

Abstract
Background
Setipiprant, a tetrahydropyridoindole derivative, is a CRTH2 (chemoattractant receptor-homologous molecule expressed on T-helper [Th]-2 cells) antagonist that has the potential to be effective in the treatment of patients with diseases with an allergic etiology, such as allergic rhinitis and asthma.
Objectives
This study investigated the disposition, metabolism, and elimination of setipiprant.
Study design
In this open-label study, a single oral dose of 1,000 mg 14C-labeled setipiprant was administered.
Participants
Six healthy male subjects were enrolled in this study.
Results
The radioactive dose was almost completely recovered in feces (88.2 %) and to a smaller extent in urine (11.7 %). The main recovery route for unchanged setipiprant was feces (50 % of the radioactive dose). The recovered amount of unchanged setipiprant in urine accounted for 3.7 %. The two main metabolites were M7 and M9 with the intact tetrahydropyridoindole core of setipiprant. M7 and M9 are supposedly two distinct dihydroxy-dihydronaphthalene isomers assumed to be formed by intermediate epoxidation of the naphthyl ring followed by a hydrolytic epoxide ring-opening. M7 and M9 accounted for 20.0 and 15.3 % of the administered radioactive dose. Both metabolites were mainly excreted via feces and to a lesser extent via urine. M7 was the only metabolite quantifiable in plasma, but at concentrations consistently below 10 % of those of the parent drug.
Conclusion
Setipiprant is mainly excreted in feces in the form of the parent drug and in smaller amounts as its metabolites M7 and M9.
Introduction
Setipiprant (ACT-129968, 2-(2-(1-naphthoyl)-8-fluoro-3,4-dihydro-1H-pyrido[4,3-b]indol-5(2H)-yl)acetic acid) is an orally available, selective CRTH2 (chemoattractant receptor-homologous molecule expressed on T helper [Th]-2 cells) antagonist. CRTH2 is a G protein-coupled receptor for prostaglandin (PGD2). PGD2 is produced by the mast cells and is a key mediator in various inflammatory diseases, including allergy and asthma [1–3]. Binding of PGD2 to CRTH2, which are expressed on the surface of blood-borne cells, induces chemotaxis of Th2 cells, basophils, and eosinophils, and stimulates cytokine release from these cells [2, 4]. Thus, antagonism of CRTH2 receptors is considered to be a promising therapeutic target for various allergic diseases and asthma.
Preclinical data showed that setipiprant potently inhibits migration of eosinophils towards PGD2 in vitro as well as in an in vivo rat model of lung eosinophilia (Actelion Pharmaceuticals Ltd, data on file). In the entry-into-man study in healthy male subjects, single and multiple doses of setipiprant of up to 1,000 mg twice daily (bid) for 6 days showed excellent tolerability and a favorable pharmacokinetic profile (Sidharta et al., unpublished data). The pharmacokinetics of setipiprant were characterized by a rapid absorption with a time to maximum plasma concentration (t max) of 2–4 h, followed by a biphasic elimination pattern. The terminal elimination half-life (t ½) was around 10 h following single-dose administration and 14–18 h following multiple-dose administration. Steady-state conditions were reached after 2–3 days. Setipiprant concentrations did not accumulate following 5.5 days of bid administration (Sidharta et al., unpublished data).
In a phase IIa proof-of-mechanism study in patients with mild to moderate allergic asthma, setipiprant (1,000 mg bid) significantly improved the forced expiratory volume in 1 second (FEV1) after a bronchial allergen challenge when compared with placebo during the late allergic reaction (3–10 h) [5]. Another phase IIa study showed significant efficacy versus placebo in seasonal allergic rhinitis [6]. Additional phase II studies with setipiprant in asthma and seasonal allergic rhinitis did not confirm efficacy and therefore the company decided to focus clinical development on the more potent follow-up compound [7].
In this article, we present the results from the absorption, distribution, metabolism, and excretion (ADME) study in healthy male subjects following administration of a single oral dose of 1,000 mg of 14C-labeled setipiprant.
Materials and Methods
Reference Compounds and Other Materials
Setipiprant (ACT-129968, 2-(2-(1-naphthoyl)-8-fluoro-3,4-dihydro-1H-pyrido[4,3-b]indol-5(2H)-yl)acetic acid) was synthesized at Almac Pharma Services, Craigavon, UK. The 14C-label of [14C]setipiprant was located at the carbonyl of the naphthoyl group and the labeled compound was also synthesized by Almac Pharma Services (Fig. 4). [14C]setipiprant was mixed in a ratio of approximately 1:2,200 with nonlabeled setipiprant and filled in hard gelatin capsules of 250 mg for oral administration. [14C]stearic acid for quality control purposes was obtained from ARC-Inc., St. Louis, MO, USA.
Fig. 4.
