

Abstract
The role of the venous system in the pathogenesis of inflammatory neurological/neurodegenerative diseases remains largely unknown and underinvestigated. Aside from cerebral venous infarcts, thromboembolic events, and cerebrovascular bleeding, several inflammatory central nervous system (CNS) diseases, such as multiple sclerosis (MS), acute disseminated encephalomyelitis (ADEM), and optic neuritis, appear to be associated with venous vascular dysfunction, and the neuropathologic hallmark of these diseases is a perivenous, rather than arterial, lesion. Such findings raise fundamental questions about the nature of these diseases, such as the reasons why their pathognomonic lesions do not develop around the arteries and what exactly are the roles of cerebral venous inflammation in their pathogenesis. Apart from this inflammatory-based view, a new hypothesis with more focus on the hemodynamic features of the cerebral and extracerebral venous system suggests that MS pathophysiology might be associated with the venous system that drains the CNS. Such a hypothesis, if proven correct, opens new therapeutic windows in MS and other neuroinflammatory diseases. Here, we present a comprehensive review of the pathophysiology of MS, ADEM, pseudotumor cerebri, and optic neuritis, with an emphasis on the roles of venous vascular system programming and dysfunction in their pathogenesis. We consider the fundamental differences between arterial and venous endothelium, their dissimilar responses to inflammation, and the potential theoretical contributions of venous insufficiency in the pathogenesis of neurovascular diseases.
Keywords: Venous, MS, CNS, ADEM, Inflammation
Introduction
The human central nervous system (CNS) can be affected by a number of inflammatory demyelinating diseases. This covers a wide range of clinically and neuropathologically heterogeneous conditions, which share some clinical characteristics, but possess distinguishing immunopathological features. Multiple sclerosis (MS) and acute disseminated encephalomyelitis (ADEM) are two of the most prominent of these inflammatory diseases. Although these conditions have different root causes, mechanisms, and courses, their underlying neuropathologies both exhibit perivenular demyelination. This strikingly significant important finding points to significant contributions by veins in these conditions, and suggests that venous dysfunction or vein-targeted disease processes, (rather than arterial pathology or injury) contributes to the development of these inflammatory CNS diseases. Unlike the cerebral arterial system, the spatial organization of cerebral venous networks is more complex and more often asymmetric, with greater structural heterogeneity than cerebral arterial anatomy. Consequently, this half of the circulatory system has been far less studied and understood [1].
Several reviews [2] have evaluated clinical and structural factors in venous contributions to neurologic diseases. In addition to the inflammatory-based view of the pathogenesis of these demyelinating diseases, the past few years has witnessed the emergence of a controversial view about MS. Could neurological disease processes such as MS be triggered or intensified in part through venous vascular disturbances? Although venous disturbances in particular have long been recognized in several forms of neurological disease, we are only recently appreciating how venous structure, programming, and responses contribute to specific features of these diseases. The concept that neurologic disease can be influenced by structural or functional abnormalities of the CNS venous system has raised intense worldwide debate among researchers, with many investigators arguing against its existence. Controlled, careful clinical studies are needed to validate when and how vascular alterations can contribute to forms of CNS injury and inflammation. Here, we provide a discussion on the potential pathogenesis of these diseases, with emphasis on venous endothelial dysfunction in MS, ADEM, and other forms of neuroinflammation.
