

The authors set out to determine the mode of action of different miRNAs in complex diseases. The main conclusion of this article is that miRNAs act primarily in a complementary rather than synergistic manner to regulate functionally related, disease-relevant mRNAs.
Keywords: microRNA, diseases, regulation, modules
Abstract
MicroRNAs (miRNAs) play a key role in regulating mRNA expression, and individual miRNAs have been proposed as diagnostic and therapeutic candidates. The identification of such candidates is complicated by the involvement of multiple miRNAs and mRNAs as well as unknown disease topology of the miRNAs. Here, we investigated if disease-associated miRNAs regulate modules of disease-associated mRNAs, if those miRNAs act complementarily or synergistically, and if single or combinations of miRNAs can be targeted to alter module functions. We first analyzed publicly available miRNA and mRNA expression data for five different diseases. Integrated target prediction and network-based analysis showed that the miRNAs regulated modules of disease-relevant genes. Most of the miRNAs acted complementarily to regulate multiple mRNAs. To functionally test these findings, we repeated the analysis using our own miRNA and mRNA expression data from CD4+ T cells from patients with seasonal allergic rhinitis. This is a good model of complex diseases because of its well-defined phenotype and pathogenesis. Combined computational and functional studies confirmed that miRNAs mainly acted complementarily and that a combination of two complementary miRNAs, miR-223 and miR-139-3p, could be targeted to alter disease-relevant module functions, namely, the release of type 2 helper T-cell (Th2) cytokines. Taken together, our findings indicate that miRNAs act complementarily to regulate modules of disease-related mRNAs and can be targeted to alter disease-relevant functions.
INTRODUCTION
The functional understanding of gene expression changes in complex diseases is complicated by the involvement of thousands of genes in such diseases (Petretto et al. 2007). Previous studies suggested that this complexity can be reduced by identifying modules of correlated and functionally related genes, which often are coregulated by the same transcription factors (TFs) (Segal et al. 2004). Such modules have been used to find novel candidate genes in complex diseases, such as atherosclerosis and allergy (Hagg et al. 2009; Mobini et al. 2009; Barrenas et al. 2012).
In this study, we hypothesized that like TFs, microRNAs (miRNAs) could also be used to find modules of coregulated disease genes and could be targeted to alter the functions of such modules. miRNAs are small RNAs that generally have a post-transcriptional inhibitory effect on gene expression (Guo et al. 2010). Increasing evidence suggests key roles of miRNAs in many biological processes such as growth, cell proliferation, and apoptosis. miRNAs have been implicated in several diseases, including cancer and coronary, neurodegenerative, and autoimmune diseases (Esquela-Kerscher and Slack 2006; van Rooij and Olson 2007; Xiao et al. 2008; Weinberg and Wood 2009). Recent analyses of mRNA- and miRNA-expression microarrays have suggested that miRNAs regulate modules of genes in prostate cancer (Wang et al. 2009b; Bonnet et al. 2010a,b). Thus, miRNAs might be therapeutically targeted to alter module function in cancer. However, it is not clear whether miRNAs regulate disease-relevant modules of genes in other diseases. A problem with therapeutic intervention may be that multiple miRNAs act synergistically to regulate the same genes (Tsang et al. 2010; Ulitsky et al. 2010; Sass et al. 2011; Su et al. 2011; Gennarino et al. 2012). This suggests that targeting of multiple miRNAs may be required for therapeutic purposes. However, it is not clear if the synergistic actions of miRNAs in the healthy state are applicable to disease, in which a small portion of all miRNAs may show relatively large changes in expression. Those changes could, in turn, lead to disease-associated changes in mRNA expression. By inference, rather than acting synergistically on the same mRNAs, those miRNAs could act complementarily on different mRNAs, thereby regulating functionally related, disease-relevant modules of mRNAs. If so, this could mean that single, or combinations of, miRNAs could be exploited for therapeutic manipulation of genes or pathways in disease modules. Such manipulations could be further facilitated if there were a few disease-associated miRNAs that acted not only complementarily but also as hubs, which regulate very large numbers of disease-associated mRNAs. Hub miRNAs have been recently described under normal physiological conditions in the mouse (Su et al. 2011), in which a small portion of all miRNAs each regulated thousands of mRNAs. It is, however, not clear if there are disease-associated hub miRNAs.
The aims of this study were to examine if disease-associated miRNAs (1) regulated disease-relevant modules, (2) were hubs and acted synergistically or complementarily, and (3) could be targeted to alter the functions of a disease module. We first analyzed publicly available miRNA- and mRNA-expression data for five highly diverse, complex diseases and found that, based on sequence-based predictions and correlation patterns, miRNAs regulated modules of disease-relevant mRNAs complementarily. However, unlike normal conditions, in which a few miRNAs act as hubs, most of the miRNAs tend to regulate multiple mRNAs. These findings were further tested using in-house–generated miRNA- and mRNA- expression data from patients with seasonal allergic rhinitis (SAR). This is a good model of complex diseases because the disease process can be mimicked in allergen-challenged CD4+ T cells from patients and healthy controls in order to identify disease relevant miRNAs and mRNAs. Functional studies can be performed in type 2 helper T-cell (Th2) polarized T cells from healthy controls, using Th2 cytokines as a read-out for disease-relevant functions. We found that, similar to the previously analyzed diseases, the miRNAs acted complementarily to regulate functionally related, disease-relevant modules of mRNAs in SAR. Instead of a few hubs, we found that most of the miRNAs each showed a tendency to regulate multiple mRNAs. Functional studies, using individual or combined silencing or overexpression of two candidate miRNAs, showed that combinations of complementarily acting miRNAs that regulated multiple mRNAs could be targeted for altering disease-relevant functions.
