

The RNA recognition motif (RRM) is the most common eukaryotic RNA-binding domain, with differences in its amino acid sequence affecting binding specificity and affinity. This study applied a deep mutational scanning approach to score the functional activity of more than 100,000 variants of the RRM2 domain of the yeast poly(A)-binding protein, Pab1. The authors use these data to refine the current understanding of the Pab1 domain and of RRM domains in general.
Keywords: epistasis, Pab1, RNA recognition motif, RNA-binding protein, structure-function analysis
Abstract
The RNA recognition motif (RRM) is the most common RNA-binding domain in eukaryotes. Differences in RRM sequences dictate, in part, both RNA and protein-binding specificities and affinities. We used a deep mutational scanning approach to study the sequence-function relationship of the RRM2 domain of the Saccharomyces cerevisiae poly(A)-binding protein (Pab1). By scoring the activity of more than 100,000 unique Pab1 variants, including 1246 with single amino acid substitutions, we delineated the mutational constraints on each residue. Clustering of residues with similar mutational patterns reveals three major classes, composed principally of RNA-binding residues, of hydrophobic core residues, and of the remaining residues. The first class also includes a highly conserved residue not involved in RNA binding, G150, which can be mutated to destabilize Pab1. A comparison of the mutational sensitivity of yeast Pab1 residues to their evolutionary conservation reveals that most residues tolerate more substitutions than are present in the natural sequences, although other residues that tolerate fewer substitutions may point to specialized functions in yeast. An analysis of ∼40,000 double mutants indicates a preference for a short distance between two mutations that display an epistatic interaction. As examples of interactions, the mutations N139T, N139S, and I157L suppress other mutations that interfere with RNA binding and protein stability. Overall, this study demonstrates that living cells can be subjected to a single assay to analyze hundreds of thousands of protein variants in parallel.
INTRODUCTION
The RNA recognition motif (RRM) is one of the most common protein domains in eukaryotes, encoded in ∼2% of all human genes (Maris et al. 2005). This ∼90-amino acid domain is present in proteins with roles in post-transcriptional processes such as pre-mRNA processing, mRNA nuclear export, translational regulation, and mRNA decay (Mangus et al. 2003; Erkmann and Kutay 2004; Deschenes-Furry et al. 2006; Kuhn et al. 2009). About half of the proteins containing an RRM have multiple copies of this domain (Maris et al. 2005; Clery et al. 2008), with the spatial arrangement of the domains, their sequence variation, and the presence of auxiliary domains dictating the affinity, specificity, and function of these proteins (Lunde et al. 2007).
A typical RRM folds into a four-stranded antiparallel β sheet, packed against two α helices, with RNA binding usually achieved by contacts made between the β sheet surface and a single-stranded RNA (Maris et al. 2005; Clery et al. 2008; Muto and Yokoyama 2012). Two highly conserved motifs, RNP1 (consensus K/R-G-F/Y-G/A-F/Y-V/I/L-X-F/Y, where X is any amino acid) and RNP2 (consensus I/V/L-F/Y-I/V/L-X-N-L), in the central two β strands, are the primary mediators of RNA binding (Adam et al. 1986; Swanson et al. 1987; Dreyfuss et al. 1988).
The poly(A)-binding protein (PABP) is a well-characterized RRM-containing protein (Dreyfuss et al. 2002; Maris et al. 2005; Lunde et al. 2007; Muto and Yokoyama 2012) and was the first member of the RRM family to be identified (Adam et al. 1986; Sachs et al. 1986). There are two major forms of PABP, which differ both in structure and in function. A nuclear poly(A)-binding protein (PABPN) is required for efficient polyadenylation of mRNA tails in the nucleus (Kuhn et al. 2009). A cytoplasmic poly(A)-binding protein (PABPC) plays roles in mRNA translation and decay, with each protomer associating with ∼27 nucleotides of poly(A) (Baer and Kornberg 1983).
The PAB1 gene of the yeast Saccharomyces cerevisiae encodes an essential cytoplasmic poly(A)-binding protein of 577 amino acids (Adam et al. 1986; Sachs et al. 1986). Pab1 consists of four tandem RRM domains that are highly conserved among cytoplasmic PABP members, as well as a proline-rich linker and a C-terminal domain (Adam et al. 1986; Sachs et al. 1986). The RRM domains associate directly with the RNA molecule, while the C-terminal region is not required for RNA binding or yeast viability (Sachs et al. 1987; Burd et al. 1991). In addition to poly(A) binding, all Pab1 RRM domains mediate protein–protein interactions (Kessler and Sachs 1998; Yao et al. 2007; Richardson et al. 2012). In particular, binding of Pab1 RRM2 to the eukaryotic initiation factor 4G (eIF4G) (Kessler and Sachs 1998) is presumed to promote the formation of a closed-loop structure between the mRNA cap and the poly(A) tail (Jacobson and Favreau 1983; Wells et al. 1998; Amrani et al. 2008) and to stimulate mRNA translation (Tarun et al. 1997; Imataka et al. 1998; Park et al. 2010).
The modular arrangement of Pab1 RRM domains shows functional redundancy. Fragments composed of RRM1–RRM2, RRM2–RRM3, and RRM3–RRM4 can bind independently to RNA in vitro (Sachs et al. 1987; Burd et al. 1991). In vivo, yeast survive most Pab1 deletions that remove large parts from either single or two adjacent RRM domains (Sachs et al. 1987), and a mutation in each RNP1 motif of the four RRM domains is required to reduce poly(A) binding sufficiently to abolish growth of yeast (Deardorff and Sachs 1997).
We sought to define the determinants of an RRM domain of the yeast Pab1 protein by the use of a method known as deep mutational scanning (Fowler et al. 2010; Araya and Fowler 2011). This method allows a large number of mutant versions of a protein to be scored for function in a single experiment. It combines high-throughput DNA sequencing with a selection in which a physical association is maintained between each protein variant and the DNA that encodes it. The sequence analysis provides the frequency of each variant in an input population and in a population after selection, with this ratio serving as a proxy for the function of each variant (Fowler et al. 2010). We demonstrated that a Pab1 construct carrying the first three RRM domains was sufficient for near wild-type growth of yeast, yet was highly sensitive to a point mutation in RRM2. This result allowed us to generate plasmid libraries containing mutations in RRM2, and to score more than 100,000 unique variants, including 1246 with single amino acid substitutions, for their ability to support the growth of yeast. Using these data, we measured the contribution of each structural element in RRM2 to Pab1 performance, dissected the in vivo effects of mutations at known RNA-binding residues and other interaction sites, and identified non-RNA-binding residues essential to RRM2 function.
RESULTS
Mutagenesis of the Pab1 RRM2 domain
We sought to establish an in vivo assay for scoring the function of variants of the Pab1 RRM2 domain based on complementation of the pab1Δ mutation. We deleted the endogenous wild-type PAB1 gene from the BY4741 strain, and because PAB1 is essential, the cells were maintained via expression of this gene from a plasmid under the control of a tetracycline-off promoter (Fig. 1A). We transformed these cells with a second plasmid that constitutively expressed a variant of PAB1 (Fig. 1A). Addition of a tetracycline analog (doxycyline) to the culture shut off the expression of the wild-type gene, making the cells completely dependent on the mutated PAB1 for their growth.
FIGURE 1.