Pathophysiology of MS with emphasis on venous dysfunction
MS is a group of immune-mediated demyelinating syndromes associated with neurodegeneration in the human CNS, which causes significant neurological disability in largely younger adults (Noseworthy [3], Compston and Coles [4]). MS can affect both gray and white matter in any region of the CNS. Four distinct clinical patterns of MS are recognized: relapsing-remitting (RRMS), primary progressive MS (PPMS), secondary progressive MS, and progressive relapsing MS. To date, vascular studies in MS have investigated cerebrovascular capillary and large vessel venous endothelial cells that are not always derived from (or strictly relevant to) the CNS [5-7]. There has been less research into the arterial and venous differences in MS. Despite these limitations, vascular contributions in MS do appear to support the notion of the vasculature being an initiating target in MS etiology and not simply a bystander presentation of other disease processes. Perhaps the strongest support for this is the number of MS therapies that have been developed, which target leukocyte binding to activated endothelial cells, a central component of the blood-brain barrier (BBB). Vascular abnormalities in MS also include evidence of increased circulating markers of vascular inflammation, [8-10], which can lead to inflammatory challenges that initiate or exacerbate CNS injury. Magnetic resonance imaging (MRI) studies in MS also indicate longer mean blood-flow transit times, which indicates relatively lower cerebral blood flow in MS plaques, as well as decreased cerebral blood flow and prolonged mean transit time in normal-appearing white matter (NAWM). Decreases in brain blood flow increase with age in MS, with severity and form of MS (PPMS > RRMS) both of which may intensify ischemic injury [2,9,11]. Importantly, in apparently NAWM, the state of ischemia appears to occur before the appearance of plaques [10]. It is unclear whether diminished cerebral flow represents restricted perfusion (arterial-sided) or outflow restriction (venous influences). Further, venous blood exiting the cerebral veins of patients with MS in susceptibility-weighted imaging (also known as blood oxygen level dependent imaging) suggests lower net tissue oxygen consumption compared with controls [12], which points to disturbances in energy metabolism. These findings suggest an early role for vascular disturbances in MS, which may trigger later injury processes, but do not specifically indicate underlying vascular defects as their basis.
There are several significant differences between venous and arterial endothelial cells, which may play a role in increased susceptibility of the venous compartment as related to MS, ADEM, or chronic venous insufficiency (CVI); these differences include arteriovenous programming, flow shear-dependent gene expression, hemodynamic effects on autacoids and venous valve organization.
Arterial versus venous differences in response to inflammation
The adhesive qualities of arterial and venous endothelial cells can be modified by inflammation or disease. In comparison with the arterial environment, lower venous shear stresses combined with increased venous endothelial permeability and responsiveness (to at least some inflammatory mediators) may make venules and veins more susceptible to developing inflammation. For example, Kalogeris et al., [13] showed that cytokine-responsive endothelial cell adhesion molecule (ECAM) responses toward cytokine exposure were higher in venous endothelium than in corresponding (umbilical) arterial endothelium, and also supported higher (venous) endothelial rates of binding of monocytes. Tumor necrosis factor (TNF)-α and lipopolysaccharide (LPS) were seen to significantly increase monocyte binding to venous, but not arterial endothelium in vitro. Furthermore, neither TNF-α nor LPS induced surface expression of vascular cell adhesion molecule (VCAM)-1 or E-selectin in arterial endothelium, and TNF did not induce VCAM-1 mRNA in arterial endothelium. Lastly, as a VLA-4 blocking antibody prevented about 75% of TNF-α-stimulated monocyte adhesion in venous endothelium, VCAM-1 dependent adhesion may be particularly important in TNF-α response. Interestingly, despite a TNF-α-mediated increase in surface-expressed intercellular adhesion molecule (ICAM)-1 in arterial endothelium, TNF-α did not increase monocyte adhesion to arterial endothelium. Amberger et al. [14] also found that venous endothelium (umbilical and saphenous veins) expressed higher levels of ICAM-1, VCAM-1, and E-selectin than arterial endothelium in response to TNF-α, interleukin (IL)-1β, and LPS, but lower levels of adhesion molecule responses to low density lipoprotein. Therefore, venous endothelium appears to be innately programmed for higher adhesive responses compared with arterial endothelium. Similarly, Wang and Feuerstein [15] showed that ischemia is a potent, albeit slower stimulus for ICAM-1 and E-selectin expression in the brain, potentially linking reduced blood flow in lesions and NAWM with immune cell infiltration.