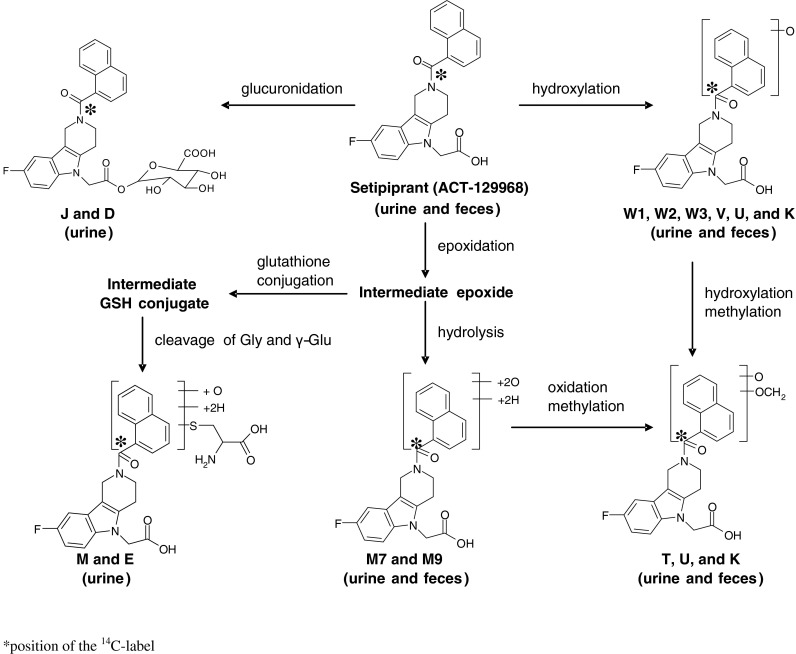
Proposed metabolic scheme for setipiprant
Subjects and Dosing
The clinical part of this study was conducted at Covance (Allschwil, Switzerland), formerly called Swiss Pharma Contract. All subjects gave written informed consent. The study was conducted in accordance with good clinical practice (GCP) and the Declaration of Helsinki. Six healthy male Caucasian subjects, with a mean age of 59.3 years (range 51–65) and a mean body mass index (BMI) of 24.4 kg/m2 (range 21.1–27.8), participated in this study. Subjects remained fasted for 10 h before, and for up to 4 h after, study drug administration. All subjects received a single dose of 1,000 mg setipiprant administered as four capsules of 250 mg with a total radioactivity of 2.60–2.62 MBq (approximately 71 μCi).
Safety and Tolerability
Safety and tolerability were evaluated by monitoring of adverse events, clinical laboratory tests, 12-lead electrocardiograph (ECG) recordings, and measuring of supine vital signs.
Sample Collections
For determination of 14C-radioactivity in whole blood and plasma, for the determination of the plasma concentrations of setipiprant, and for metabolic profiling, blood was collected in ethylenediaminetetraacetic acid (EDTA)-containing tubes at the following time points: predose and 0.67, 1.33, 2, 2.66, 3.33, 4, 5, 6, 7, 8, 10, 13, 18, 24, 48, 72, and 96 h post-dose. After sample preparation, the samples were immediately stored at −70 °C until analysis. An acidified aliquot (acidified with 0.1 M HCl [1:10 v/v]) was obtained from each plasma sample. Expired air samples (used for analysis of radioactivity recovery only) were collected at the same time points. Subjects were instructed to gently blow through a straw into a trapping solution containing 2 mL 1 N hyamine hydroxide and 2 mL ethanol with thymolphthalein as pH indicator until the indicator had become completely colorless (i.e., neutralization of hyamine hydroxide by an equimolar amount of CO2). Subsequently, the collection vials were stored at +4 °C pending analysis of total radioactivity. Urine samples were collected in light-protected tubes on day 1 over 8-h intervals post-dosing and then on days 2–6 at 24-h intervals. All feces were collected over 4 days post-dosing and, after weighing, immediately stored at −70 °C. Radioactivity was measured in daily collected urine and feces until day 4. Where the individual recovery of the total radioactivity was <85 % of the administered dose, daily sample collection was continued until the threshold was reached or until the total daily radioactive excretion was ≤1 % of the administered dose.
Measurement of Total Radioactivity
Radioactivity in samples of whole blood, plasma, urine, feces, and expired air was determined in triplicate using a TRI-CARB 2800TR liquid scintillation counter (Perkin Elmer Life and Analytical Sciences, Waltham, MA, USA). Whole blood samples were prepared by incubation for 10 min at 20 °C with an ethanol/tissue solubilizer mixture (1:1) and then for 30 min at 40 °C after addition of hydrogen peroxide. Liquid scintillation fluid (Ultima Gold®, Perkin Elmer Life and Analytical Sciences) was added and vials counted after having been allowed to stand in the dark at 5 °C for at least 48 h and subsequently at 20 °C for at least 30 min. Liquid scintillation fluid was added to urine (Ultima Gold®), plasma, and expired air (Aerosol-2, Perkin Elmer Life and Analytical Sciences, Downers Grove, IL, USA) samples, kept for at least 30 min at 20 °C in the dark and counted for 10 or 120 min, depending on sample radioactivity. Fecal extracts were homogenized in 1–2 equivalents of water (w/w) and three aliquots of approximately 300 mg were transferred to a porcelain cup and combusted using an OX-700 oxidizer (Zinsser Analytic GmbH, Frankfurt, Germany). The combusted material was taken up in scintillation fluid (Oxysolve-C-400, Zinsser Analytic, Berkshire, UK) and radioactivity determined. The performance of the radioactivity counting was monitored by running simultaneous quality control samples containing known activities of 14C-stearic acid (ARC-Inc., St. Louis, MO, USA).