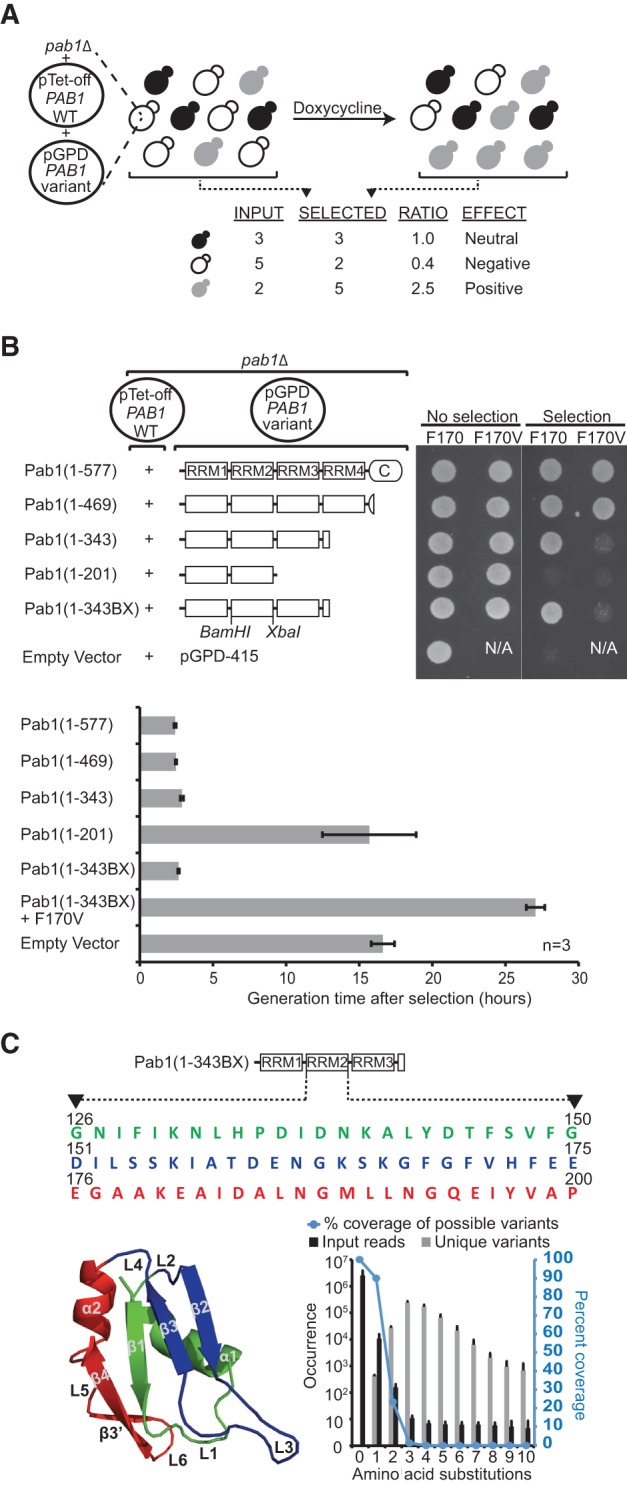
Experimental design of the deep mutational scan for the Pab1 RRM2 domain. (A) Protocol to assess the effects of RRM2 mutations on the in vivo function of Pab1. pab1Δ cells carry two plasmids, one expressing the full-length Pab1 protein under a tetracycline-off promoter (pTet-PAB1 WT) and the other expressing one of many variants of Pab1 from a constitutively active promoter (pGPD-PAB1 variant). The cells are grown to logarithmic phase in liquid culture, and a tetracycline analog (doxycycline) is added to the media. Cells expressing variants of Pab1 that cannot fully complement the loss of the wild-type protein grow slower than cells expressing neutral variants of Pab1. Sequencing the mutated fragments of the variant population before (input) and after selection (output) can be used to quantify these effects as the ratio of frequencies in each pool for each variant. (B) Selection of a Pab1 fragment that displays growth-rate sensitivity to a single point mutation. pab1Δ cells carrying the two plasmids specified in A with the variant plasmid expressing truncated forms of Pab1 with or without F170V substitution were tested for growth in the presence of doxycycline (20 μg/mL) on plates (top) and in liquid culture (bottom). Generation time was calculated starting from 8 h after doxycycline addition to eliminate cell divisions due to residual Pab1 activity. Note that cells carrying an empty vector grow at a low rate, probably due to leaky expression of the wild-type protein. (C) RRM2 mutagenesis. Shown are the Pab1(1–343BX) construct and the RRM2 sequence that was mutagneized. Each colored sequence and the corresponding elements on the structure of the human RRM2 domain (PDB_ID 1CVJ) represents a 25-amino acid-long RRM2 sequence that was doped with an average of three DNA base substitutions per variant. The graph at the bottom right depicts averages values of the listed properties of the three input libraries with respect to variants carrying a specified number of amino acid substitutions.
We required a Pab1 construct in which single amino acid changes in RRM2 would affect the activity of the protein. Point mutations in one of the RRM domains, designed to disrupt the domain's ability to bind RNA, are suppressed by the redundant function of the other three RRM domains (Deardorff and Sachs 1997). Therefore, we tested a series of Pab1 C-terminal truncations both for their ability to support cell growth and for their sensitivity to a single amino acid substitution, F170V, which disrupts RNA binding (Deardorff and Sachs 1997). Like the full-length protein Pab1(1–577), a construct lacking most of the C-terminal domain, Pab1(1–469), was sufficient for growth and was insensitive to the F170V mutation (Fig. 1B). However, a further truncation, Pab1(1–343), which includes RRM1–RRM2–RRM3 and the N-terminal 25 amino acids of RRM4, resulted in good growth upon doxycycline treatment only when F170 was present; the substitution F170V in this construct resulted in almost no growth (Fig. 1B). In liquid culture, cells carrying the Pab1(1–343) fragment grew at a slightly decreased rate, which might be due to the loss of RRM4 rather than to the absence of the C-terminal region, as deletion of the C-terminal region alone did not affect growth (Fig. 1B, bottom). Based on these observations, we chose the Pab1(1–343) fragment as the scaffold into which mutations would be introduced. To avoid unwanted PCR amplification of the wild-type RRM2 domain encoded on the tetracycline-off promoter plasmid, we introduced a total of 18 synonymous changes including BamHI and XbaI sites in the eight codons N-terminal and C-terminal to RRM2, and designated this construct as Pab1(1–343BX). These mutations had no effect on growth (Fig. 1B).
We created three separate libraries from DNA oligonucleotides that were made double stranded and cloned into the Pab1(1–343BX) construct (Fig. 1C). Each library spanned 75 bases (i.e., 25 amino acids) in RRM2, with an average of three mutations per variant. Yeast carrying each one of the library pools were grown to logarithmic phase and then diluted into doxycycline-containing media. We collected samples before (input) and after 22 h of growth in the presence of doxycyline (selected), extracted plasmids, PCR amplified the segment that had been mutated, and carried out sequence analysis. The enrichment score for each variant, based on the change in frequency from input to selection, serves as a proxy for the function of the variant (Fig. 1A).
Enrichment scores were generated for hundreds of thousands of DNA and protein variants (Supplemental Table 1). Input read counts ranged from a single read for variants with multiple base substitutions to tens of thousands of reads for variants with a single-base substitution. Assuming that most synonymous mutations have a negligible effect on Pab1 activity in this assay, we used the enrichment score distribution of ∼5000 synonymous variants (carrying either single or multiple synonymous substitutions) to assess the effect of input read depth on the reliability of the enrichment scores. Based on a variance cutoff of 0.25 (Supplemental Fig. 1), we required a read depth of at least 40 input reads for inclusion of a variant in further analysis. This cutoff provided data for 110,745 protein variants, including 1246 single amino acid substitutions (∼83% of all possible ones in each library); 39,912 double amino acid substitutions (∼11% of all possible ones in each library); and many other variants with three or more mutations. The enrichment score distribution of all variants (normalized to the wild-type enrichment score) revealed that, in general, most mutations were deleterious for RRM2 function. Unlike variants with missense mutations, variants with synonymous mutations had enrichment scores that were concentrated around the wild-type score (Supplemental Fig. 2). Assuming that neutral variants with missense mutations have an enrichment score distribution similar to that of synonymous mutants, we used the enrichment score distribution of synonymous variants to calculate an empirical false discovery rate (FDR) of neutral variants among variants carrying missense mutations. For all three segments, the distribution of synonymous variants suggested that all missense variants with an enrichment score >1 are likely to be neutral (Supplemental Fig. 2). For variants with an enrichment score <1, the FDR of neutral variants dropped sharply as enrichment scores decreased (Supplemental Fig. 2), with an average estimate of ∼25%, 8.5%, and 3.25% of variants with log2 enrichment scores of −0.5, −1.0, and −2.0 being neutral, respectively, for the three library segments. These distributions indicate that low enrichment scores arise mostly from the failure of Pab1 to function rather than from stochastic variation in measurements. After correcting for the enrichment score distribution of neutral variants with missense mutations, we estimated the fraction of variants carrying deleterious mutations (i.e., enrichment scores <1) to be ∼83%, 81%, and 63% of total variants for libraries 1, 2, and 3, respectively.
FIGURE 2.

Effect of single amino acid substitutions on the in vivo function of the Pab1 RRM2 domain. (A) A heat map displaying the enrichment scores (log2 transformed) of single amino acid substitutions in the RRM2 domain. Each column represents an RRM2 sequence position and each row a substitution to a specific amino acid. An asterisk designates the row of nonsense mutations. Color ranges from blue for the most deleterious mutations to red for the most beneficial ones. Substitutions that were not sequenced in the input or selected pools or that were eliminated by subsequent quality filtration steps are shown in gray; wild-type residues are marked with white dots. The secondary structure of the RRM2 domain aligned to the sequence is shown below the heat map as well as the average enrichment score for each position. (B) The average enrichment scores are projected on the crystal structure of the human RRM2 (PDB_ID 1CVJ). RRM1 and the connecting linker are shown in black and the poly(A) RNA in orange.