With respect to underlying BBB differences between venous and arterial endothelium, we have previously reported that, compared with arterial endothelial cells, venous endothelial cells expressed more vascular endothelial (VE)-cadherin at the mRNA and protein levels Kevil et al., [16]. Conversely, arterial endothelial cells were found to express eighteen-fold more occludin at the protein and nine-fold more at the mRNA level. Occludin was also seen to be more organized at inter-endothelial junctions in umbilical arterial endothelial cells compared with umbilical venous endothelial cells Kevil et al., [16]. Interestingly, disturbances in flow direction, but not necessarily shear, modulate claudin-5, another component of tight junctions, which also shows arteriovenous endothelial heterogeneity, with arterial endothelium expressing higher levels of claudin-5 than venous endothelium [17]. Claudin-5 is also induced by estradiol [18], which could be a factor in gender-specific differences in BBB or disease incidence. Endothelial expression of junctional components (and barrier) also depends on other cells in the neurovascular complex, such as astrocytes [19], as well as the effects of inflammatory mediators on these support cells and the endothelium [20].
Although arterial and venous endothelial cells are heterogeneous, ‘arterialization’ of venous endothelial grafts (for example, in coronary artery bypass grafts), also suggests that fluid shear, oxygen tension differences, and other environmental factors can remodel transplanted veins into arterial homologs, and significant post-natal arteriovenous plasticity may be induced under different circumstances. Because each vascular type exhibits different relative responses to different types of injury and inflammatory stimuli, chronically altered shear stress or retrograde flow may lead to injury, which could reflect the mechanical trauma of the intima, as well as a shear-dependent remodeling of vessels exposed to shear forces. Adamson et al. recently showed that retrograde flow, rather than shear forces, diminishes the venous endothelial solute barrier by decreasing the organization of endothelial junctional VE-cadherin and occludin, a finding supporting the concept that abnormal flow patterns can dysregulate endothelial barrier properties Adamson et al., [21]. It is still unclear whether transvenular leukocyte extravasation is also enhanced by retrograde flow.
Zakkar et al. reported that induction of a pro-inflammatory phenotype on venous endothelial cells involves the phosphorylation-dependent activation of p38 mitogen-activated protein kinase (MAPK), which leads to the production of chemokines, including IL-8 and monocyte chemotactic protein-1 Zakkar et al., [22]. Thus, veins exposed to shear undergo activation of p38 MAPK, which may lead to inflammation. By contrast, shear exposure in arteries has been shown to lead to induction of MAPK phosphatase (MKP)-1, which decreases MAPK signaling. In that report, Sakkar et al. demonstrated that dexamethasone could induce expression of MKP-1 in venous endothelium, effectively recapitulating the protective effect of shear seen in arterial endothelium exposed to laminar shear stress. These events require extracellular regulated kinase (Erk)1 and Erk 2, cAMP response element binding, and oxidant signaling. The current use of dexamethasone in MS might therefore correct an abnormal flow-mediated activation of venous inflammatory programs and fully integrate components of the BBB.
Is there altered hemodynamic signaling in venous inflammation?