Quantitative LC–MS/MS Analysis
Plasma concentrations of setipiprant in non-acidified and acidified plasma were determined using validated liquid chromatography–mass spectrometry (LC–MS)/MS methods at Swiss BioAnalytics (Birsfelden, Switzerland). The method for acidified plasma was established to prevent the degradation of potentially formed acyl-glucuronides in plasma. The method for analysis in non-acidified plasma had two calibration ranges, a low calibration from 1.00 to 1,000 ng/mL and a high calibration range from 20.0 to 20,000 ng/mL. Following protein precipitation with acetonitrile containing the stable labeled internal standard and centrifugation, 10 μL of the diluted sample was injected onto the analytical column. Solid-phase extraction was used for processing of acidified plasma samples. Plasma samples were fortified with the labeled internal standards and applied under a gentle vacuum onto 96-well SPEC C18 AR SPE plates (Agilent Technologies, Palo Alto, CA, USA). After washing with 0.1 % formic acid and a mixture of 0.1 % formic acid/methanol (9:1, v/v) samples were eluted with 200 μl methanol, diluted with 0.1 % formic acid (1:1, v/v), mixed, and aliquots of 10 μl were injected onto the chromatographic system.
The chromatographic system consisted of a Rheos 2200 pump (Thermo Fisher Scientific, Waltham, MA, USA), an analytical column (method for non-acidified plasma: Atlantis d C18, 2.1 × 20 mm, 3 μm (Waters, Milford, MA, USA); method for acidified plasma: Zorbax XDB-C18, 2.1 × 50 mm, 5 μm (Agilent Technologies, Palo Alto, CA, USA), and an autosampler (PAL; CTC Analytics, Zwingen, Switzerland). For the chromatographic separation, the solvents were solvent A (0.1 % formic acid) and solvent B (0.1 % formic acid in acetonitrile). The flow rate was set to 0.4 mL/min. A gradient was used in which solvent B was held at 10 % for 0.2 min and that increased linearly up to 90 % within 0.8 min.
Mass spectrometric analysis was performed with a triple quadrupole mass spectrometer TSQ Quantum (Thermo Fisher Scientific, Waltham, MA, USA) operating in positive electrospray ionization mode with capillary temperature at 350 °C and spray voltage at 4.0 kV.
The inclusion of quality control samples throughout the complete study assured method integrity. An analytical run was accepted when at least two-thirds of the quality control samples (analyzed in duplicate) were within ±15 % of their nominal value and when not more than 50 % of the quality control samples at the same concentration were outside this limit. In non-acidified plasma, the accuracy and precision of the quality control samples were 88.0–100.7 and 2.3–18.0 % in the low calibration range and 102.3–105.3 and 1.8–6.2 % in the high calibration range. In acidified plasma, the accuracy and precision of the quality control samples were 99.1–110.7 and 2.0–8.0 %, respectively.
Pharmacokinetic Analysis
The pharmacokinetic parameters of total radioactivity and setipiprant were calculated by noncompartmental analysis from the plasma or blood concentration–time data using Phoenix WinNonlin version 6.1 (Pharsight, Mountain View, CA, USA).
Sample Preparation for In Vivo Metabolic Profiling
Plasma samples (acidified and non-acidified) from all six subjects at the same time point were pooled (predose, 1.33, 2.66, 3.33, 4, 7, 10 h [only for acidified plasma]) to have sufficient radioactivity for detection. Acetonitrile 7.5 mL was added to an aliquot of 2.5 mL plasma pool. After protein precipitation at room temperature, plasma samples were centrifuged for 20 min at 4,000 rpm and 8 °C and the supernatant collected. The protein pellet was resuspended with 7.5 mL of acetonitrile and the resulting suspension vortexed and centrifuged for 20 min at 4,000 rpm and 8 °C. This procedure was repeated twice. The supernatants were combined and evaporated to dryness and reconstituted with 1,000 μL of 0.05 % formic acid in water/methanol/acetonitrile/dimethyl sulfoxide (1:1:1:1, v/v/v/v). Aliquots of 90 μL were injected onto the high-performance liquid chromatography (HPLC) system. Aliquots of 25 μL were taken for liquid scintillation counting to determine the procedural recovery, which was 87.5 % (acidified plasma) and 85.6 % (non-acidified plasma). Recovery of radioactivity from the HPLC system was 79.7 %.
Urine sample pools were prepared with the representative percentage of urine volume of all subjects for the following sampling time intervals: predose, 0–8, 8–16, 16–24, and 24–48 h post-dose. Aliquots of the urine pools were evaporated to dryness under a gentle stream of nitrogen, reconstituted in 10 % of the initial sample volume of water and analyzed without additional sample preparation. A 90-μL aliquot of each pool was injected onto the HPLC system. Procedural recovery of sample preparation was 83.6 %, and recovery of radioactivity from the HPLC system was 94.0 %.
Pooled feces samples containing the representative percentage of feces weight of all subjects were prepared for each sampling day. Pooled feces were extracted by addition of three equivalents (w/v) of methanol and vortex-mixing for approximately 10 min. Samples were then centrifuged for 20 min at 4,000 rpm and 8 °C. After centrifugation, the supernatant was decanted off. The pellet was extracted two more times as described above. Supernatants were combined and evaporated to dryness and reconstituted in 0.05 % formic acid in water/methanol/acetonitrile/dimethyl sulfoxide (1:1:1:1, v/v/v/v). A 50-μL aliquot was injected onto the HPLC system. Duplicate aliquots of 50 μL were used for liquid scintillation counting to determine procedural recovery which was 84.9 %. Recovery of radioactivity from the HPLC system was 92.8 %.