Effect of single amino acid substitutions
We generated a mutational sensitivity map for single mutations that shows the enrichment scores of 1190 missense and 56 nonsense mutations (Fig. 2A; Supplemental Table 2). Several observations suggest that these enrichment scores correlate with the function of the Pab1 RRM2 domain. First, missense mutations led to a wide range of growth, whereas nonsense mutations uniformly resulted in extremely poor growth (median enrichment score of 0.06). Second, proline was the most harmful missense substitution (median enrichment score of 0.22) compared with all other missense substitutions (median enrichment score of 0.8), consistent with the disruptive nature of a proline mutation on α helices and β sheet structures (Chou and Fasman 1978). Third, we found a good correspondence between the effect of previously characterized RRM2 mutations and enrichment scores; RRM2 F170V (enrichment score of 0.04) reduces binding of the protein to poly(A) by >97% (Deardorff and Sachs 1997) and RRM2 K166Q (enrichment score of 0.65), if combined with mutations to the equivalent residues in the other three RRMs, reduces binding to poly(A) by >70% (Deardorff and Sachs 1997).
While the enrichment scores of the single amino acid substitutions indicate that most mutations were deleterious for RRM2 function, a few mutations had enrichment scores that were greater than that of the wild type. In particular, the enrichment score for Q194C was 2.9. However, we measured the growth rate of yeast cells carrying Q194C and found that it was the same as those carrying the wild-type version (data not shown). This observation agrees with our finding that enriched variants follow the distribution of synonymous mutants and therefore are likely to be neutral (Supplemental Fig. 2).
The distribution of the single amino acid substitution enrichment scores along the sequence and structure of RRM2 points to the β sheet as the element most sensitive to mutation (Fig. 2A,B). In particular, strands β1 and β3, which carry the RNP motifs, show the highest sensitivity to mutation (both with a median enrichment score of 0.09), suggesting that RNA binding mediated by these two motifs is the most important in vivo function of the RRM2 domain. Strands β2 and β4, which assist poly(A) binding in vitro (Deo et al. 1999), appear to contribute less to this function (median enrichment scores 0.38 and 0.90, respectively). In helices α1 and α2, residues with side chains oriented toward the core showed greater sensitivity to mutation than surface residues (Fig. 2B). Additionally, helix α2 was less sensitive to mutation (median enrichment score 0.90) than helix α1 (median enrichment score 0.76). In particular, mutations at residues 180–181 (KE) and 184–186 (DAL), which are part of the eIF4G binding site in helix α2 (Otero et al. 1999), had only minor effects on cell growth (median enrichment score 0.89). In vitro, mutations at these sites result in complete loss of eIF4G binding and diminished mRNA translation (Otero et al. 1999), but in vivo, a weak affinity of RRM1 for eIF4G may compensate for loss of eIF4G binding by RRM2 (Kessler and Sachs 1998; Richardson et al. 2012). Lastly, of the loop regions, L2, which connects helix α1 to strand β2 by a four amino acid turn, was the least resistant to mutation (median enrichment score 0.19), making it the most sensitive element after strands β1 and β3.
Clustering mutation sensitivity profiles identifies structurally related residues
We clustered both RRM2 positions (along the x-axis) and the substituting amino acids (along the y-axis) by the similarity in their sensitivity profiles (Fig. 3A). This clustering grouped together amino acids that have similar chemical properties such as hydrophobic, aromatic, positively charged, and negatively charged. That replacements of amino acids with similar ones resulted in correspondingly similar enrichment scores argues that the deep mutational scanning assay provides a sensitive and accurate readout of mutational sensitivity.
FIGURE 3.

Clustering the effects of single amino acid substitutions groups structurally related residues. (A) Pab1 RRM2 positions and substituting amino acids were clustered based on enrichment score values and color coded as shown in Figure 2. The dotted line creates three clusters of RRM2 residues. Positions corresponding to RNA-binding residues in the human RRM2 are colored for their clustering (magenta) or lack of clustering (cyan) to group I. Positions corresponding to aliphatic residues are colored for their clustering (green) or lack of clustering (yellow) to group II. (B) Clustered residues displayed on the structure of the human RRM2 domain (PDB_ID 1CVJ) and color coded as in A. (Left) RNA-binding surface of RRM1–RRM2 with poly(A) shown in orange; (middle) RNA-binding residues with the poly(A) and the RNA-binding residues removed to observe the RRM2 core residues; (right) image as at left rotated 90° at the horizontal axis.
The clustering revealed three major groups of RRM2 positions that have distinct profiles. The first group showed sensitivity to nearly all amino acid substitutions (Fig. 3A, cluster I). Seven of the 11 residues in this group (N127, F129, K131, S154, F168, F170, and H172) interact directly with RNA, based on the structure of the human protein in complex with poly(A) (Deo et al. 1999). Three other residues whose human equivalents were shown to associate with poly(A), N132, Y197, and P200, did not cluster with this group, displaying lower sensitivities to mutations. N132 and P200 tolerated multiple amino acid substitutions without affecting Pab1 function, while Y197 showed moderate sensitivity to mutations and could not be substituted by any amino acid without reducing function by >10%. Unlike the mutation-sensitive RNA-binding residues that make substantial contacts with the adenine bases and the backbone phosphates, these three residues are situated more peripherally to the RNA path (Fig. 3B, left and right). N132 is part of the RNP2 motif (Maris et al. 2005) and is highly conserved. The equivalent residue in the human protein forms a hydrogen bond with an adenine base (Deo et al. 1999), but this base is also specified and stabilized by the residues equivalent to K131 and F168, which also form hydrogen bonds with the RNA phosphate groups that surround the adenine base. Similarly, the residues in the human protein equivalent to Y197 and P200 make contacts with another adenine base, which also interacts with three other residues (Deo et al. 1999). Thus, the minimal effect of substitutions to N132, Y197, and P200 may be due to their limited contributions to RNA binding relative to other residues that associate with the same adenine bases.
The second group of clustered residues showed sensitivity to most amino acid substitutions except for hydrophobic ones (Fig. 3A, cluster II). Ten of the 12 residues in this group are aliphatic, with most of them inaccessible to solvent (average Accessible Surface Area = 2.6 Å2) and constituting part of the RRM2 core structure (Fig. 3B, middle). I152 in loop L2 was an exception as it showed the highest sensitivity to hydrophobic substitutions and the highest solvent accessibility area (ASA = 24.2 Å2) relative to the other aliphatic residues within this group. These observations suggest a specialized role for I152 that requires features other than hydrophobicity. All other aliphatic residues that were not clustered with this group were more exposed to solvent (average ASA = 51.1 Å2) (Fig. 3B, middle). Taken together, these results show that mutational profiles can accurately distinguish different classes of aliphatic residues.
We found K156 to cluster together with core residues, with leucine, isoleucine, and arginine being its least detrimental replacements, although none of them was able to fully compensate for loss of K156. These results suggest that both polarity and the nonpolar neck of K156 are required for its function (Dyson et al. 2006). In the human PABP-1 RRM2, the residue equivalent to K156 is involved in packing interactions between RRM1 and RRM2, which stabilize the RNA-binding trough (Deo et al. 1999). Compaction of RRM2 against RRM1 buries the large surface at their interface from solvent access, likely providing an explanation for why hydrophobic residues at this position are tolerated.
The third cluster is composed of the remaining positions, which show moderate to low mutation sensitivity. These residues are found mostly at the outer shell of RRM2 (Fig. 3A, cluster III) and contribute to Pab1 activity by functions that do not produce a clear mutational profile.
Mutation at G150 destabilizes RRM2 structure
Four positions other than the RNA-binding residues were clustered as highly sensitive to most amino acid substitutions (Fig. 3A, cluster I). Of these, G169 and F173 are adjacent to RNA-binding residues in RNP1 and, therefore, the deleterious effect of mutations at these positions could result at least in part from interference with RNA binding. Of the other two, F149 is situated at the end of helix α1 and G150 is in a β-turn structure that follows this helix, remote from the RNA-binding trough and in close proximity to the side chain of F173 (Fig. 4A). The high sensitivity may suggest an additional function for F173, together with F149 and G150, that does not involve RNA binding.
FIGURE 4.

Mutational sensitivity of residues in the helices α1 and α2 interface suggests a role for these residues in Pab1 stability. (A) Three of the four residues highly sensitive to mutation but not RNA binding (F149, G150, and F173) are found in close proximity in the RRM2 opening between the two helices. (B) Cold-suppressible phenotype of mutants carrying G150T, F149T, and F173A. pab1Δ cells carrying the two plasmids shown in Figure 1A with the variant plasmid expressing the specified mutations from Pab1(1–343BX) were grown in the absence or in the presence of 5-fluoro-orotic acid (5FOA) to follow the survival of the mutants upon wild-type plasmid loss. (C) Protease sensitivity of a G150T mutant. Western blot showing GST fusions of Pab1(1–343BX) constructs following treatment with increasing concentrations of proteinase K. (D) Conservation scores for multiple sequence alignment positions created from 119 RRM sequences in the protein data bank. The RNP elements and the four mutation sensitive residues are shown.
