kinase. Mol Cell Biol 16: 3169–3178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiuchi S, Tsuzuki K, Yoshida Y, Yamada N, Hagimura N, Okado H, Miwa A, Kurihara H, Nakazato Y, Tamura M, et al. 2002. Blockage of Ca2+-permeable AMPA receptors suppresses migration and induces apoptosis in human glioblastoma cells. Nat Med 8: 971–978 [DOI] [PubMed] [Google Scholar]
- Kallman AM, Sahlin M, Ohman M 2003. ADAR2 A → I editing: Site selectivity and editing efficiency are separate events. Nucleic Acids Res 31: 4874–4881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y, Ito K, Sun H, Aizawa H, Kanazawa I, Kwak S 2004. Glutamate receptors: RNA editing and death of motor neurons. Nature 427: 801. [DOI] [PubMed] [Google Scholar]
- Kawahara Y, Zinshteyn B, Chendrimada TP, Shiekhattar R, Nishikura K 2007a. RNA editing of the microRNA-151 precursor blocks cleavage by the Dicer–TRBP complex. EMBO Rep 8: 763–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y, Zinshteyn B, Sethupathy P, Iizasa H, Hatzigeorgiou AG, Nishikura K 2007b. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science 315: 1137–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosravi-Far R, Solski PA, Clark GJ, Kinch MS, Der CJ 1995. Activation of Rac1, RhoA, and mitogen-activated protein kinases is required for Ras transformation. Mol Cell Biol 15: 6443–6453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DD, Kim TT, Walsh T, Kobayashi Y, Matise TC, Buyske S, Gabriel A 2004. Widespread RNA editing of embedded alu elements in the human transcriptome. Genome Res 14: 1719–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann KA, Bass BL 2000. Double-stranded RNA adenosine deaminases ADAR1 and ADAR2 have overlapping specificities. Biochemistry 39: 12875–12884 [DOI] [PubMed] [Google Scholar]
- Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB 2003. Prediction of mammalian microRNA targets. Cell 115: 787–798 [DOI] [PubMed] [Google Scholar]
- Liang H, Landweber LF 2007. Hypothesis: RNA editing of microRNA target sites in humans? RNA 13: 463–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundmark R, Doherty GJ, Howes MT, Cortese K, Vallis Y, Parton RG, McMahon HT 2008. The GTPase-activating protein GRAF1 regulates the CLIC/GEEC endocytic pathway. Curr Biol 18: 1802–1808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas S, Patt S, Schrey M, Rich A 2001. Underediting of glutamate receptor GluR-B mRNA in malignant gliomas. Proc Natl Acad Sci 98: 14687–14692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melcher T, Maas S, Herb A, Sprengel R, Higuchi M, Seeburg PH 1996a. RED2, a brain-specific member of the RNA-specific adenosine deaminase family. J Biol Chem 271: 31795–31798 [DOI] [PubMed] [Google Scholar]
- Melcher T, Maas S, Herb A, Sprengel R, Seeburg PH, Higuchi M 1996b. A mammalian RNA editing enzyme. Nature 379: 460–464 [DOI] [PubMed] [Google Scholar]
- Miranda KC, Huynh T, Tay Y, Ang YS, Tam WL, Thomson AM, Lim B, Rigoutsos I 2006. A pattern-based method for the identification of microRNA binding sites and their corresponding heteroduplexes. Cell 126: 1203–1217 [DOI] [PubMed] [Google Scholar]
- Miyamura Y, Suzuki T, Kono M, Inagaki K, Ito S, Suzuki N, Tomita Y 2003. Mutations of the RNA-specific adenosine deaminase gene (DSRAD) are involved in dyschromatosis symmetrica hereditaria. Am J Hum Genet 73: 693–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikura K 2010. Functions and regulation of RNA editing by ADAR deaminases. Annu Rev Biochem 79: 321–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz N, Levanon EY, Amariglio N, Heimberger AB, Ram Z, Constantini S, Barbash ZS, Adamsky K, Safran M, Hirschberg A, et al. 2007. Altered adenosine-to-inosine RNA editing in human cancer. Genome Res 17: 1586–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasanth KV, Prasanth SG, Xuan Z, Hearn S, Freier SM, Bennett CF, Zhang MQ, Spector DL 2005. Regulating gene expression through RNA nuclear retention. Cell 123: 249–263 [DOI] [PubMed] [Google Scholar]
- Qian Z, Lin J, Qian J, Yao DM, Wang YL, Han LX, Zhu ZH, Xiao GF 2010. [Quantification of GRAF gene expression in patients with acute myeloid leukemia using EvaGreen real time quantitative PCR.] Zhonghua Yi Xue Yi Chuan Xue Za Zhi 27: 290–293 [DOI] [PubMed] [Google Scholar]
- Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, et al. 2012. Mutations in ADAR1 cause Aicardi-Goutières syndrome associated with a type I interferon signature. Nat Genet 44: 1243–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedmann EM, Schopoff S, Hartner JC, Jantsch MF 2008. Specificity of ADAR-mediated RNA editing in newly identified targets. RNA 14: 1110–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueter SM, Dawson TR, Emeson RB 1999. Regulation of alternative splicing by RNA editing. Nature 399: 75–80 [DOI] [PubMed] [Google Scholar]
- Saunders LR, Barber GN 2003. The dsRNA binding protein family: Critical roles, diverse cellular functions. FASEB J 17: 961–983 [DOI] [PubMed] [Google Scholar]
- Schmauss C 2005. Regulation of serotonin 2C receptor pre-mRNA editing by serotonin. Int Rev Neurobiol 63: 83–100 [DOI] [PubMed] [Google Scholar]
- Schumacher JM, Lee K, Edelhoff S, Braun RE 1995. Distribution of Tenr, an RNA-binding protein, in a lattice-like network within the spermatid nucleus in the mouse. Biol Reprod 52: 1274–1283 [DOI] [PubMed] [Google Scholar]
- Sun Q, Zhang Y, Yang G, Chen X, Zhang Y, Cao G, Wang J, Sun Y, Zhang P, Fan M, et al. 2008. Transforming growth factor-β-regulated miR-24 promotes skeletal muscle differentiation. Nucleic Acids Res 36: 2690–2699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W, Fei Y, Page M 2012. Biological significance of RNA editing in cells. Mol Biotechnol 52: 91–100 [DOI] [PubMed] [Google Scholar]
- Tariq A, Garncarz W, Handl C, Balik A, Pusch O, Jantsch MF 2013. RNA-interacting proteins act as site-specific repressors of ADAR2-mediated RNA editing and fluctuate upon neuronal stimulation. Nucleic Acids Res 41: 2581–2593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner RW, Smith JE, Cooperman BS, Nishikura K 1989. A double-stranded RNA unwinding activity introduces structural alterations by means of adenosine to inosine conversions in mammalian cells and Xenopus eggs. Proc Natl Acad Sci 86: 2647–2651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong SK, Sato S, Lazinski DW 2001. Substrate recognition by ADAR1 and ADAR2. RNA 7: 846–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Chendrimada TP, Wang Q, Higuchi M, Seeburg PH, Shiekhattar R, Nishikura K 2006. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat Struct Mol Biol 13: 13–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zohrabian VM, Nandu H, Gulati N, Khitrov G, Zhao C, Mohan A, Demattia J, Braun A, Das K, Murali R, et al. 2007. Gene expression profiling of metastatic brain cancer. Oncol Rep 18: 321–328 [PubMed] [Google Scholar]