Metabolite Profiling Analysis
The metabolite profile of sample extracts was analyzed by LC–MS/MS combined with offline radioactivity detection after fraction collection. The LC–MS/MS/radioactivity detection system consisted of a PAL autosampler (CTC Analytics AG, Zwingen, Switzerland), a Rheos 2200 pump (Thermo Fisher Scientific, Waltham, MA, USA), a Gecko-2000 column oven (Cluzeau Info Lab, Courbevoie, France), a Luna C18 column (4.6 × 250 mm, 5 μm; Phenomenex, Aschaffenburg, Germany), a LTQ Orbitrap mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA), and a FC204 fraction collector (Gilson Inc., Middleton, WI, USA). The system was operated by Xcalibur 2.1 software (Thermo Fisher Scientific, Waltham, MA, USA). The post-column flow was split at a ratio of 1:5 between the mass spectrometer and the fraction collector. Fractions were sampled into 96-well plates, which were preconditioned with solid-phase scintillation material (Deepwell Luma Plates; PerkinElmer Life and Analytical Sciences, Shelton, CT, USA). The fraction collection interval was 0.15 min. After evaporation to dryness, the plates were analyzed by scintillation counting using a microplate counter (TopCount NXT; Perkin Elmer Life and Analytical Sciences, Waltham, MA, USA). Radiochromatograms were reconstructed by conversion of raw data (counts per fraction vs. fraction number) into chromatographic data (counts per fraction vs. retention time) and processed by the Laura 4.0.3 (LabLogic Systems Limited, Sheffield, South Yorkshire, UK) software. Chromatographic peaks in the reconstructed radiochromatograms were manually integrated. Metabolites were quantified by calculating the percentage of each integrated radiopeak relative to the sum of all peaks in the radiochromatogram. The mass spectrometer was operated to collect full scan and MSn data simultaneously if a predefined ion exceeded an intensity threshold. A radiochromatogram of each sample was reconstructed. All major radiopeaks were assigned and quantified considering an average background of 1 count per minute (CPM). The radiopeak areas were determined in CPM. Only radiopeaks with a signal-to-noise ratio >3 were considered detectable and only radiopeaks with signal height of 9 CPM and above were accepted as quantifiable. Co-eluting metabolites were quantified together. Each quantifiable radiopeak area expressed in percent relative to the total radiochromatogram area was transformed into disintegrations per minute (DPM)/mL or DPM/g. The transformation was done considering the total DPM/mL or DPM/g value of each sample pool. The ng eq/mL values were determined for each quantifiable plasma metabolite. The transformation of the DPM/mL value into the ng eq/mL was carried out considering the specific activity (DPM/ng) of the radiolabeled parent compound [14C]setipiprant.
Structure Elucidation of Metabolites
Structure assignments of the major metabolites were carried out by HPLC/MSn with an LTQ Orbitrap mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA), operated at a resolving power of 15,000 or higher. The capillary temperature of the instrument was kept at 275 °C and spectra in positive and negative ion mode were collected. The mass accuracy of the instrument was better than 2 ppm and unequivocally allowed the determination of the sum formula of the metabolites. Comparison of MSn data of the known parent and the unknown metabolites allowed further structural assignments, even though differentiation between possible isomers is not possible this way. No reference standards were available to verify the assignments.
Results
Safety and Tolerability of Setipiprant
All six subjects completed the study. Single-dose treatment with 1,000 mg [14C]setipiprant was well tolerated. Four subjects (67 %) reported seven adverse events, all of mild intensity. Headache and diarrhea, both reported by two subjects (33 %), were the adverse events considered by the investigator to be related to study drug. The adverse events considered to be unrelated to study drug were feces discolored (two subjects, 33 %) and abdominal discomfort (one subject). No clinically significant abnormalities were observed in clinical laboratory, vital signs, or ECG variables.
Mass Balance and Excretion in Feces and Urine
The cumulative recovery of radioactivity expressed as percentage of the administered dose in feces, urine, and total (mass balance) is shown in Fig. 1. None of the subjects had quantifiable amounts of radioactivity in any expired air sample. Hence, expired air was not a relevant excretion route and was therefore not considered for the calculation of total recovery. No subject vomited during the study. Thus, no corrections for losses by this route were needed. Excretion of the 14C-related radioactivity was virtually complete within 5–6 days. The recovery was relatively quick in the initial days after dosing. Additional recovery was slower in the collection fractions from 72 h onwards as total recovery reached values close to 100 %. Most of the urine recovery occurred within the initial 24 h after dosing. The mean (range) recovery of the administered radioactive dose was 99.96 % (97.04–102.90). The majority of the radioactivity was recovered in the feces (which consists of absorbed and non-absorbed dose), with a mean recovery of 88.2 % (83.1–94.8) of the administered dose. The recovered mean radioactivity in urine accounted for 11.7 % (8.2–14.3) of the administered dose.
Fig. 1.