Based on their sensitivity to mutation, we examined the roles of F149, G150, G169, and F173 in more detail. From the RRM2 structure (Fig. 4A), we hypothesized that F149 and G150 act with F173 to stabilize the RRM structure by bridging helix α1 and helix α2 to bury the hydrophobic core from solvent. Mutations at these positions should therefore destabilize RRM2, a phenotype that might be suppressed at low temperature. Yeast expressing a variant with any of the F149T, G150T, G169M, F173A mutations or with F170V, which interferes with RNA binding (all with enrichment scores <0.1) did not grow at 30°C (Fig. 4B). However, the growth defects due to the F149, G150, and F173 mutations were suppressed at 20°C (Fig. 4B). In support of the role of G150 in RRM2 stability, the G150T protein showed higher sensitivity to protease cleavage and a different cleavage pattern from that of the wild-type protein (Fig. 4C). In contrast, the protease sensitivity and cleavage pattern of the RNA-binding defective mutant F170V were similar to that of the wild-type protein. A comparison of RRM sequences from various RRM-containing proteins present in the protein database revealed that F149, G150, and F173 are highly conserved (Fig. 4D), with G150 (glycine in 102 of the 119 RRM sequences) and F173 (phenylalanine or tyrosine at 99 of the 119) having the highest conservation score among all RRM residues. Taken together, the temperature-sensitive phenotype, protease sensitivity, and conservation suggest a role for G150 and the two phenylalanines in stabilizing RRM structure.
A comparison of functional data to evolutionary conservation
The mutational sensitivity and the evolutionary conservation of a residue are strongly correlated (Bottema et al. 1991; Stone and Sidow 2005), but discordances between these two properties may help to characterize function. For example, a residue with low evolutionary conservation, but high mutational sensitivity, may participate in a function specific to the organism being tested, or these discordant properties may suggest that the assay conditions were harsher than the forces applied by natural selection.
As a first approach, we sought to compare neutral amino acid substitutions from this study with naturally occurring substitutions in other PABP sequences. The degree of tolerance to homologous substitutions may indicate the functional constraint on each residue in yeast. To this end, we created function-based logo plots for the four β strands of the RNA-binding surface and flanking residues (Fig. 5A), which display wild-type residues and amino acid substitutions that resulted in a neutral effect on Pab1 RRM2 function (defined as >0.95 of the wild-type residue performance, see Materials and Methods). The logo plots show that of the RNA-binding residues, N127, K131, S154, F168, H172, and Y197 could not be replaced by any other amino acids without loss of Pab1 activity. Tyrosine substitution of F129 and F170 and multiple substitutions of N132 and P200 were tolerated.
FIGURE 5.

Discrepancy between mutation sensitivity data and evolutionary conservation provides functional insights. (A, top) Logo plots generated for all amino acid substitutions that resulted in no more than a 5% reduction in performance compared with the wild type. Presented are only the four β-strands and flanking residues. (Bottom) Logo plots generated for the same RRM2 elements from a multiple sequence alignment of 306 Pab1 homologous sequences. The yeast RRM2 sequence corresponding to these logo plots is shown below. Residues shown to bind RNA in the human RRM2 domain (Deo et al. 1999) are underlined. (B) Comparison of the property entropy of each Pab1 RRM2 position in the multiple sequence alignment created from homologous sequences to the property entropies that were derived from all amino acid substitutions that showed no more than a 5% reduction in performance. The trendline is shown in a solid line, together with the Pearson's R2 value. Dotted line represents perfect correlation. (C) The ratio between the functional conservation score to the evolutionary conservation score is color coded on the structure of the human RRM2 (PDB ID 1CVJ). From top, left in clockwise direction: lateral (facing RRM3), lateral (facing RRM1), dorsal, and ventral views of RRM2. RRM1 is shown in transparent green and the poly(A) in gray.
A comparison of the function-based logo plot to the logo plot derived from 306 poly(A)-binding protein homologs (listed in Supplemental Table 4) reveals a general agreement between the two, with 24 out of the 32 positions sharing at least one amino acid of the two most frequent amino acids that occupy these positions in each plot. However, most positions of the yeast Pab1 functionally tolerated more substitutions than are present in the natural sequences, a feature that might be due to the limited selective force applied on yeast Pab1 function in this assay.
For 11 positions, the amino acid in the yeast RRM2 sequence was different from the most frequent amino acid in the naturally occurring sequences. Of these, in nine cases (D151N, S155C, I157V, G193D, I196V, A199G, P200H, Q194K, and E195K) a change of the yeast amino acid to the most frequent amino acid in natural PABP sequences had a neutral effect. These observations suggest that adaptation of poly(A)-binding proteins to various eukaryotes involves minor functional consequences of single amino acid substitutions. However, some substitutions to amino acids that are present in natural PABP proteins were less tolerated by yeast Pab1. For example, the RNA-binding residue H172 is a glutamine in some poly(A)-binding proteins (Fig. 5A), but H172Q cannot fully complement for the loss of H172 (enrichment score 0.73). This result indicates that poly(A)-binding proteins with histidine at position 172 differ in how they bind RNA compared with PABP proteins with glutamine at this position.
To further study the correlation between our functional data set and the evolutionary record, we sought to compare the degree of conservation for each RRM2 position. To this end, we used the “property entropy” method (Capra and Singh 2007), which scores the variation (Shannon entropy) at each position in a multiple sequence alignment with respect to the stereochemical similarity between the amino acids that populate it (Williamson 1995; Mirny and Shakhnovich 1999; Capra and Singh 2007). This measure allowed us to assess the conservation of natural as well as engineered variants by applying identical criteria for the two data sets without introducing corrections that are commonly applied by other methods to score conservation of natural sequences (such as phylogenetic tree construction or amino acid background frequencies). We found a moderate correlation in property entropy (R2 = 0.43) between naturally occurring and engineered sequences (Fig. 5B). Specifically, for most residues, the higher the evolutionary conservation, the higher the functional conservation was. As found for the logo plots of four of the β strand sequences, most positions could tolerate more mutations than would be expected by their evolutionary conservation.
Color-coding the ratio between the functional conservation score and the evolutionary conservation score on the human RRM2 structure allowed us to identify regions whose scores do not match (Fig. 5C). This comparison reveals that the RNA-binding residues N127, F129, K131, S154, F170, H172, and Y197, as well as some of the adjacent residues, G126, I128, I130, L153, S155, V171, V196, and V198, are functionally more conserved than suggested by their evolutionary conservation. A second region in which function shows greater conservation than evolutionary conservation encompasses certain residues that face the RRM1 interface. Contacts between helix α2 of RRM1 and helix α1 of RRM2, and between strand β4 of RRM1 and strand β2 of RRM2, stabilize the RNA-binding trough formed by the two tandem β sheets and facilitate the binding to eIF4G and poly(A) (Deo et al. 1999; Safaee et al. 2012). Strand β2 residues that mediate these interdomain interactions (K156 and L153) are functionally more important than evolutionary conservation would suggest. A third region that shows that divergence between function and evolutionary conservation comprises the eIF4G-binding site (Otero et al. 1999) including residues E181 and A185.
For these three regions, the high ratio of functional conservation to evolutionary conservation may reflect a sensitized activity of Pab1 in this assay. For example, decreased RNA binding due to lack of RRM4 may result in oversensitivity to mutations that further degrade this activity, either directly (such as sensitivity to mutations to RNA-binding residues) or indirectly (such as sensitivity to mutations that destabilize the trough formation between the RRM1 and RRM2 RNA-binding surfaces). Alternatively, the high functional conservation to evolutionary conservation ratio may suggest a specialized function for these residues in yeast that cannot be complemented by equivalent residues from other species. The failure of human PABP-1 segments to complement for eIF4G binding (Otero et al. 1999) supports this possibility.
Epistatic interactions between two mutations
An epistatic interaction between two mutations describes an observed gain or loss of function of a double mutant that exceeds predictions based on the functional consequences of each of the constituent single mutations alone (Horovitz 1996). To study epistasis in Pab1 RRM2, we used the enrichment scores of 39,609 trios for which the scores of the two single mutants and the corresponding double mutant were available. We used a product interaction model previously applied to large-scale mutational data (Fowler et al. 2010; Araya et al. 2012) such that in the absence of epistasis, the observed enrichment score of the double mutant should equal the product of the two single mutants’ enrichment scores. The observed enrichment scores correlated well with the products of the single mutants (R2 = 0.76), suggesting that, in general, no substantial epistasis occurs in double mutants (Supplemental Fig. 3A). We used LOESS function to correct for input read counts effects (Supplemental Fig. 3B) and selected those double mutants whose epistasis scores exceeded two standard deviations from the mean as candidates for displaying strong epistatic interactions. Of the 39,609 double mutants, 411 showed positive epistasis (i.e., performance higher than expected) and 1444 negative epistasis (i.e., performance lower than expected).
The distribution of the spacing along the primary sequence between the mutations in double mutants revealed enrichment for short distances in the case of mutation pairs that showed either positive or negative epistatic interactions (Fig. 6A). Variants with positive epistasis showed a preference for the inclusion of interacting mutations that lie no more than three residues apart. Variants with negative epistasis also showed a preference for short distances between interacting mutations, with zero to five residues apart being the most significant range (wilcoxon P-value = 0.0024). From these distributions (Fig. 6A), we estimate that primary sequence proximity is responsible for ∼8.6% of the variants showing positive epistasis and 7.4% of those showing negative epistasis.