In what other ways might flow disturbances lead to hyperactivation of inflammatory responses in the venous circulation? Krueppel-like factor (KLF)2 and KLF4 are shear-dependent transcription factors that suppress endothelial responses to inflammatory stimuli, such as TNF-α [23], and several important shear-sensing mechanisms rely on KLF2 and KLF4 to provide important links between laminar fluid shear and the maintenance of a quiescent endothelial phenotype (Table 1). Conversely, disturbances in normal flow patterns might increase inflammation through KLF2/4 dysregulation. Laminar shear regulates KLF2 by promoting the phosphorylation and nuclear export of histone deacetylase (HDAC)5, a process under the control of Ca2+/calmodulin. This process partitions HDAC5 from myocyte enhancer factor-2, which then triggers KLF2 expression. Wu et al., [24] also showed that laminar shear suppressed miRNA-92a, an endogenous inhibitor of KLF2 [24] and KLF4 [25]. KLF4 expression is also suppressed by HDACs, and is de-repressed by the HDAC inhibitor trichostatin-A (Table 1) [26]. KLF2 expression varies dramatically between arteries and veins, with arteries expressing about four-fold more KLF2 than their corresponding veins [23]. Liu et al. also found that freshly isolated arterial endothelium expressed higher levels of KLF2 than did venous endothelium, consistent with greater KLF2 arterial responses to fluid shear stress [23]. KLF2 was shown to be suppressed by inflammatory stimuli such as IL-1β [27], unlike KLF4, which paradoxically was activated by TNF-α, IL-1β and interferon (IFN)-γ [28], as well as shear. Venous cells exposed to shear also increase KLF2 expression (compared with static cultures) [29]. KLF2 is an important shear-activated transcription factor which upregulates endothelial nitric oxide synthase (eNOS) and thrombomodulin (TM) expression and reduces plasminogen activator inhibitor-1 (PAI-1) expression [30]. KLF2 also suppresses IL-1β induced endothelial VCAM-1 and E-selectin expression and TNF-α induction of tissue factor (TF) [27,30,31]. Shear-induced expression of KLF2 also suppresses activation of the pro-inflammatory transcription factors activator protein-1, nuclear factor κB Das et al., [32], and activating transcription factor 2 Fledderus et al., [33]. Importantly, induction of KLF2 in venous endothelium reduced TNF-α-induced E-selectin and VCAM-1. Shear-activated KLF2 also maintains endothelial quiescence by suppression of TNF-α receptors, upregulation of eNOS [27] and by decreasing angiopoietin-2 content in endothelial Weibel-Palade bodies [34]. KLF4 is similarly induced in endothelial cells by laminar shear stress and interestingly by inflammatory cytokines. Like KLF2, KLF4 also induces eNOS and TM, and suppresses endothelial VCAM-1 expression. KLF4 activation also decreases thrombus formation by downregulating tissue factor expression [28], and KLF4 also downregulates plasminogen activator inhibitor (PAI)-1. Therefore venous hemodynamic flow disturbances that lead to silencing of anti-inflammatory KLF2/KLF 4 programs might increase inflammation through altered endothelial barrier, leukocyte binding, and hemostasis [35]. Interestingly, 3-hydroxy-3-methylglutaryl-coenzyme A reductase statin drugs have recently been described as activators of KLF2 [36] and KLF4 [37], and may restore or maintain atheroprotective programs suppressed by abnormal venous flow fluid shear patterns. Statin activation of KLF2 also induces hemoxygenase-1, an important suppressor of inflammation [38]. Similarly, other drugs that maintain KLF2/4, such as HDAC inhibitors, might represent novel treatments for treating abnormal signaling in venous (and also arterial) endothelium produced by flow abnormalities.
Table 1.
Pathophysiology of venous abnormalities in multiple sclerosis and potential therapeutic strategies
| Pathophysiology | Involved molecules | Potential intervention | Potential treatments | References |
|---|---|---|---|---|
| Higher venous endothelial responses to inflammation |
Cytokines, chemokines, adhesion molecules, occludin |
Induction of MKP-1, protection against shear stress responses |
Dexamethasone |
[16,39] |
| Altered hemodynamic signaling in venous inflammation |
KLF2, KLF4, eNOS, VCAM-1, PAI-I, TNF-α |
Activation of KLF2 and KLF4 |
Statin drugs, HDAC inhibitors (for example, trichostatin-A) |
[8,30,36,40] |
| BBB dysregulation |
NMDA receptor, MMP-8, MMP-9, p38 MAPK |
MMP inhibitor, p38 MAPK inhibitor |
Doxycycline, minocycline, SB 239063 |
[41-44] |
| Venous remodeling |
Collagens, iron, TGF-β1, p38 MAPK, VEGF, TIMP, MMP |
p38 MAPK inhibitor, TGF modifier, angiotensin antagonist, anti-angiogenic drug, MMP inhibitor |
Drugs (dilamapimod, avotermin, candesartan, bevacizumab, cavtratin, doxycycline, desferrioxamine) |
[8,45-47] |
| Hemodynamic abnormality, CCSVI | PGI2, NO, EDHF | Venous pressure reduction | venoplasty | [48,49] |
Abbreviations:BBB blood-brain barrier, CCSVI chronic cerebrospinal venous insufficiency, EDHF endothelium-derived hyperpolarizing factor, eNOS Endothelial nitric oxide synthase, HDAC histone deacetylase, KLF Krueppel-like factor, MAPK mitogen-activated protein kinase, MKP mitogen-activated protein kinase phosphatase, MMP matrix metalloproteinase, MS multiple sclerosis, NMDA N-methyl-D-aspartate, NO nitrous oxide, PAI plasminogen activator inhibitor, PGI2 prostaglandin I2 (prostacyclin), TGF transforming growth factor, TIMP tissue inhibitor of metalloproteinase, TNF tumor necrosis factor, VCAM vascular cell adhesion molecule, VEGF Vascular endothelial growth factor.