Mean (SD) time course of cumulative recovery of setipiprant-associated 14C-radioactivity in feces, urine, and total. SD standard deviation
Pharmacokinetics and Disposition of Setipiprant
The mean whole blood and plasma concentration–time profiles of setipiprant-associated 14C-radioactivity are shown in Fig. 2a. After a relatively rapid increase with maximum concentrations of total radioactivity attained after 2.0–2.3 h, whole blood and plasma concentrations of setipiprant-associated 14C-radioactivity initially quickly declined in a multi-exponential manner. The last recorded value above the lower limit of quantification with the radioactive method in whole blood and plasma was at 24 and 72 h post-dose, respectively. The pharmacokinetic parameters in plasma and whole blood of setipiprant-associated 14C-radioactivity are summarized in Table 1. Peak plasma concentration (C max), area under the concentration–time curve (AUC)0–t, and AUC0–∞ in whole blood were 45–60 % lower than in plasma, indicating that most of the radioactivity was associated to plasma. The t ½ of 14C-radioactivity in whole blood (6.7 h) was also shorter than in plasma (24.2 h).
Fig. 2.

a Arithmetic mean and SD whole blood and plasma (non-acidified) concentration–time profiles of setipiprant-associated 14C-radioactivity (linear scale) (n = 6). b Arithmetic mean and SD plasma (non-acidified) concentration–time profile of parent setipiprant (linear and semi-logarithmic scale) (n = 6). SD standard deviation
Table 1.
Pharmacokinetic parameters of setipiprant in plasma (non-acidified) and total radioactivity in plasma and whole blood
| C max (µg/mL)a | t max (h) | t ½ (h) | AUC0–∞(µg × h/mL)b | |
|---|---|---|---|---|
| Setipiprant | 15.6 (12.6, 19.4) | 2.33 (2.00–5.00) | 12.5 (10.3, 15.2) | 61.1 (44.9, 83.1) |
| Radioactivity in plasma | 15.1 (12.4, 18.4) | 2.33 (2.00–5.00) | 24.2 (17.6, 33.3) | 83.9 (61.6, 114) |
| Radioactivity in whole blood | 8.47 (6.88, 10.4) | 2.00 (2.00–5.00) | 6.7 (4.14, 10.8) | 38.6 (27.8, 53.5) |
Data are expressed as median (range) for t max and geometric mean (95 % CI) for C max, t ½, and AUC0–∞; N = 6
AUC area under the concentration–time curve, CI confidence interval, C max peak plasma concentration, t max time to C max, t ½ terminal elimination half-life
aUnit for radioactivity in whole blood and plasma is µg equivalents/mL
bUnit for radioactivity in whole blood and plasma is µg equivalents × h/mL
The mean plasma concentration–time profile of setipiprant (cold method) is depicted in Fig. 2b. The pharmacokinetic parameters are summarized in Table 1. Following a rapid absorption with a median t max of 2.33 h, plasma concentrations of parent setipiprant initially quickly declined, followed by several slower decline phases. The last recorded value above the lower limit of quantification with the cold method was at 144 h post-dose.
The plasma concentration–time profiles of setipiprant-associated 14C-radioactivity and setipiprant (cold method) were almost identical, suggesting that the amount of circulating metabolites is small. However, the t ½ of setipiprant was 12.5 h, which is shorter than the t ½ for the radioactivity in plasma, suggesting that there were at least some metabolites formed.
Quantitative Profiles of [14C]setipiprant and Metabolites in Plasma and Excreta
Representative radiochromatograms in plasma, urine, and feces are shown in Fig. 3. The radioactivity associated with parent setipiprant and its metabolite M7 in plasma (Table 2) and excreted in feces and urine expressed as a percent of the administered dose on each of the evaluated days is shown in Tables 3 and 4. Similar results were obtained for acidified and non-acidified plasma. Only parent setipiprant and its metabolite M7 were detected in plasma at quantities above the limit of quantification (Table 2). Small, not quantifiable, amounts of the acyl-glucuronides J and D were detected in the acidified plasma samples (but not in the non-acidified plasma).
Fig. 3.








Representative radiochromatograms. a Representative HPLC radiochromatograms of human (acidified) plasma pools: 1.33 h (I), 2 h (II), 4 h (III), 7 h (IV). b Representative HPLC radiochromatograms for feces pools: 0–24 h (I), 24–48 h (II), 48–72 h (III), 72–96 h (IV), 96–120 h (V). c Representative HPLC radiochromatograms for urine pools: 0–8 h (I), 8–16 h (II), 16–24 h (III), 24–48 h (IV), 48–72 h (V). CPM count per minute, HPLC high-performance liquid chromatography
Table 2.
Concentrations of circulating setipiprant metabolites in plasma (acidified)
| Metabolite ID | RTRD (min) | C eq (MWparent) of metabolite | ||||
|---|---|---|---|---|---|---|
| 80 min | 160 min | 200 min | 240 min | 7 h | ||
| Unknown | 2.6 | ND | ND | ND | ND | ND |
| M9 (m/z 437) | 26.2 | ND | BLQ | BLQ | BLQ | ND |
| M7 (m/z 437) | 27.8 | ND | 477 | 457 | 379 | BLQ |
| J (m/z 579) | 35.9 | BLQ | BLQ | BLQ | BLQ | BLQ |
| V (m/z 419) | 36.5 | ND | BLQ | BLQ | BLQ | ND |
| D (m/z 579) | 36.7 | |||||
| Setipiprant (m/z 403) | 42.4 | 7,520 | 14,200 | 11,100 | 10,200 | 1,780 |
BLQ below limit of quantification, ND not detected, RD radio detection, RT retention time
Concentrations (C eq [ng equivalents/mL]) are corrected for dilution and molecular weight of the respective analyte
Table 3.