RESULTS
miRNAs regulate disease-relevant mRNA modules in human diseases
We investigated five complex diseases, namely, type 2 diabetes (T2D), chronic obstructive pulmonary disease (COPD), renal cell carcinoma (RCC), acute lymphoblastic leukemia (ALL), and pancreatic cancer (PC) for which both miRNA- and mRNA-expression microarray data were publicly available. We constructed miRNA–mRNA regulatory networks for each disease, in which miRNAs and mRNAs were connected by links. The miRNAs and mRNAs in these networks were differentially expressed in the particular disease, and miRNAs and mRNAs were connected if the miRNA was predicted to target the mRNA and their expression profiles were anti-correlated (Fig. 1A–E). Modules were observed in each disease, although they were less distinct in RCC. Gene Ontology (GO) term and pathway enrichment analysis showed the disease relevance of the identified modules (Supplemental Table 1; Supplemental Fig. 1). Most of the miRNAs tend to regulate multiple mRNAs, while each mRNA might be regulated by few miRNAs. Because the number of mRNA regulated by each miRNA did not follow power law in any of the diseases, there were no formal grounds to define hub miRNAs (Supplemental Fig. 2; Barabasi and Albert 1999). In cancers, each miRNA regulated 84.8 ± 7.2 mRNAs (RCC, ALL, and PC). The corresponding figure in noncancerous complex diseases was 16.1 ± 2.2 mRNAs (T2D and COPD), while each mRNA was regulated by 12.3 ± 10.8 and 1.3 ± 0.54 miRNAs in the two disease categories. When we considered only ALL and PC in the cancers, each miRNA regulated 33.3 ± 4.1 mRNAs, whereas each mRNA was regulated by 2.31 ± 1.1 miRNAs. Next, we examined if the miRNAs acted synergistically or complementarily. For this purpose, we calculated the Jaccard similarity index, which indicates the extent of overlap of the mRNA targets regulated by any two miRNAs in the networks. We found that except in RCC, the miRNAs in each disease had few overlapping targets, supporting a complementary mechanism of regulation. Specifically, the miRNAs in T2D, COPD, ALL, and PC had significantly fewer overlaps (P < 3 × 10−3 for the four diseases) among their shared targets compared with random networks (Supplemental Fig. 3). However, in RCC this trend was not significant. Taken together, computational analysis of miRNA regulation in different diseases indicated that miRNAs regulated modules of disease-relevant mRNAs in human diseases in a complementary way. Most miRNAs seemed to regulate multiple mRNAs but had limited common targets.
FIGURE 1.

miRNA–mRNA regulatory networks in different human diseases. (A) Type 2 diabetes miRNA–mRNA network: The network consisted of 80 nodes, comprising five differentially expressed (DE) miRNAs and 75 DE mRNA targets, with 78 edges showing the miRNA–mRNA regulation. (B) Chronic obstructive pulmonary disease miRNA–mRNA network: The network consisted of 167 nodes comprising 13 DE miRNAs and 154 DE mRNA targets, with 221 edges showing the miRNA–mRNA regulation. (C) Acute lymphoblastic leukemia (B-lineage) miRNA–mRNA network: The network consisted of 264 nodes comprising 24 DE miRNAs and 240 DE mRNA targets, with 480 edges showing the miRNA–mRNA regulation. (D) Pancreatic cancer miRNA–mRNA network: The network consisted of 862 nodes comprising 48 DE miRNAs and 814 DE mRNA targets, with 1921 edges showing the miRNA–mRNA regulation. (E) Renal cell carcinoma miRNA–mRNA network: The network consisted of 1566 nodes comprising 94 DE miRNAs, and 1472 DE mRNA targets, with 11,683 edges showing the miRNA–mRNA regulation. (F) miRNA–mRNA regulatory networks in seasonal allergic rhinitis (allergomiR-target network). The network consisted of 1270 nodes comprising 43 allergomiRs, and 1227 ΔΔ mRNA targets, with 2934 edges showing the miRNA–mRNA regulation. miRNAs are represented as red nodes, while their predicted mRNA targets are represented in blue. The links between the nodes represent regulation of the mRNAs by miRNAs.
miRNAs act complementarily to regulate modules of disease-relevant mRNAs in SAR
To determine if miRNAs acted complementarily to regulate modules of mRNAs in disease and if combinations of miRNAs could be used to alter disease-relevant functions, we repeated the analyses using in-house–generated miRNA and mRNA expression data in SAR. SAR is an ideal model for complex diseases as the external cause (pollen) and key cells (CD4+ T cells) are known. Unlike in most complex diseases, in SAR it is possible to mimic the disease process by obtaining CD4+ T cells outside of the pollen season, when the patients are asymptomatic, and challenge in vitro with pollen or diluent. Thus, it is possible to identify disease-specific changes by comparing expression differences from allergen- and diluent-challenged cells from patients with those observed in controls (Benson et al. 2006a; Bosco et al. 2009). This comparison will henceforth be referred to as a delta-delta (ΔΔ) expression analysis. In contrast, in other diseases, it is generally only possible to compare samples from patients with active disease to samples from healthy controls, which may or may not be exposed to the disease-causing agent(s).
We performed 80 mRNA and 80 miRNA microarray analyses of allergen- or diluent-challenged CD4+ T cells from patients with SAR and healthy controls. Since allergen induces altered expression of immune-related proteins in CD4+ T cells from both patients and healthy controls (Akdis et al. 2004; Bosco et al. 2009), we hypothesized that miRNAs would change in both patients and controls. Indeed, 199 miRNAs were differentially expressed in patients and 77 miRNAs in controls (fold change ≥1.5, P < 0.05). Seventy miRNAs were shared between patients and controls. To define allergy-specific miRNAs (henceforth referred to as allergomiRs), we derived the ΔΔ miRNA expression differences between allergen- and diluent-challenged CD4+ T cells from patients and controls (for details, see Materials and Methods). This resulted in the identification of 44 allergomiRs (Fig. 2; Supplemental Table 2). Next, we performed ΔΔ expression analysis for mRNAs in allergen- and diluent-challenged CD4+ T cells from the same patients and controls. This resulted in the identification of 2094 mRNAs that differed significantly in expression.