FIGURE 6.

Epistatic interactions in double mutants. (A) Contribution of spacing between residues in the primary sequence corresponding to the two mutations in each double mutant to epistatic interactions. A sequence distance of 0 corresponds to adjacent residues. (B) Contribution of physical distance between the two mutations forming each double mutant to epistatic interactions. Shown is the distribution of distances between the center of the masses of the two mutated residues based on the human RRM2 structure (PDB ID 1CVJ). Only variants with five or more residues separating the two mutated residues in the RRM2 sequence were included. For A and B, the P-values of wilcoxon-rank tests for the differences between the positive and negative epistasis groups to the no-epistasis group are specified. (C) Arc diagrams displaying the interactions between mutation pairs in variants with positive (red) or negative (blue) epistatic interactions. The sequence and the secondary structure of each segment are shown. The color of each node represents substitution to a specific amino acid and the size represents the fraction of variants with that particular mutation. A color map describing the identity of substituting amino acids is shown. (D) Presumed effects of N139 and I157 substitutions on strand β2 and helix α1 association may account for suppression of deleterious mutations. (Top, left) Structure of the human RRM2 in green with N112 and V130 residues shown in color-coded sticks (carbon, light blue; nitrogen, blue; oxygen, red). The hydrogen bond between the carbonyl group of the asparagine side chain and the amino group of the valine backbone is shown by a black dotted line, along with the distance. Hydrophobic residues found in close proximity to Val130 are also shown. (Top, right) Replacing the specified residues with the yeast residues supports a similar association between I157 and N139 and between I157 to the conserved aliphatic residues. (Bottom, left) Substitution N139T (as well as N139S) may weaken the hydrogen bonding with I157 by increasing the distance between the hydrogen donor and acceptor groups. (Bottom, right) Substitution I157L may cause a similar effect by destabilizing Loop L1 and L3 association.
We also examined epistasis with respect to the distribution of physical distance using the structure of the human PABP-1 RRM2 domain as a proxy for the yeast domain structure. Residues can be in close physical distance either because they reside nearby in the primary sequence or because they are distant in the primary sequence and come together due to protein folding. To eliminate effects due to sequence proximity, we followed the distribution of physical distances between mutations that are five residues or more apart. This distribution revealed a similar association between short distance and epistasis. In particular, positively interacting mutations showed enrichment for distances shorter than ∼12 Å between the centers of mass of the two residues, and negatively interacting mutations showed enrichment for distances between ∼10 and 15 Å between these centers of mass (Fig. 6B). Based on these distributions, we estimate the upper limit of 12 Å accounts for ∼17% of variants displaying positive epistatic interaction, and the range of 10–15 Å accounts for ∼7% of variants displaying negative epistatic interaction. Although the two physical distances slightly overlap, no pair of residues was shared between the two groups, suggesting that physical distance acts on different sets of residues with respect to positive and negative epistasis.
Specific residues and mutations serve as hot spots for epistatic interaction (Hinkley et al. 2011; Araya et al. 2012). In particular, substitutions N139S and N139T were responsible for >30% of the positive epistasis interactions in segment 1. Though N139S and N139T as single mutations had only a slightly negative effect on function (enrichment scores 0.88 and 0.86, respectively), these mutations partly rescued deleterious mutations at RNA-binding sites (e.g., F129I and K131N), core residues (e.g., I128N and I130S), and G150 (e.g., G150A and G150S). Similarly, I157L (enrichment score 0.96) was present in >13% of positive epistatic interactions in segment 2, and suppressed deleterious mutations in an RNA-binding residue (F168S, F168I, and F168C) and in RRM1–RRM2 interface residues (K156R, K156M, and L153V).
In the human RRM2 structure, the residues corresponding to N139 and I157 form a hydrogen bond between the carbonyl group of an asparagine side chain and the backbone amino group of a valine that connect helix α1 and strand β2 (Fig. 6D). N139S and N139T may slightly destabilize the association between the two elements by weakening the presumed hydrogen bond between N139 and I157. I157L may cause a similar outcome by destabilizing the hydrophobic interactions between loops L1 and L3. Although as single mutations these had only a minor effect on RRM2 function, they may confer flexibility to the RRM2 structure to allow this domain to adjust its structure to accommodate other mutations that interfere with RNA binding or protein stability.
While the enrichment score of the G150T mutation, which destabilizes RRM2 (Fig. 4), was too low to detect negatively interacting mutations, we found G150A and G150S substitutions comprised ∼15% of all negative epistatic interactions in segment 1. These two substitutions had only a moderate effect on Pab1 function (enrichment scores 0.62 and 0.66, respectively) relative to other substitutions at this position, probably due to the small size of their side chains. G150A and G150S negatively interacted with a similar set of mutations (P-value of Fisher exact test = 1.9e−23) listed in Supplemental Table 5. This set of mutations may enhance the destabilizing effect of G150 mutations, pointing to loop 1, which carries most of the G150A and G150S interacting mutations, as an important element for RRM stability. Moreover, mutations at N132, an RNA-binding residue in loop 1, negatively interacted with G150A and G150S, suggesting an additional role for N132 in supporting the integrity of RRM2 structure.
DISCUSSION
By assaying variants of the RRM2 domain of the yeast Pab1 in high throughput, we scored most (83%) of the possible 1500 single amino acid substitutions (including stop codons), and more than 100,000 variants with multiple substitution events, in a 75-residue-long sequence. The results highlight the RNA-binding surface of RRM2 as the most important element for its function, although each position in the RRM2 shows a nearly unique pattern of mutational sensitivity. We clustered the data to reveal other residues highly sensitive to mutation, as well as core hydrophobic residues that tolerated substitution only by other hydrophobic amino acids. By comparing the evolutionary conservation of RRM residues with their ability to function in the context of the yeast Pab1 protein, we could implicate some residues in yeast-specific functions. Finally, we used epistasis analysis to identify interacting residues in Pab1.
Beside the two RNP motifs, the deep mutational scan suggests that residue G150, present in the loop L2 between helix α1 and strand β2, is an additional signature of the RRM domain family. This residue was one of the few non-RNA-binding residues to display extreme sensitivity to mutations and to be highly conserved within the RRM family. The cold-suppressible phenotype of yeast carrying Pab1 with the G150T mutation, along with the proteinase sensitivity of the mutant protein, suggests a critical role for this residue in stabilizing the RRM structure. G150, which is in the L2 β-turn, may be essential to maintain the gap between the two RRM helices inaccessible to solvent. In agreement with this general function, a mutation at the corresponding residue (G53S) in the RRM1 domain of the C. elegans UNC-75 protein eliminated activity (Kuroyanagi et al. 2013). Given the similar mutational sensitivity, cold-suppressible phenotypes of mutants, and the close proximity of their side chains to the gap between the two helices, residues F149 and F173 may act with G150 in the same structural role. Indeed, a temperature-sensitive mutant (F87A) in the residue corresponding to F173 in the RRM1 domain of the splicing factor Prp24 (Kwan and Brow 2005) suggests that the presumed role for this RNP1 consensus residue in RRM2 stabilization might be general in other RRM sequences. Another loop L2 residue, I152, may contribute to the solvent inaccessibility of the gap between helix α1 and helix α2. This proposed function is supported both by I152 having the highest sensitivity to mutations of the RRM2 aliphatic residues and by its partial solvent accessibility, which distinguish it from other core residues.
While the consensus residues of the RNP motifs are the most commonly found in nature, it is not known how well RRM consensus residues can substitute for wild-type residues in a single, specified RRM domain. The Pab1 RRM2 mutational data provide evidence both for the functional redundancy of some of these consensus residues and for the inability of other consensus residues to support Pab1 activity. In particular, the RRM consensus residues of the two RNP elements that were tolerated in the yeast RNP motifs appear in some PABP sequences, while consensus residues that were not tolerated are absent from all PABP sequences. However, for some RNP positions (such as H172), the yeast RRM2 tolerated neither the general consensus residue nor a PABP consensus residue, suggesting a highly specific function in yeast for these residues.
We found that clustering RRM2 positions based on their mutational sensitivity could distinguish between core and non-core aliphatic residues and could identify other residues, such as K156, that function within the hydrophobic core. These data are consistent with a long history of mutagenesis (e.g., Lim and Sauer 1989) that has found hydrophobicity to be the most essential feature of residues in a protein's core. Although a structure of the RRM2 domain is available only for the human PABP-1 protein (Deo et al. 1999), the match between the mutational sensitivity of the yeast residues to their positions in the human protein suggests that the in vivo structure of the yeast domain resembles the in vitro structure of the human one. Given the striking signature in the mutational profile of core residues, these profiles can serve as a general approach to evaluate structures that have been defined in vitro, as previously shown (Adkar et al. 2012) or to refine folding predictions for proteins whose structure is unknown.