Radioactivity associated to setipiprant and each of its metabolites expressed as percentage of the administered dose excreted in feces
| Metabolite ID | RTRD (min) | % of administered dose excreted in feces | ||||
|---|---|---|---|---|---|---|
| 0–24 h | 24–48 h | 48–72 h | 72–96 h | 96–120 h | ||
| Unknown | 2.6 | 0.65 | ND | ND | ND | ND |
| L | 17.5 | ND | ND | ND | ND | ND |
| M (m/z 540) | 20.3 | ND | ND | ND | ND | ND |
| E (m/z 540) | 22.1 | ND | ND | ND | ND | ND |
| P | 23.9 | ND | ND | ND | ND | ND |
| M9 (m/z 437) | 26.2 | 0.78 | 2.92 | 2.76 | 1.30 | 0.48 |
| M7 (m/z 437) | 27.8 | 1.70 | 5.25 | 5.22 | 2.24 | 0.85 |
| Q | 29.9 | ND | ND | ND | ND | ND |
| R | 33.1 | ND | ND | ND | ND | ND |
| C (m/z 579) | 34.0 | ND | ND | ND | ND | ND |
| W1 (m/z 419) | 34.6 | 0.09 | 0.26 | 0.27 | 0.15 | 0.10 |
| W2 (m/z 419) | 35.0 | |||||
| W3 (m/z 419) | 35.5 | 0.08 | 0.16 | 0.22 | 0.10 | BLQ |
| I (m/z 579) | 35.2 | ND | ND | ND | ND | ND |
| J (m/z 579) | 35.9 | ND | ND | ND | ND | ND |
| T (m/z 449) | 36.1 | 0.10 | 0.54 | 0.40 | 0.19 | 0.14 |
| V (m/z 419) | 36.5 | 0.10 | 0.29 | 0.31 | 0.14 | BLQ |
| D (m/z 579) | 36.7 | ND | ND | ND | ND | ND |
| U (m/z 449; m/z 419) | 37.0 | 0.08 | 0.27 | 0.23 | 0.09 | BLQ |
| X | 37.4 | 0.05 | ND | ND | ND | ND |
| Z (m/z 579) | 37.7 | ND | ND | ND | ND | ND |
| K (m/z 449; m/z 419) | 38.3 | 0.11 | 0.43 | 0.34 | 0.16 | BLQ |
| Y | 40.3 | ND | 0.08 | ND | ND | ND |
| Setipiprant (m/z 403) | 42.4 | 13.73 | 17.57 | 9.98 | 7.04 | 1.72 |
| G | 58.3 | BLQ | 0.13 | 0.09 | BLQ | ND |
| H | 59.5 | 0.16 | 0.22 | 0.16 | 0.12 | ND |
BLQ below limit of quantification, ND not detected, RD radio detection, RT retention time
Table 4.
Radioactivity associated to setipiprant and each of its metabolites excreted in urine expressed as percentage of the administered dose for the respective urine collection intervals
| Metabolite ID | RTRD (min) | % of administered dose excreted in urine | ||||
|---|---|---|---|---|---|---|
| 0–8 h | 8–16 h | 16–24 h | 24–48 h | 48–72 h | ||
| Unknown | 2.6 | 0.10 | ND | ND | ND | ND |
| L | 17.5 | 0.09 | ND | ND | ND | ND |
| M (m/z 540) | 20.3 | 0.06 | 0.02 | BLQ | ND | ND |
| E (m/z 540) | 21.2 | 0.12 | 0.03 | BLQ | ND | ND |
| P | 23.9 | 0.10 | BLQ | ND | ND | ND |
| M9 (m/z 437) | 26.2 | 0.84 | 0.14 | 0.06 | BLQ | ND |
| M7 (m/z 437) | 27.8 | 3.29 | 0.81 | 0.26 | 0.33 | 0.09 |
| Q | 29.9 | 0.05 | ND | ND | ND | ND |
| R | 33.1 | 0.23 | 0.04 | BLQ | ND | ND |
| C (m/z 579) | 34.0 | 0.08 | BLQ | ND | ND | ND |
| W1 (m/z 419) | 34.6 | 0.05 | ND | ND | ND | ND |
| W2 (m/z 419) | 35.0 | |||||
| W3 (m/z 419) | 35.5 | BLQ | ND | ND | ND | ND |
| I (m/z 579) | 35.2 | |||||
| J (m/z 579) | 35.9 | 0.03 | ND | ND | ND | ND |
| T (m/z 449) | 36.1 | |||||
| V (m/z 419) | 36.5 | 0.32 | 0.07 | BLQ | ND | ND |
| D (m/z 579) | 36.7 | |||||
| U (m/z 449; m/z 419) | 37.0 | ND | ND | ND | ND | ND |
| X | 37.4 | ND | ND | ND | ND | ND |
| Z (m/z 579) | 37.7 | 0.05 | BLQ | ND | ND | ND |
| K (m/z 449; m/z 419) | 38.3 | |||||
| Y | 40.3 | ND | ND | ND | ND | ND |
| Setipiprant (m/z 403) | 42.4 | 3.13 | 0.37 | 0.11 | 0.12 | BLQ |
| G | 58.3 | ND | ND | ND | ND | ND |
| H | 59.5 | ND | ND | ND | ND | ND |
BLQ below limit of quantification, ND not detected, RD radio detection, RT retention time
Parent setipiprant was the main moiety recovered from feces in all evaluated collection periods, accounting for a daily excretion of up to 17.6 % of the radioactivity dose on a given study day (day 2), followed by M7 (accounting for a daily excretion of up to 5.3 % (day 2) of the radioactivity dose) and M9 (accounting for a daily excretion of up to 2.9 % (day 2) of the radioactivity dose) (Table 3). The unknown early peak (retention time [RT] 2.6 min) accounted for 0.65 % of the radioactivity dose on the first day after dosing and was not detected thereafter. Metabolite T accounted for more than 0.5 % of the radioactivity dose on the second day after dosing and was also the most prevalent moiety after parent setipiprant, M7, and M9 on the third to fifth day after dosing.