FIGURE 2.

Expression profiles of allergomiRs in SAR. Unsupervised hierarchical clustering of allergomiRs identified by ΔΔ expression analysis in the CD4+ T cells obtained from patients samples challenged with allergen and diluent, represented as sample numbers with prefixes A and D, respectively. AllergomiRs distinctly separate the allergen-challenged patient samples from that of diluent-challenged, implying disease-specific response.
By using integrated target prediction and network analysis, we found that the allergomiRs regulated two mRNA modules (Fig. 1F). The disease relevance of the two modules was supported by analyses of their functional characteristics (Supplemental Fig. 4A). The modules were enriched for known disease genes (Supplemental Fig. 4B) and contained novel candidate genes, which were validated by an independent mRNA microarray study of allergen-challenged CD4+ T cells (Supplemental Fig. 5). The two modules consisted of 43 allergomiRs, which putatively regulated 1227 mRNAs. Thus, 59% of all the 2094 ΔΔ mRNAs were regulated by allergomiRs. We found that similar to the other four previously analyzed diseases, rather than a few allergomiRs acting as hubs, most of the allergomiRs tend to regulate multiple mRNAs. Furthermore, most of the allergomiRs acted complementarily. Each allergomiR on average regulated 68.2 ± 6.2 mRNAs, while each mRNA was regulated by 2.4 ± 0.05 allergomiRs. Only six allergomiRs regulated less than 10 mRNAs, and the majority regulated more than 50 mRNAs. Around 54% of the allergomiRs did not share any mRNA targets with other allergomiRs. AllergomiRs had an average Jaccard index of 0.027 ± 0.001 compared with a random distribution of 0.036 ± 0.001 (P< 1×10−5 based on comparison to random networks) (Supplemental Fig. 3F), indicating that allergomiRs acted complementarily in SAR. Since most allergomiRs acted complementarily to regulate many mRNAs, this supported that single or combinations of allergomiRs could be targeted to alter module functions.
Candidate allergomiRs miR-223 and miR-139-3p acted complementarily to regulate the Th2 cytokines IL-5 and IL-13
Allergic inflammation is associated with Th2 cell polarization, which results in increased release of the Th2 cytokines IL-5 and IL-13. These cytokines activate eosinophils and contribute to IgE production and mucous hypersecretion (Boyce et al. 2012). Th2 cell polarization can be induced in naïve T cells from healthy donors, and the effects of allergomiRs can be tested, using IL-5 and IL-13 as read-outs (note that IL-4 was not analyzed because it was used for the Th2 polarization). We tested if allergomiRs would affect these two cytokines, by acting either individually or in combination, and if the allergomiRs acted synergistically or complementarily. We analyzed two allergomiRs, namely, miR-223 and miR-139-3p. They were selected because they (1) were among the allergomiRs that regulated most mRNAs (Fig. 1F), (2) had immune system genes as their targets (but not IL5 and IL13), and (3) had high expression levels (a prerequisite for performing knock-down experiments). The allergomiRs were overexpressed and silenced by transient transfection of the respective miRNAs or antagomirs individually. We also performed combined overexpression and silencing experiments.
The overexpression and the decrease in the levels of the respective miRNAs were confirmed by qPCR (Fig. 3). For IL-5, the overexpression of miR-223 resulted in a 1.3-fold inhibition (P = 0.3), while the inhibition increased IL-5 levels by 2.5-fold (P = 0.03). The corresponding figures for miR-139-3p were 1.7-fold (P = 0.02) and 1.5-fold (P = 0.07), respectively. Thus, each allergomiR influenced IL-5 levels with varying degree of magnitude (Fig. 4). This suggested that the individual effects of the allergomiRs might not be strong enough to affect the cytokine levels. To test if the two allergomiRs acted in consort, we analyzed the effects of combined overexpression and silencing on IL-5. The combined overexpression decreased the IL-5 levels by 2.5-fold (P = 0.01), while the combined silencing increased the levels by 2.2-fold (P < 0.05). This supported that the two allergomiRs acted in consort. We also analyzed the effect of individual and combined overexpression and silencing of these miRNAs on IL-13. The combined overexpression showed a significant decrease of IL-13 levels (P < 0.05) (Fig. 4).
FIGURE 3.

Relative expression levels of miR-223 and miR-139-3p upon overexpression and silencing. qPCR analysis of miR-223 and miR-139-3p in total CD4+ T cells polarized toward Th2 for 12 and 48 h (n = 5–7) upon (A) overexpression of individual miRNAs, (B) combined overexpression of miR-223 and miR-139-3p, (C) silencing of individual miRNAs, and (D) combined silencing of miR-223 and miR-139-3p. The bars in the graph show mean ± SEM of gene expression as the fold change normalized to respective paired controls. The black bar represents the normalized expression values of the paired controls (hence a value of one). Expression levels of overexpression or silencing of miRNAs and controls were compared using a two-tailed paired t-test. (*) P< 0.05; (**) P< 5×10−3; (***) P< 5×10−4.
FIGURE 4.