The deep mutational scanning approach has been used to study the in vivo effect of all possible 171 single amino acid substitutions across a 9-amino acid-long stretch of the yeast Hsp90 (Hietpas et al. 2011) and many of the possible 1425 single amino acid substitutions in the 75-amino acid-long ubiquitin sequence (Roscoe et al. 2013). However, we found that current yeast methods and sequencing technology allow an in vivo assessment of nearly two orders of magnitude more variants, which enables an analysis of variants with multiple mutations while still maintaining high coverage for variants with a single-point mutation. Using these data to study the epistatic interactions between two mutations in ∼40,000 double mutants revealed ∼2000 double mutants with extreme epistasis scores. We identified a small but significant preference for sequence proximity, up to three residues (positive epistasis) and five residues (negative epistasis), and for short physical distance (up to 12 Å for positive epistasis and 10–15 Å for negative epistasis). Overall, we found that short sequence and physical distance play a role in ∼25% of the positive epistasis events and 14% of the negative epistasis events.
Though further study will be needed to determine the extent to which sequence and physical proximity govern positive and negative epistasis in other proteins, in a comprehensive analysis of drosophilid genomes Callahan et al. (2011) found a correlation between amino acid substitutions undergoing positive selection and their separation within primary protein sequences, with a second substitution strongly enhanced within ∼10 residues of the first. Among the explanations provided for this correlation was epistasis. Thus, both the evolutionary study and our mutational one point to a role in positive epistasis of residues nearby in the primary sequence.
Epistasis analysis revealed mutations N139T, N139S, and I157L as suppressors of multiple deleterious mutations in residues associated with RNA binding, protein stability, and interdomain interactions. Residues corresponding to N139 and I157 interact in the human RRM2 domain, implying that the association between these two residues is critical for suppression. We suggest that weakening of the hydrogen bonding between these two residues by the three substitutions slightly interferes with the association of helix α1 and strand β2. However, the flexibility that results from the substitutions may allow RRM2 to adjust to mutations that damage its structure. Given the structural conservation of the RRM domain family, the contact site between helix α1 and strand β2 may serve as a general target for mutations that can suppress the effects of disruptive mutations.
This mutational analysis of Pab1 RRM2 has allowed us to assess the in vivo function of residues in this domain. A similar approach with the three other Pab1 RRM domains could highlight both common and unique properties of the sequence–function relationship of each Pab1 RRM domain.
MATERIALS AND METHODS
Plasmids
To create a tetracycline-regulated Pab1 expression system, we cloned the complete coding sequence of PAB1 into the BamHI and NotI sites of pCM188 (URA3, tetO2 promoter, CEN) (Gari et al. 1997). Truncated variants Pab1(1–469) and Pab1(1–343) were generated by cloning the complete coding sequence of PAB1, Pab1(1–577), into the XmaI and XhoI sites of p415GPD (LEU2, GPD1 promoter, CEN) (Mumberg et al. 1995) and removing the 3′ terminal sequences by treating the plasmid with either SphI and XhoI or with NdeI and XhoI, respectively. The fragment encoding Pab1(1–201) was cloned into the XmaI and XhoI sites of p415GPD. For bacterial expression, the Pab1(1–343BX) fragment carrying either wild-type, F170V, or G150T mutation was cloned into the XmaI and XhoI sites of pGEX4T2 (Addgene).
Yeast strains and growth conditions
The pab1 knockout strain (MATa ura3Δ0 leu2Δ0 met15Δ0 his3Δ1 pab1Δ::NatMX) was created by replacing the endogenous PAB1 gene in strain BY4741 with a NatMX cassette from a PUG6 plasmid (Guldener et al. 1996) and selecting for clonNAT-resistant transformants. To maintain cell viability, we expressed the complete coding sequence of PAB1 from the Tet-off vector pCM188 prior to gene disruption. We refer to the pab1Δ strain that expresses PAB1 from pCM188 as pab1Δ [PAB1]. Truncated and mutated PAB1 variants were cloned into p415GPD and transformed into pab1Δ[PAB1]. Cells were grown at 30°C in synthetic complete (SC) media lacking leucine and uracil and supplemented with 2% glucose. The effect of the PAB1 mutations on growth was tested by adding the tetracycline analog, doxycycline (Sigma, D-9891), to a final concentration of 20 μg/mL, unless otherwise indicated.
Construction of PAB1 RRM2 libraries in yeast
The DNA encoding the complete Pab1 protein followed by two stop codons was cloned into the XmaI and XhoI sites of p415GPD. After disrupting the BamHI and XbaI sites in the multiple cloning site, we introduced a series of synonymous mutations at eight codons on either side of RRM2 (codons 118–125 [CCTTCCCTACGTAAAAAAGGATCC] and 203–210 [TCTAGAAAAGAGAGGGATTCCCAG] with synonymous mutations shown in bold and restriction sites underlined) to create silent and unique BamHI and XbaI restriction sites. The changes also allowed specific amplification of the PAB1 RRM2 insert for high-throughput sequencing. Three oligonucleotides covering codons 126–150, 151–175, and 176–200 in the PAB1 coding sequence were synthesized with a 4% error rate by TriLink Biotechnologies, filled in, and cloned into the BamHI and XbaI sites that flanked the RRM2 domain.
Following propagation in bacteria, the pab1Δ[PAB1] strain was transformed by each library by a modified version of the LiAc-PEG method (Gietz and Woods 2002). Specifically, an overnight culture was diluted into 50 mL of fresh YPD medium to an OD600 of 0.45 and cultured at 30°C for two cell divisions. Cells were washed and resuspended in 2 mL of LiSORB solution (100 mM LiAc, 1 M Sorbitol in TE) and incubated for 30 min at room temperature with constant shaking; 1 mL of LiPEG solution (100 mM LiAc, 40% PEG 3350 in TE) was mixed with 1 μg of plasmid and 50 μg of salmon sperm DNA (Sigma, D1626) and added to 200 μL of cell suspension. After a 30-min incubation at room temperature with constant shaking, 100 μL of DMSO was added to the sample, followed by heat shock at 42°C for 15 min. Cells were recovered in 10 mL of YPD supplemented with 0.5 M Sorbitol at 30°C for 1 h, providing transformation efficiency of 3 × 105 transformants per 1 μg of plasmid DNA.
Yeast selection
Yeast carrying one of the three libraries were grown to log phase in SC medium lacking leucine and uracil, supplemented with 2% glucose, and diluted into fresh medium containing 20 μg/mL of doxycycline to a final concentration of 4 × 104 cells/mL. Selection was carried out for 22 h with the culture growing to a density of 5 × 106–1 × 107 cells/mL. Next, 2.5 × 108 cells from each culture were collected before (“input”) and after selection (“selected”).
Library preparation for high throughput sequencing
Cells were resuspended in miniprep buffer P1 (Qiagen, 27106) and treated with 100 μg of Zymolase 20T (ImmunO, 320921) in the presence of 50 mM DTT for 2 h at 37°C to digest yeast cell walls. After one freeze and thaw cycle from −80°C to 30 sec at 42°C, yeast DNA was recovered in 50 μL of 10 mM Tris-HCl (pH 8.0) by the standard Qiagen miniprep protocol (Qiagen, 27106). DNA was treated with 60 units of Exonuclease I (USB, 70073X) and with 15 units of Lambda exonuclease (NEB, M0262S) for 2 h at 37°C to remove excess of yeast genomic DNA, and plasmid DNA was purified and concentrated using a Zymo Research kit (D4004). Library fragments were amplified by 18 PCR cycles using primers specific to the synonymous changes that flank the RRM2 domain, and sequenced by an Illumina GAIIx sequencer by pair-end reads.
Scoring the performance of library variants
We used the Enrich software package (Fowler et al. 2011) to remove low-quality reads (discarding reads with base Q score <20); to determine the location and identity of mutations, while filtering out variants with insertions or deletions; to calculate the frequency of each variant appearing in the input and selected pools; and to provide an enrichment score for each variant appearing in both pools by calculating the ratio between the two frequencies (selected/input). Enrichment scores were further normalized to the wild-type score.
Use of synonymous mutations to set input read cutoff and enrichment score distribution of neutral variants
Enrichment scores for variants carrying missense mutations were arranged from low to high. At each enrichment score X, the proportion of synonymous variants with a score at least as extreme as X was multiplied by the total number of missense variants and divided by the number of missense variants with a score at least as extreme as X to yield an estimate of the False Discovery Rate. Missense variants with less extreme enrichment scores but lower estimated FDRs have their FDRs set to the highest FDR among the set of variants with more extreme enrichment scores as used for the calculation of Q-values. Final estimated FDR values were multiplied by two to account for the two extremes.