Metabolite M7 was the main urinary moiety present in all collected fractions and the only moiety detected in urine on the third day after dosing (Table 4). The second urinary moiety in all fractions was parent setipiprant and the third moiety was M9. In successive fractions, the number of detected peaks gradually decreased. By the second day, only M7 and parent setipiprant were still quantifiable; M9 was detectable but below the limit of quantification, and the other moieties were no longer detectable.
The overall metabolic profile of setipiprant in the excreta of the study subjects is provided in Table 5. Unchanged setipiprant was recovered in an amount accounting for 53.8 % of the administered radioactive dose. The proposed metabolic scheme for setipiprant, including the proposed molecular structure of the metabolites, is provided in Fig. 4. The precise molecular structure of the metabolites was not elucidated. The two main metabolites were M7 and M9 with the intact tetrahydropyridoindole core of setipiprant. M7 and M9 are supposedly two distinct dihydroxy-dihydronaphthalene isomers assumed to be formed by intermediate epoxidation of the naphthyl ring followed by a hydrolytic epoxide ring-opening. M7 and M9 are further metabolized by oxidation and methylation to form T, U, and K. The same intermediate epoxide leads by glutathione conjugation to M and E, which are found in urine only. J and D are supposed to be formed by glucuronidation and subsequently excreted via urine. Hydroxylation of the naphthyl moiety of setipiprant leads to various metabolites (W1, W2, W3, V, U, K). These metabolites were found in urine and feces.
Table 5.
Overall metabolic profile of setipiprant in the excreta of the study subjects
| Metabolite ID | RTRD (min) | % of Aadministered excreted | % of Aadministered excreted | |
|---|---|---|---|---|
| Urine | Feces | Urine and feces | ||
| Unknown | 2.6 | 0.10 | 0.65 | 0.75 |
| L | 17.5 | 0.09 | ND | 0.09 |
| M (m/z 540) | 20.3 | 0.08 | ND | 0.08 |
| E (m/z 540) | 21.2 | 0.15 | ND | 0.15 |
| P | 23.9 | 0.10 | ND | 0.10 |
| M9 (m/z 437) | 26.2 | 1.04 | 8.24 | 9.28 |
| M7 (m/z 437) | 27.8 | 4.78 | 15.26 | 20.04 |
| Q | 29.9 | 0.05 | ND | 0.05 |
| R | 33.1 | 0.27 | ND | 0.27 |
| C (m/z 579) | 34.0 | 0.08 | ND | 0.08 |
| W1 (m/z 419) | 34.6 | 0.05 | 0.87 | 0.92 |
| W2 (m/z 419) | 35.0 | |||
| W3 (m/z 419) | 35.5 | BLQ | 0.56 | 0.56 |
| I (m/z 579) | 35.2 | ND | BLQ | |
| J (m/z 579) | 35.9 | 0.03 | 1.37 | 1.40 |
| T (m/z 449) | 36.1 | |||
| V (m/z 419) | 36.5 | 0.39 | 0.84 | 1.23 |
| D (m/z 579) | 36.7 | |||
| U (m/z 449; m/z 419) | 37.0 | ND | 0.67 | 0.67 |
| X | 37.4 | ND | 0.05 | 0.05 |
| Z (m/z 579) | 37.7 | 0.05 | 1.04 | 1.09 |
| K (m/z 449; m/z 419) | 38.3 | |||
| Y | 40.3 | ND | 0.08 | 0.08 |
| Setipiprant (m/z 403) | 42.4 | 3.73 | 50.04 | 53.77 |
| G | 58.3 | ND | 0.22 | 0.22 |
| H | 59.5 | ND | 0.66 | 0.66 |
BLQ below limit of quantification, ND not detected, % of A administered % of administered radioactive dose, RD radio detection, RT retention time
Unchanged setipiprant was mainly recovered in feces (50.0 % of the radioactive dose). Small amounts of unchanged setipiprant were also found in urine (3.8 % of the radioactive dose). The second moiety by excreted amount, accounting for 20.0 % of the administered radioactivity dose, was metabolite M7. M7 was also mostly excreted by feces (15.3 % of the administered dose). However, it was the quantitatively most important setipiprant-derived radioactive moiety in urine, accounting for 4.8 % of the administered dose. M7 was the only radioactive moiety in addition to parent setipiprant that was quantifiable in plasma, but M7 concentrations were consistently below 10 % (maximum: 6.3 % at 240 min post-dose in non-acidified plasma) of those of the parent drug. The third moiety by excreted amount, accounting for 9.3 % of the administered radioactive dose, was metabolite M9. M9 was also mostly excreted by feces (8.2 % of the administered dose). M9 was not quantifiable in plasma. The other moieties accounted for smaller amounts of the administered radioactive dose, with only metabolites J/T, V/D, and Z/K accounting for the excretion of more than 1 % but <1.5 % of the administered dose.