Relative levels of IL-5 and IL-13 cytokines upon individual and combined overexpression or silencing of miR-223 and miR-139-3p. (A,B) Relative levels of IL-5 (A) and IL-13 (B) upon individual or combined overexpression of miRNAs miR-223 and miR-139-3p. (C,D) Relative levels of IL-5 (C) and IL-13 (D) upon individual or combined silencing of miRNAs miR-223 and miR-139-3p. Cytokine levels were measured with ELISA in supernatants from total CD4+ T cells polarized toward Th2 for 12 and 48 h (n = 7) upon overexpression or silencing of miRNAs. The bars in the graph show mean ± SEM. Relative levels were calculated by normalizing the abundance levels of cytokines upon silencing or overexpression of miRNAs with their respective paired nontargeting controls. The black bar represents the normalized cytokine levels of the nontargeting controls (hence a value of one). Cytokine levels upon overexpression or silencing of miRNAs and nontargeting controls were compared using a two-tailed paired t-test. (*) P < 0.05; (**) P < 5 × 10−3.
mRNA microarray expression analyses following individual and combined miRNA silencing supported that the allergomiRs acted complementarily
To test if the observed effects on the secreted cytokine levels were synergistic or complementary, we performed mRNA expression microarray analysis upon individual and combined silencing of miR-223 and miR-139-3p. Interestingly, miR-223 silencing affected the levels of TFs REL and HDAC2, which regulate IL-13 levels, and CIITA, a regulator of both IL-5 and IL-13 expression. In contrast, miR-139-3p silencing affected the levels of JUND and NFACT1, regulators of IL-5 expression, and SMAD3, which regulates the expression of both IL-5 and IL-13. This showed that both these miRNAs acted complementarily to affect IL-5 and IL-13 levels through intermediate targets.
We then examined the magnitude of effects of these miRNA on their targets. We observed that significantly more predicted targets of miR-223 and miR-139-3p were up-regulated compared with random in the individual silencing (P < 1 × 10−5 for both; P-values based on permutation tests; see Materials and Methods). In the combined silencing, we found that significantly more targets of miR-223 alone were up-regulated (permutation P = 5 × 10−3) (Supplemental Fig. 6). Similarly, miR-223 targets had higher fold changes compared with nontargets in both the miR-223 and combined silencing microarrays (Fig. 5). However, no such effect was observed for miR-139-3p targets. This indicated a stronger influence of miR-223 in Th2 cells compared with miR-139-3p in allergic response. Taken together, these results show a complementary mode of regulation and differential effects on their targets by both these miRNAs.
FIGURE 5.

Distribution of fold changes for up-regulated genes in gene expression analysis following miRNA silencing. (A) miR-223 (T-223) targets were more up-regulated than the nontarget (NT) genes upon miR-223 silencing. (B) miR-139-3p (T-139) targets had comparable up-regulation to NT genes upon miR-139-3p silencing. (C) T-223 targets were more up-regulated compared with NTs upon combined silencing of miR-223 and miR-139-3p. T-139 targets and the common targets of both the miRNAs (T-Both) had comparable fold changes to the respective NTs. The box and horizontal black line show the first and third quartiles and the median; whiskers show the most extreme data points up to 1.5 times the interquartile range (IQR) from the box. Notches around the median are proportional to the IQR and inversely proportional to the square root of n; notches that do not overlap indicate that two distributions have significantly different medians.
DISCUSSION
Recent studies demonstrate the potential use of miRNAs as diagnostic or therapeutic candidates (van Rooij and Olson 2007; Love et al. 2008). Such applications are complicated by the large numbers of miRNAs and mRNAs that are involved in common diseases. Another problem is that, at least under normal physiological conditions, multiple miRNAs seem to act synergistically (Tsang et al. 2010; Gennarino et al. 2012). If a synergistic action also applies to disease, this implies that targeting multiple miRNAs may be required for therapeutic purposes. In this study, we first examined if miRNAs could be used to find modules of coregulated genes in five diverse human diseases. We reasoned that if miRNA-regulated modules could be found in these highly diverse diseases, such modules would also be found in other complex diseases. The advantage of modules is that they are thought to contain the most disease-relevant genes (out of the many identified by high-throughput techniques). Thus, targeting regulators of such modules could have both diagnostic and therapeutic potential (Barabasi et al. 2011; Barrenas et al. 2012). By using computational analyses of mRNA and miRNA microarray data from these diseases, we found that miRNAs acted complementarily to regulate modules of disease-related genes in four diseases (Fig. 6). Most of the miRNAs regulated multiple mRNA targets. As the four diseases are heterogeneous and have partially known disease mechanisms, we turned to SAR to validate our findings. This is a good model of complex diseases in that it is common and has a well-defined phenotype and pathogenesis. Moreover, the main disease process can be studied in allergen- or diluent-challenged CD4+ T cells obtained from both patients and controls. Thus, unlike most other complex diseases, it is possible to identify disease-specific responses by performing ΔΔ expression analyses. The importance of this lies in that many external disease-associated substances, such as allergens, induce the release of inflammatory proteins not only in patients but also in controls (Akdis et al. 2004). In this study, we found that both patients and controls responded to the allergen challenge with changes in the expression of miRNAs. However, similar to the previously analyzed diseases, a relatively limited number of disease-specific miRNAs (allergomiRs) acted in a complementary way to regulate a large number of mRNAs in disease-relevant modules in SAR. To functionally test if the allergomiRs acted complementarily, we examined the effects of overexpressing and silencing two allergomiRs, miR-223 and miR-139-3p. We analyzed their effects on the secreted cytokines, IL-5 and IL-13, which have critical roles in allergy (Woodfolk 2007). These analyses showed variable effects of the individual allergomiRs but consistent effects of the combined allergomiRs. Further analyses with mRNA microarrays after individual and combined silencing of the two miRNAs showed that this complementarity was due to altered regulation of TFs known to regulate IL-5 and IL-13. Taken together, our results support that targeting pairs of complementary miRNAs may have therapeutic potential. We also found that the allergomiR-regulated modules contained novel candidate genes. Further, allergomiRs and mRNA modules derived in this study are an important resource for allergy research. miRNAs are known to play a key role in immune responses (O'Connell et al. 2010). Few studies have focused on miRNAs in allergic disease, and these have predominantly evaluated the effects of isolated miRNAs (Chiba et al. 2009; Lu et al. 2009; Mattes et al. 2009; Goncharova et al. 2010). A recent miRNA microarray study of mild asthmatics could not identify miRNAs related to the disease (Williams et al. 2009). Thus the roles of miRNAs in response to an allergen and their mode of transcriptional regulation in allergy are unclear. However, recently, altered expression of miR-223 was described in eosinophilic esophagitis, a disease with pathophysiological similarities with allergy (Lu et al. 2012). One of the main outcomes of this study is the identification of a relatively limited number of allergomiRs. Those allergomiRs regulated modules consisting of functionally related genes and previously known disease genes. The modules could also be used to identify novel candidate genes likely to be relevant to allergy. These findings show that functionally related genes tend to be coregulated by the same miRNAs. Thus, in analogy to what has previously been shown for TFs and to recent studies of miRNAs in cancers (Gennarino et al. 2012), miRNAs can be used to find modules of coregulated disease-relevant genes. It is also of note that the analytical principles may be generally applicable to similar clinical applications in other diseases. The increasing availability of public mRNA and miRNA expression data will facilitate such applications.