Clustering of enrichment scores
Enrichment scores of single amino acid substitutions were log2 transformed and visualized using Matrix2png (Pavlidis and Noble 2003). Complete linkage hierarchical clustering with a Euclidean distance similarity metric for both RRM2 residues and substituting amino acids was performed using Gene Cluster 3.0 (de Hoon et al. 2004) and visualized by Java TreeView 1.1.6r2 (Saldanha 2004). Accessible Surface Areas (ASA) of hydrophobic residues from human RRM2 structure (1CVJ) were obtained from PDBePISA (Krissinel and Henrick 2007).
GST–Pab1 purification and Proteinase K sensitivity assay
Glutathione S-transferase (GST) fusions of Pab1(1–346) were overexpressed in Escherichia coli strain DE3. Cells were collected and lysed by sonication in lysis buffer (20 mM Tris-HCl at pH 7.6, 200 mM NaCl, 0.2 mM EDTA, 1 mM DTT) in the presence of protease inhibitor cocktail (Roche, 05056489001). Proteins were bound to Glutathione-Sepharose beads (GE Healthcare, 17-5132-01), washed (20 mM Tris-HCl at pH 7.6, 1 M NaCl, 0.2 mM EDTA, 1 mM DTT), and eluted (20 mM Tris-HCl at pH 7.6, 1 M NaCl, 0.2 mM EDTA, 10 mM glutathione) according to the manufacturer's instructions. Proteins were dialyzed overnight at 4°C in PBS (25 mM NaPO4 at pH 7.0, 150 mM NaCl) containing 20% glycerol. To assess Proteinase K sensitivity, 10 μg of GST and GST fusion proteins were treated with 0, 0.004, 0.02, 0.1, and 0.5 ng/μL of Proteinase K (NEB, P8102S) in a 20-μL reaction buffer (25 mM Tris-HCl at pH 7.5, 2.5 mM CaCl2) for 1 h at 37°C. Digestion was stopped by adding PMSF to a final concentration of 5 mM.
Calculating RRM conservation
To evaluate the general conservation of residues in RRMs by an unbiased approach, we searched the protein databank (PDB) for RRM domains using the terms “RRM” and “RNA Recognition Motif” and collected the PDB-ID of all proteins with a known RRM structure. Using these IDs, we extracted all of the sequences of the structurally defined RRMs from the UniProt Knowledge Base with the exception of proteins with multiple structurally resolved RRM domains, where we randomly selected a single domain for the analysis. Taking this approach provided us with 119 RRM sequences, all from unique proteins (see Supplemental Table 3 for the list of sequences). Multiple sequence alignment was performed using the MAFFT program (Katoh and Toh 2008), and a conservation score for each site was determined by the Protein Residue Conservation Prediction program using the Jensen-Shannon divergence (JSD) scoring method (Capra and Singh 2007).
Comparing functional conservation to evolutionary conservation
To create a consensus sequence that represents every mutation tolerated in the β-sheet structure, all mutations were unlinked from the input and the selected sequence pools and their frequencies were determined. For each position in each pool, the frequency of every mutation was normalized to the frequency of the wild-type residue, which was set to 1.0. Hence, for every mutation the ratio of frequencies (selected/input) indicates its enrichment relative to the enrichment score of the wild-type residue at the same position, which is equal to 1.0. Amino acid substitutions with an enrichment ratio below 0.95 were assumed to be deleterious for Pab1 RRM2 function and were removed from the analysis, while the other mutations and wild-type residues were used to create 1000 arbitrary sequences that represent their relative enrichment scores. Logo plots from these sequences were created using WebLogo 3.0 (Crooks et al. 2004). Logo plots for natural Pab1 homologs were created by providing WebLogo a MAFFT-based multiple sequence alignment of 306 sequences selected by ConSurf server (Ashkenazy et al. 2010) from the UniRef90 database (Pruitt et al. 2011) showing a maximal 95% identity between sequences and a minimum of 35% identity with Pab1 (Supplemental Table 4). To calculate functional and evolutionary conservations, we measured the property entropy for each site in the library sequences and UniRef90-based multiple sequence alignment that were used to create the logo plots by the Protein Residue Conservation Prediction tool (Capra and Singh 2007). We used a window size of two residues, which incorporates the estimated conservation of adjacent residues into the score of each site.
Epistasis analysis
Epistasis scores were calculated using the product model formula ((ɛ = WAB–WA × WB), where W symbolizes a variant's enrichment score, and A and B represent two different amino acid substitutions). Variants carrying stop codons as one of the single amino acid substitutions and others with predicted read counts lower than 1 (WA × WB × input_read_counts of variantAB) were eliminated. To correct for input read effect on epistasis data, the R package locfit was used to fit a local regression to the graph of epistasis versus input reads. We used the standard local polynomial model with cubic decay and a nearest neighbor fraction of 0.7, which provides an estimate of the mean epistasis score as a function of input reads. An additional local regression was fitted to the squared residuals of the epistasis scores from their estimated mean and double mutants, with a local estimated z-score greater than 2 or less than −2 were collected as highly positively and highly negatively epistatic mutants, respectively.
Structure visualization
PyMol visualization software (v1.5.0.5) was used to create all figures of PABP-1 structure.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Charlie Lee for assistance with DNA sequencing. We thank Eric Phizicky, Marvin Wickens, Roy Parker, Douglas Fowler, and Osnat Penn for helpful discussions and comments on the manuscript. This work was supported by grant P41 GM103533 from the National Institute of General Medical Sciences of the NIH. S.F. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Freely available online through the RNA Open Access option
REFERENCES
- Adam SA, Nakagawa T, Swanson MS, Woodruff TK, Dreyfuss G 1986. mRNA polyadenylate-binding protein: Gene isolation and sequencing and identification of a ribonucleoprotein consensus sequence. Mol Cell Biol 6: 2932–2943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adkar BV, Tripathi A, Sahoo A, Bajaj K, Goswami D, Chakrabarti P, Swarnkar MK, Gokhale RS, Varadarajan R 2012. Protein model discrimination using mutational sensitivity derived from deep sequencing. Structure 20: 371–381 [DOI] [PubMed] [Google Scholar]
- Amrani N, Ghosh S, Mangus DA, Jacobson A 2008. Translation factors promote the formation of two states of the closed-loop mRNP. Nature 453: 1276–1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araya CL, Fowler DM 2011. Deep mutational scanning: Assessing protein function on a massive scale. Trends Biotechnol 29: 435–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araya CL, Fowler DM, Chen W, Muniez I, Kelly JW, Fields S 2012. A fundamental protein property, thermodynamic stability, revealed solely from large-scale measurements of protein function. Proc Natl Acad Sci 109: 16858–16863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N 2010. ConSurf 2010: Calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res 38: W529–W533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baer BW, Kornberg RD 1983. The protein responsible for the repeating structure of cytoplasmic poly(A)-ribonucleoprotein. J Cell Biol 96: 717–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottema CD, Ketterling RP, Ii S, Yoon HS, Phillips JA III, Sommer SS 1991. Missense mutations and evolutionary conservation of amino acids: Evidence that many of the amino acids in factor IX function as “spacer” elements. Am J Hum Genet 49: 820–838 [PMC free article] [PubMed] [Google Scholar]
- Burd CG, Matunis EL, Dreyfuss G 1991. The multiple RNA-binding domains of the mRNA poly(A)-binding protein have different RNA-binding activities. Mol Cell Biol 11: 3419–3424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan B, Neher RA, Bachtrog D, Andolfatto P, Shraiman BI 2011. Correlated evolution of nearby residues in Drosophilid proteins. PLoS Genet 7: e1001315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capra JA, Singh M 2007. Predicting functionally important residues from sequence conservation. Bioinformatics 23: 1875–1882 [DOI] [PubMed] [Google Scholar]
- Chou PY, Fasman GD 1978. Empirical predictions of protein conformation. Annu Rev Biochem 47: 251–276 [DOI] [PubMed] [Google Scholar]
- Clery A, Blatter M, Allain FH 2008. RNA recognition motifs: Boring? Not quite. Curr Opin Struct Biol 18: 290–298 [DOI] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE 2004. WebLogo: A sequence logo generator. Genome Res 14: 1188–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Hoon MJ, Imoto S, Nolan J, Miyano S 2004. Open source clustering software. Bioinformatics 20: 1453–1454 [DOI] [PubMed] [Google Scholar]
- Deardorff JA, Sachs AB 1997. Differential effects of aromatic and charged residue substitutions in the RNA binding domains of the yeast poly(A)-binding protein. J Mol Biol 269: 67–81 [DOI] [PubMed] [Google Scholar]