Discussion
The aim of this study was to characterize the disposition and metabolism of setipiprant, a selective CRTH2 antagonist, in humans. The setipiprant-associated 14C-radioactivity (converted to µg eq/mL setipiprant) and setipiprant concentrations in plasma obtained by two different methods were almost identical, indicating that most of the drug in plasma is unchanged setipiprant. The administered radioactive dose was almost completely recovered (99.96 %) within 5–6 days, with 88.2 % of the administered radioactive dose recovered in feces and 11.7 % in urine. Negligible not quantifiable amounts of radioactivity were found in expired air. Based on the results obtained, the bioavailability of setipiprant appears to be at least 50 %, which is in line with animal data. Setipiprant exhibited an oral bioavailability of 32–55 % in rats and of 26–46 % in dogs. Setipiprant does not appear to be extensively metabolized. Unchanged setipiprant made up 53.8 % of the administered radioactive dose. None of the metabolites was found in plasma accounting for more than 10 % of setipiprant. The two main metabolites were M7 and M9, two distinct dihydroxy-dihydronaphthalene isomers assumed to be formed by intermediate epoxidation of the naphthyl ring followed by a hydrolytic epoxide ring-opening. M7 and M9 were both mainly excreted via feces and to a smaller extent via urine. The only difference in the metabolic profiling of the acidified compared with the non-acidified plasma was that small not quantifiable amounts of acyl-glucuronides were detected (J and D). Because setipiprant-associated 14C-radioactivity and setipiprant concentrations in plasma were similar, and only low amounts of M7 and M9 were detected, it is likely that there are no other yet non-identified metabolites. Due to the low abundance of the metabolites, no specific toxicology studies were conducted with any metabolite.
Conclusion
Setipiprant is metabolized to a moderate extent. Setipiprant is mainly excreted in feces as parent drug and in smaller amounts as metabolites M7 and M9.
Acknowledgments
The authors thank Covance (Allschwil, Switzerland) with Thierry Kamtchoua as principal investigator for the clinical conduct of the study and Luis López Lázaro for writing parts of the clinical study report. The authors also thank Julien Pothier and Heinz Fretz from Actelion Pharmaceuticals Ltd for their careful manuscript review.
Declaration of interest
This study was sponsored by Actelion Pharmaceuticals Ltd. Matthias Hoch and Jasper Dingemanse are full-time employees of Actelion Pharmaceuticals Ltd. Swiss BioAnalytics received funding from Actelion Pharmaceuticals Ltd. Janine Wank and Ina Kluge were full-time employees of Swiss BioAnalytics at time of study conduct and data analysis. Winfried Wagner-Redeker is full-time employee of Swiss BioAnalytics.
References
- 1.Arima M, Fukuda T. Prostaglandin D2 receptors DP and CRTH2 in the pathogenesis of asthma. Curr Mol Med. 2008;8(5):365–375. doi: 10.2174/156652408785160970. [DOI] [PubMed] [Google Scholar]
- 2.Schuligoi R, Sturm E, Luschnig P, Konya V, Philipose S, Sedej M, et al. CRTH2 and D-type prostanoid receptor antagonists as novel therapeutic agents for inflammatory diseases. Pharmacology. 2010;85(6):372–382. doi: 10.1159/000313836. [DOI] [PubMed] [Google Scholar]
- 3.Satoh T, Moroi R, Aritake K, Urade Y, Kanai Y, Sumi K, et al. Prostaglandin D2 plays an essential role in chronic allergic inflammation of the skin via CRTH2 receptor. J Immunol. 2006;177(4):2621–2629. doi: 10.4049/jimmunol.177.4.2621. [DOI] [PubMed] [Google Scholar]
- 4.Kostenis E, Ulven T. Emerging roles of DP and CRTH2 in allergic inflammation. Trends Mol Med. 2006;12(4):148–158. doi: 10.1016/j.molmed.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 5.Diamant Z, Singh D, O’Connor B, Zuiker R, Ponnarambil S, Leaker B, et al. Effect of multiple-dose setipiprant, a selective oral CRTH2 antagonist, on allergen-induced airway responses in allergic asthmatic patients. Am J Respir Crit Care Med 185;2012:A3957.
- 6.Company press release: Actelion’s novel CRTH2 antagonist meets primary endpoint in Phase II study in patients with seasonal allergic rhinitis. http://www.actelion.com. Accessed 23 May 2011.
- 7.Company press release: Actelion provides update on CRTH2 program. http://www.actelion.com. Accessed 2 April 2012.