FIGURE 6.

A proposed model of miRNA regulation in diseases. Under normal conditions, miRNAs act synergistically regulating multiple common targets involved in a biological function, represented in panel A as functional module. In pathological conditions (panel B), miRNAs act complementarily to regulate mRNA modules, with limited overlap among their targets.
Deriving a miRNA regulatory network for a cell type or a disease has been challenging owing to (1) miRNAs have different target genes in different cell types; (2) miRNAs act in groups, rather than in isolation; (3) miRNAs have overlapping targets; and (4) there is limited specificity of sequence-based target predictions for miRNAs (Sioud and Cekaite 2010). To address these, we constructed miRNA regulatory networks for different diseases by integrating different layers of information, namely, inverse correlations between mRNA and miRNA expression, target predictions, and significant differential or ΔΔ expression changes for both mRNAs and miRNAs. We note that estimation of inverse correlations between miRNA and mRNA pairs might have a tendency to underestimate the multiple miRNAs acting on an mRNA. Nevertheless, this provides a good framework to focus our network on mRNAs whose expression changes are more likely to be explained by miRNAs than by other regulatory factors, such as TFs and other epigenetic marks. Sequence-based prediction of targets for miRNAs is done by several methods, none of which is complete or accurate. To address this, we combined three different prediction methods, including the in-house–devised support vector machine (SVM) method and derived integrated scores for the predictions. These target predictions are an important resource for future studies.
The modules of coregulated genes showed an enrichment of pathways and GO terms of known relevance to the respective diseases. In SAR, to find novel candidate genes, we searched among the first-degree neighbors of known disease genes. The rationale behind this was that previous studies have shown that novel candidate genes can be identified among the close neighbors of known disease genes (Lim et al. 2006; Lage et al. 2007). To our knowledge, this is the first time this principle has been applied to mRNA targets coregulated by miRNAs. We found that the majority of the coregulated targets were in fact candidate genes, based on their involvement in other inflammatory diseases as well as an independent gene expression microarray study of allergen-challenged CD4+ T cells from patients with SAR. An important implication for future studies is that miRNA regulatory networks representing coregulation may be used to find novel candidate genes in complex diseases.
MATERIALS AND METHODS
Diseasome-level analysis
We obtained miRNA and mRNA microarray data for T2D (GSE26168), COPD (GSE38974), B-cell ALL (GSE14834), PC (GSE32688), and RCC (GSE16441) from NCBI Gene Expression Omnibus. We identified differentially expressed mRNAs using an unpaired, moderated t-test as implemented in limma in R, and miRNAs were identified by performing a Welch t-test. For each disease, we tested if miRNAs acted complementarily in the miRNA–mRNA regulatory networks, by generating 10,000 degree-preserved random networks. For each random network, we calculated the Jaccard similarity index for every pairwise combination of miRNAs and calculated the average of these values to represent the average Jaccard index of that network. P-values were estimated as the ratio of the number of random networks that have average Jaccard index lower than or equal to the average in the real network to the total number of random networks (10,000).
miRNA–mRNA correlations, target predictions, and examining disease-relevance
We then calculated Pearson correlations between differentially expressed miRNAs and mRNAs to find pairs that showed significant inverse correlations (P-value <0.05). Target predictions for miRNAs listed in miRBase release 14.0 were done using three predictions systems, including a two-step SVM classifier that models the individual characteristics of functional binding sites and the global characteristics of miRNA-targeted mRNAs (Saito and Saetrom 2010), Target scan (Lewis et al. 2003), and MicroCosm, the database with miRBase predicted targets generated using the miRanda algorithm (Griffiths-Jones et al. 2008).
We integrated the predictions made by the three prediction methods by generating a combined score for each prediction across all the three methods. For this, first we normalized all the scores from each of the predictions between zero and one using
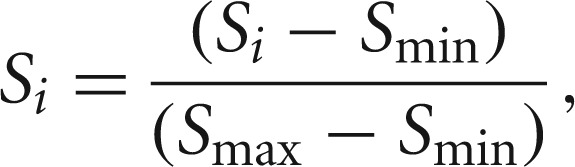 |
where Si is the score of miRNA-target pair prediction from a method, Smin is the minimum score, and Smax is the maximum score given by the method for any miRNA–target pair prediction. Note that for targetscan predictions, a lower score signified better prediction than a higher score. Accordingly, the normalization was undertaken.