- Deo RC, Bonanno JB, Sonenberg N, Burley SK 1999. Recognition of polyadenylate RNA by the poly(A)-binding protein. Cell 98: 835–845 [DOI] [PubMed] [Google Scholar]
- Deschenes-Furry J, Perrone-Bizzozero N, Jasmin BJ 2006. The RNA-binding protein HuD: A regulator of neuronal differentiation, maintenance and plasticity. Bioessays 28: 822–833 [DOI] [PubMed] [Google Scholar]
- Dreyfuss G, Swanson MS, Pinol-Roma S 1988. Heterogeneous nuclear ribonucleoprotein particles and the pathway of mRNA formation. Trends Biochem Sci 13: 86–91 [DOI] [PubMed] [Google Scholar]
- Dreyfuss G, Kim VN, Kataoka N 2002. Messenger-RNA-binding proteins and the messages they carry. Nat Rev Mol Cell Biol 3: 195–205 [DOI] [PubMed] [Google Scholar]
- Dyson HJ, Wright PE, Scheraga HA 2006. The role of hydrophobic interactions in initiation and propagation of protein folding. Proc Natl Acad Sci 103: 13057–13061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkmann JA, Kutay U 2004. Nuclear export of mRNA: From the site of transcription to the cytoplasm. Exp Cell Res 296: 12–20 [DOI] [PubMed] [Google Scholar]
- Fowler DM, Araya CL, Fleishman SJ, Kellogg EH, Stephany JJ, Baker D, Fields S 2010. High-resolution mapping of protein sequence-function relationships. Nat Methods 7: 741–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler DM, Araya CL, Gerard W, Fields S 2011. Enrich: Software for analysis of protein function by enrichment and depletion of variants. Bioinformatics 27: 3430–3431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gari E, Piedrafita L, Aldea M, Herrero E 1997. A set of vectors with a tetracycline-regulatable promoter system for modulated gene expression in Saccharomyces cerevisiae. Yeast 13: 837–848 [DOI] [PubMed] [Google Scholar]
- Gietz RD, Woods RA 2002. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol 350: 87–96 [DOI] [PubMed] [Google Scholar]
- Guldener U, Heck S, Fielder T, Beinhauer J, Hegemann JH 1996. A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic Acids Res 24: 2519–2524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hietpas RT, Jensen JD, Bolon DN 2011. Experimental illumination of a fitness landscape. Proc Natl Acad Sci 108: 7896–7901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinkley T, Martins J, Chappey C, Haddad M, Stawiski E, Whitcomb JM, Petropoulos CJ, Bonhoeffer S 2011. A systems analysis of mutational effects in HIV-1 protease and reverse transcriptase. Nat Genet 43: 487–489 [DOI] [PubMed] [Google Scholar]
- Horovitz A 1996. Double-mutant cycles: A powerful tool for analyzing protein structure and function. Fold Des 1: R121–R126 [DOI] [PubMed] [Google Scholar]
- Imataka H, Gradi A, Sonenberg N 1998. A newly identified N-terminal amino acid sequence of human eIF4G binds poly(A)-binding protein and functions in poly(A)-dependent translation. EMBO J 17: 7480–7489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson A, Favreau M 1983. Possible involvement of poly(A) in protein synthesis. Nucleic Acids Res 11: 6353–6368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Toh H 2008. Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform 9: 286–298 [DOI] [PubMed] [Google Scholar]
- Kessler SH, Sachs AB 1998. RNA recognition motif 2 of yeast Pab1p is required for its functional interaction with eukaryotic translation initiation factor 4G. Mol Cell Biol 18: 51–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, Henrick K 2007. Inference of macromolecular assemblies from crystalline state. J Mol Biol 372: 774–797 [DOI] [PubMed] [Google Scholar]
- Kuhn U, Gundel M, Knoth A, Kerwitz Y, Rudel S, Wahle E 2009. Poly(A) tail length is controlled by the nuclear poly(A)-binding protein regulating the interaction between poly(A) polymerase and the cleavage and polyadenylation specificity factor. J Biol Chem 284: 22803–22814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroyanagi H, Watanabe Y, Hagiwara M 2013. CELF family RNA-binding protein UNC-75 regulates two sets of mutually exclusive exons of the unc-32 gene in neuron-specific manners in Caenorhabditis elegans. PLoS Genet 9: e1003337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan SS, Brow DA 2005. The N- and C-terminal RNA recognition motifs of splicing factor Prp24 have distinct functions in U6 RNA binding. RNA 11: 808–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim WA, Sauer RT 1989. Alternative packing arrangements in the hydrophobic core of λ repressor. Nature 339: 31–36 [DOI] [PubMed] [Google Scholar]
- Lunde BM, Moore C, Varani G 2007. RNA-binding proteins: Modular design for efficient function. Nat Rev Mol Cell Biol 8: 479–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangus DA, Evans MC, Jacobson A 2003. Poly(A)-binding proteins: Multifunctional scaffolds for the post-transcriptional control of gene expression. Genome Biol 4: 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maris C, Dominguez C, Allain FH 2005. The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS J 272: 2118–2131 [DOI] [PubMed] [Google Scholar]
- Mirny LA, Shakhnovich EI 1999. Universally conserved positions in protein folds: Reading evolutionary signals about stability, folding kinetics and function. J Mol Biol 291: 177–196 [DOI] [PubMed] [Google Scholar]
- Mumberg D, Muller R, Funk M 1995. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156: 119–122 [DOI] [PubMed] [Google Scholar]
- Muto Y, Yokoyama S 2012. Structural insight into RNA recognition motifs: Versatile molecular Lego building blocks for biological systems. Wiley Interdiscip Rev RNA 3: 229–246 [DOI] [PubMed] [Google Scholar]
- Otero LJ, Ashe MP, Sachs AB 1999. The yeast poly(A)-binding protein Pab1p stimulates in vitro poly(A)-dependent and cap-dependent translation by distinct mechanisms. EMBO J 18: 3153–3163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park EH, Walker SE, Lee JM, Rothenburg S, Lorsch JR, Hinnebusch AG 2010. Multiple elements in the eIF4G1 N-terminus promote assembly of eIF4G1*PABP mRNPs in vivo. EMBO J 30: 302–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlidis P, Noble WS 2003. Matrix2png: A utility for visualizing matrix data. Bioinformatics 19: 295–296 [DOI] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, Brown GR, Maglott DR 2011. NCBI Reference Sequences (RefSeq): Current status, new features and genome annotation policy. Nucleic Acids Res 40: D130–D135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson R, Denis CL, Zhang C, Nielsen ME, Chiang YC, Kierkegaard M, Wang X, Lee DJ, Andersen JS, Yao G 2012. Mass spectrometric identification of proteins that interact through specific domains of the poly(A) binding protein. Mol Genet Genomics 287: 711–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roscoe BP, Thayer KM, Zeldovich KB, Fushman D, Bolon DN 2013. Analyses of the effects of all ubiquitin point mutants on yeast growth rate. J Mol Biol 425: 1363–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs AB, Bond MW, Kornberg RD 1986. A single gene from yeast for both nuclear and cytoplasmic polyadenylate-binding proteins: Domain structure and expression. Cell 45: 827–835 [DOI] [PubMed] [Google Scholar]
- Sachs AB, Davis RW, Kornberg RD 1987. A single domain of yeast poly(A)-binding protein is necessary and sufficient for RNA binding and cell viability. Mol Cell Biol 7: 3268–3276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safaee N, Kozlov G, Noronha AM, Xie J, Wilds CJ, Gehring K 2012. Interdomain allostery promotes assembly of the poly(A) mRNA complex with PABP and eIF4G. Mol Cell 48: 375–386 [DOI] [PubMed] [Google Scholar]
- Saldanha AJ 2004. Java Treeview—extensible visualization of microarray data. Bioinformatics 20: 3246–3248 [DOI] [PubMed] [Google Scholar]
- Stone EA, Sidow A 2005. Physicochemical constraint violation by missense substitutions mediates impairment of protein function and disease severity. Genome Res 15: 978–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson MS, Nakagawa TY, LeVan K, Dreyfuss G 1987. Primary structure of human nuclear ribonucleoprotein particle C proteins: Conservation of sequence and domain structures in heterogeneous nuclear RNA, mRNA, and pre-rRNA-binding proteins. Mol Cell Biol 7: 1731–1739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarun SZ Jr, Wells SE, Deardorff JA, Sachs AB 1997. Translation initiation factor eIF4G mediates in vitro poly(A) tail-dependent translation. Proc Natl Acad Sci 94: 9046–9051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells SE, Hillner PE, Vale RD, Sachs AB 1998. Circularization of mRNA by eukaryotic translation initiation factors. Mol Cell 2: 135–140 [DOI] [PubMed] [Google Scholar]
- Williamson RM 1995. Information theory analysis of the relationship between primary sequence structure and ligand recognition among a class of facilitated transporters. J Theor Biol 174: 179–188 [DOI] [PubMed] [Google Scholar]
- Yao G, Chiang YC, Zhang C, Lee DJ, Laue TM, Denis CL 2007. PAB1 self-association precludes its binding to poly(A), thereby accelerating CCR4 deadenylation in vivo. Mol Cell Biol 27: 6243–6253 [DOI] [PMC free article] [PubMed] [Google Scholar]