The normalized scores for each miRNA–target pair from all or any of the prediction methods, as available, were integrated according to the method previously described (von Mering et al. 2005) using
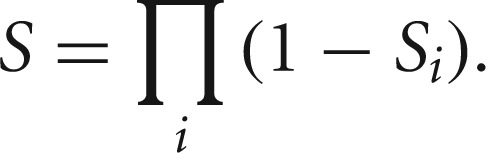 |
We considered the top 25 percentile of the integrated predictions for further analysis. Disease miRNA regulatory networks were constructed using differentially expressed mRNAs that were inversely correlated and predicted targets of differentially expressed miRNAs. Pathway analysis and the TFs regulating IL-5 and IL-13 were obtained using Ingenuity pathway analysis, and GO enrichment was carried out using The Database for Annotation, Visualization and Integrated Discovery (DAVID) bioinformatics resources 6.7 and used biological process (BP_FAT) for identifying enriched terms (Huang da et al. 2007).
SAR study subjects
We recruited patients with SAR and matched healthy controls of Swedish origin at The Queen Silvia Children's Hospital, Gothenburg. SAR was defined by a positive seasonal history and a positive skin prick test or by a positive ImmunoCap Rapid (Phadia) to birch and/or grass pollen. Patients with perennial symptoms or asthma were not included. The healthy subjects did not have any history for SAR and had negative ImmunoCap Rapid tests. The mean ± SEM age of patients and controls was 25.4 ± 1.7 and 25.7 ± 2.2, respectively. Both patients and controls had equal gender representations (10 men and 10 women in each). This study was approved by the ethics board of University of Gothenburg, and all participants provided written consent for participation. We collected 20 samples from patients and healthy controls out of season.
In vitro stimulation of CD4+ T cells with allergen
For the quantitative real-time PCR and gene expression microarray of CD4+ T cells, peripheral blood mononuclear cells (PBMCs) from patients with grass/birch pollen–induced allergic rhinitis were prepared and challenged with diluents or grass/birch pollen extract (100 μg/mL, ALK-Abello) at a density of 106 cells/mL. After 7 d of incubation at 37°C and 5% CO2, the CD4+ T cells were enriched by using anti-CD4-coated paramagnetic microbeads and a MACS system according to the instructions of the manufacturer (Miltenyi Biotech).
miRNA- and gene-expression profiling
The miRNA- and mRNA-expression microarray analyses of allergen- and diluent-challenged (addition of an equal volume of PBS) CD4+ T cells were performed in samples from 20 patients and 20 healthy controls according to the method previously described (Benson et al. 2006a,b). Paired samples were generated by culturing the CD4+ T cells with allergen or diluent according to the method previously described (Benson et al. 2006a). We performed miRNA- and mRNA-expression profiling using Illumina's Human v2 miRNA panel and HumanWG-6 v3.0 Expression BeadChips (Illumina), respectively, on the allergen- and diluent-challenged samples derived from all the 40 subjects, according to the manufacturer's instructions.
Raw data were quantile normalized, and all miRNA probes that had a detection P-value <0.05 in ≥60% samples and those that did not have annotation in miRBase 14.0 were removed. This resulted in the selection of 584 out of 1145 probes. mRNA probes with a detection score below 0.95 were discarded. We calculated delta (Δ) expression differences for miRNA and mRNA probes in patients and controls by subtracting the expression in diluent-challenged samples from the allergen-challenged samples. These Δ values from patients and controls were then compared using a Welch t-test to derive the ΔΔ expression (fold change ≥1.5, P < 0.05).
We tested the expression of novel candidates identified using allergomiR-target network in an independent gene expression microarray study. For this study of CD4+ T cells, PBMCs from 12 patients were challenged with diluent or allergen and analyzed using Human-6 Expression BeadChips (Illumina) according to the manufacturer's instructions (Wang et al. 2009a).
Quantitative RT-PCR validation
Total RNA was extracted from CD4+ T cells using the miRNeasy Mini kit and QIAzol lysis reagent (Qiagen) according to the manufacturer's protocol. cDNA was prepared using the miRCURY LNA Universal RT microRNA PCR kit (Exiqon) according to the manufacturer's protocol. We performed quantitative real-time PCR using the ABI 7900HT system (Applied Biosystems). We amplified miR-223 and miR-139-3p with PCR primer sets and miRCury LNA SYBR Green master mix from Exiqon according to the manufacturer's instructions. As an internal control, snord44 (hsa) PCR primer sets (Exiqon) was used. Data were analyzed using the comparative Ct method. Statistical analyses were performed with a paired, two-tailed Student's t-test if data were derived in pairs (control and sample from same donor) and unpaired, two-tailed Student's t-test if samples were derived from different individuals (patient and healthy control).
Overexpression and inhibition of miR-223 and miR-139-3p
CD4+ T cells were isolated from buffy coat using CD4+ T cell isolation kit II (Miltenyi). For each transfection, 2 million cells were transfected with a final pre-miR (miRNA precursors) or anti-miR (miRNA inhibitors) concentration of 100 nM. The pre-miR and anti-miR were obtained from Ambion alongside with negative controls (#1 pre-miR and #1 anti-miR miRNA inhibitor). The Ambion pre-miR miRNA precursors are chemically modified double-stranded RNAs designed to mimic endogenous mature miRNAs and are used for overexpression. The Ambion anti-miR inhibitors are chemically modified single-stranded RNA molecules which specifically bind endogenous miRNAs aiding in silencing.
The CD4+ T-cells were electroporated with the Amaxa Nucleofector II system (Lonza) using a human T-cell Nucleofecter kit (Lonza) and program U-014 (T cells, human, high viability) according to the manufacturer's manual. After transfection, the cells were incubated for 4–5 h in 2 mL RPMI medium containing 5% FBS, 5% glutamin, 3.9 µg/mL β-mercaptoethanol, and 100 µg/mL gentamycin in a humidified incubator at 37°C and 5% CO2. Post incubation, the cells were polarized toward Th2 in 1 mL polarization-medium (anti-CD28, final concentration 500 ng/mL; anti-IL-12, final concentration 5 μg/mL; IL-4, final concentration 10 ng/mL; IL-2, final concentration 17 ng/mL) for 12 or 48 h, after which the cells were lyzed with 700 μL QIazol. The total RNA was isolated using the miRNeasy Mini Kit from Qiagen.
Gene expression, upon inhibiton of miR-223, miR139-3P and their combination, was analyzed using a Human GE 4 × 44K v2 Microarray Kit (Agilent Tech). The data were quantile normalized and analyzed using GeneSpring GX 10.0 software (Agilent Tech), using a two-tailed t-test for unequal variances.
We tested for enrichment of the predicted targets of miR-223 and miR-139-3p among the up-regulated genes obtained upon individual and combined silencing using a permutation test (employing 10000 random iterations).
ELISA analysis
We analyzed the protein levels of IL-5 and IL-13 in the supernatant upon overexpression and inhibition of miR-223, miR139-3p, and both in combination relative to pre-miR or anti-miR controls with ELISA kits from R&D Systems 12 h and 48 h post transfection. Differences between the two-paired experimental groups were analyzed using a paired, two-tailed Student's t-test.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Kerstin Sandstedt for her help in collecting samples. This work was supported by the European Molecular Biology Organization (S.C.); European Commission, Framework 7, grant no. 223367 (MultiMod) and the Swedish Research Council (M.B.); and the Norwegian Functional Genomics Program (FUGE) of the Norwegian Research Council (P.S.).
REFERENCES
- Akdis M, Verhagen J, Taylor A, Karamloo F, Karagiannidis C, Crameri R, Thunberg S, Deniz G, Valenta R, Fiebig H, et al. 2004. Immune responses in healthy and allergic individuals are characterized by a fine balance between allergen-specific T regulatory 1 and T helper 2 cells. J Exp Med 199: 1567–1575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabasi AL, Albert R 1999. Emergence of scaling in random networks. Science 286: 509–512 [DOI] [PubMed] [Google Scholar]
- Barabasi AL, Gulbahce N, Loscalzo J 2011. Network medicine: A network-based approach to human disease. Nat Rev Genet 12: 56–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrenas F, Chavali S, Couto Alves A, Coin L, Jarvelin MR, Jornsten R, Langston MA, Ramasamy A, Rogers G, Wang H, et al. 2012. Highly interconnected genes in disease-specific networks are enriched for disease-associated polymorphisms. Genome Biol 13: R46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson M, Carlsson L, Guillot G, Jernas M, Langston MA, Rudemo M, Andersson B 2006a. A network-based analysis of allergen-challenged CD4+ T cells from patients with allergic rhinitis. Genes Immun 7: 514–521 [DOI] [PubMed] [Google Scholar]
- Benson M, Langston MA, Adner M, Andersson B, Torinssson-Naluai A, Cardell LO 2006b. A network-based analysis of the late-phase reaction of the skin. J Allergy Clin Immunol 118: 220–225 [DOI] [PubMed] [Google Scholar]
- Bonnet E, Michoel T, Van de Peer Y 2010a. Prediction of a gene regulatory network linked to prostate cancer from gene expression, microRNA and clinical data. Bioinformatics 26: i638–i644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet E, Tatari M, Joshi A, Michoel T, Marchal K, Berx G, Van de Peer Y 2010b. Module network inference from a cancer gene expression data set identifies microRNA regulated modules. PLoS One 5: e10162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco A, McKenna KL, Firth MJ, Sly PD, Holt PG 2009. A network modeling approach to analysis of the Th2 memory responses underlying human atopic disease. J Immunol 182: 6011–6021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce JA, Bochner B, Finkelman FD, Rothenberg ME 2012. Advances in mechanisms of asthma, allergy, and immunology in 2011. J Allergy Clin Immunol 129: 335–341 [DOI] [PubMed] [Google Scholar]
- Chiba Y, Tanabe M, Goto K, Sakai H, Misawa M 2009. Down-regulation of miR-133a contributes to up-regulation of RhoA in bronchial smooth muscle cells. Am J Respir Crit Care Med 180: 713–719 [DOI] [PubMed] [Google Scholar]
- Esquela-Kerscher A, Slack FJ 2006. Oncomirs—microRNAs with a role in cancer. Nat Rev Cancer 6: 259–269 [DOI] [PubMed] [Google Scholar]
- Gennarino VA, D'Angelo G, Dharmalingam G, Fernandez S, Russolillo G, Sanges R, Mutarelli M, Belcastro V, Ballabio A, Verde P, et al. 2012. Identification of microRNA-regulated gene networks by expression analysis of target genes. Genome Res 22: 1163–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncharova EA, Lim PN, Chisolm A, Fogle HW III, Taylor JH, Goncharov DA, Eszterhas A, Panettieri RA Jr, Krymskaya VP 2010. Interferons modulate mitogen-induced protein synthesis in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 299: L25–L35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ 2008. miRBase: Tools for microRNA genomics. Nucleic Acids Res 36(Database issue): D154–D158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Ingolia NT, Weissman JS, Bartel DP 2010. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466: 835–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagg S, Skogsberg J, Lundstrom J, Noori P, Nilsson R, Zhong H, Maleki S, Shang MM, Brinne B, Bradshaw M, et al. 2009. Multi-organ expression profiling uncovers a gene module in coronary artery disease involving transendothelial migration of leukocytes and LIM domain binding 2: The Stockholm Atherosclerosis Gene Expression (STAGE) study. PLoS Genet 5: e1000754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Tan Q, Kir J, Liu D, Bryant D, Guo Y, Stephens R, Baseler MW, Lane HC, et al. 2007. DAVID bioinformatics resources: Expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res 35(Web Server issue): W169–W175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lage K, Karlberg EO, Storling ZM, Olason PI, Pedersen AG, Rigina O, Hinsby AM, Tumer Z, Pociot F, Tommerup N, et al. 2007. A human phenome-interactome network of protein complexes implicated in genetic disorders. Nat Biotechnol 25: 309–316 [DOI] [PubMed] [Google Scholar]
- Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB 2003. Prediction of mammalian microRNA targets. Cell 115: 787–798 [DOI] [PubMed] [Google Scholar]
- Lim J, Hao T, Shaw C, Patel AJ, Szabo G, Rual JF, Fisk CJ, Li N, Smolyar A, Hill DE, et al. 2006. A protein–protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration. Cell 125: 801–814 [DOI] [PubMed] [Google Scholar]
- Love TM, Moffett HF, Novina CD 2008. Not miR-ly small RNAs: Big potential for microRNAs in therapy. J Allergy Clin Immunol 121: 309–319 [DOI] [PubMed] [Google Scholar]
- Lu TX, Munitz A, Rothenberg ME 2009. MicroRNA-21 is up-regulated in allergic airway inflammation and regulates IL-12p35 expression. J Immunol 182: 4994–5002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu TX, Sherrill JD, Wen T, Plassard AJ, Besse JA, Abonia JP, Franciosi JP, Putnam PE, Eby M, Martin LJ, et al. 2012. MicroRNA signature in patients with eosinophilic esophagitis, reversibility with glucocorticoids, and assessment as disease biomarkers. J Allergy Clin Immunol 129: 1064–1075.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattes J, Collison A, Plank M, Phipps S, Foster PS 2009. Antagonism of microRNA-126 suppresses the effector function of TH2 cells and the development of allergic airways disease. Proc Natl Acad Sci 106: 18704–18709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mobini R, Andersson BA, Erjefalt J, Hahn-Zoric M, Langston MA, Perkins AD, Cardell LO, Benson M 2009. A module-based analytical strategy to identify novel disease-associated genes shows an inhibitory role for interleukin 7 receptor in allergic inflammation. BMC Syst Biol 3: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell RM, Rao DS, Chaudhuri AA, Baltimore D 2010. Physiological and pathological roles for microRNAs in the immune system. Nat Rev Immunol 10: 111–122 [DOI] [PubMed] [Google Scholar]
- Petretto E, Liu ET, Aitman TJ 2007. A gene harvest revealing the archeology and complexity of human disease. Nat Genet 39: 1299–1301 [DOI] [PubMed] [Google Scholar]
- Saito T, Saetrom P 2010. A two-step site and mRNA-level model for predicting microRNA targets. BMC Bioinformatics 11: 612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sass S, Dietmann S, Burk UC, Brabletz S, Lutter D, Kowarsch A, Mayer KF, Brabletz T, Ruepp A, Theis FJ, et al. 2011. MicroRNAs coordinately regulate protein complexes. BMC Syst Biol 5: 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E, Friedman N, Koller D, Regev A 2004. A module map showing conditional activity of expression modules in cancer. Nat Genet 36: 1090–1098 [DOI] [PubMed] [Google Scholar]
- Sioud M, Cekaite L 2010. Profiling of miRNA expression and prediction of target genes. Methods Mol Biol 629: 257–271 [DOI] [PubMed] [Google Scholar]
- Su WL, Kleinhanz RR, Schadt EE 2011. Characterizing the role of miRNAs within gene regulatory networks using integrative genomics techniques. Mol Syst Biol 7: 490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang JS, Ebert MS, van Oudenaarden A 2010. Genome-wide dissection of microRNA functions and cotargeting networks using gene set signatures. Mol Cell 38: 140–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitsky I, Laurent LC, Shamir R 2010. Towards computational prediction of microRNA function and activity. Nucleic Acids Res 38: e160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rooij E, Olson EN 2007. MicroRNAs: Powerful new regulators of heart disease and provocative therapeutic targets. J Clin Invest 117: 2369–2376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Mering C, Jensen LJ, Snel B, Hooper SD, Krupp M, Foglierini M, Jouffre N, Huynen MA, Bork P 2005. STRING: Known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res 33(Database issue): D433–D437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Barrenas F, Bruhn S, Mobini R, Benson M 2009a. Increased IFN-γ activity in seasonal allergic rhinitis is decreased by corticosteroid treatment. J Allergy Clin Immunol 124: 1360–1362 [DOI] [PubMed] [Google Scholar]
- Wang L, Tang H, Thayanithy V, Subramanian S, Oberg AL, Cunningham JM, Cerhan JR, Steer CJ, Thibodeau SN 2009b. Gene networks and microRNAs implicated in aggressive prostate cancer. Cancer Res 69: 9490–9497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg MS, Wood MJ 2009. Short non-coding RNA biology and neurodegenerative disorders: Novel disease targets and therapeutics. Hum Mol Genet 18(R1): R27–R39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams AE, Larner-Svensson H, Perry MM, Campbell GA, Herrick SE, Adcock IM, Erjefalt JS, Chung KF, Lindsay MA 2009. MicroRNA expression profiling in mild asthmatic human airways and effect of corticosteroid therapy. PLoS One 4: e5889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodfolk JA 2007. T-cell responses to allergens. J Allergy Clin Immunol 119: 280–294 [DOI] [PubMed] [Google Scholar]
- Xiao C, Srinivasan L, Calado DP, Patterson HC, Zhang B, Wang J, Henderson JM, Kutok JL, Rajewsky K 2008. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat Immunol 9: 405–414 [DOI] [PMC free article] [PubMed] [Google Scholar]