Is the blood-brain barrier altered by factors induced in neurodegenerative disorders?
Several factors present in MS may dysregulate BBB in such a way that when presented with altered flow or pressure gradients, significant disturbances in BBB could be produced. It is now fairly well accepted that VE cells express N-methyl-D-aspartate (NMDA) and metabotropic receptor complexes, which contribute to regulation of the BBB. Glutamate is increased in the cerebrospinal fluid (CSF) in patients during relapse [50] consistent with its release during CNS injury. Binding of glutamate to endothelial NMDA receptor elevates intracellular oxidants [44] and disturbs the microvascular barrier [51], effects that may exacerbate matrix metalloproteinase (MMP)-9-mediated proteolysis of tight junctional components in the BBB, such as occludin Wachtel et al., [52] and claudin-5 [53]. Serum MMP-8 and MMP-9 are correlated with decreased numbers of T2-weighted lesions. [41]It is unclear what the sources of these MMPs are in this setting. Importantly, MMP-9 is known to proteolyze occludin, a tight junction target of the BBB Wachtel et al., [52]. Interestingly, it has been reported that, compared with laminar shear stress, oscillatory flow increases endothelial MMP-9 expression [54], and might alter the BBB in regions experiencing abnormal flow. In Alzheimer’s disease, β-amyloid appears to help activate MMP-9, and may increase permeability [55]. Other proteases, such as neutrophil elastase, may disturb the BBB Carden et al., [56] and proteolyze VE-cadherin. In this setting, generation of oxidants can inhibit endogenous anti-proteases such as α-1 anti-trypsin [57] and tissue inhibitors of metalloproteinase (TIMPs) [58], which limit junction-degrading proteases, and thus exacerbate BBB failure. The use of broad-spectrum antioxidants and MMP inhibitors (such as doxycycline and minocycline) in clinical trials [43] may preserve BBB integrity of the BBB. Several groups have described elevations of circulating inflammatory cytokines (IL-12p40, IL-17, IL-23) in patients with active MS, which decrease during remission or are reduced by IFN-β1b therapy [41]. Mechanistically, factors in sera from MS patients (in exacerbation) were found to decrease VE-cadherin and occludin expression [7], potentially contributing to the loss of BBB integrity through weaker junction organization, protein expression, and junction degradation.
Activation of p38 MAPK may influence the structural integrity of the blood brain barrier and assembly of components forming the BBB. For example, p38 MAPK activation has been shown to disturb normal assembly of occludin within tight junctions [59]. Furthermore, exposure of endothelial cells to the growth factor vascular endothelial growth factor (VEGF)-A increases permeability through phosphorylation of serine occludin (Ser490), which promotes the ubiquitination and clearance of Ser90. This loss of occludin at junctions would be expected to ‘disintegrate’ the normal junctional barrier. Interestingly, another effect of dexamethasone in ‘arterializing’ venous endothelium appears to be its effect in ‘externalizing’ cytoplasmic occludin [60], leading to a denser junction organization (Table 1). Therefore, laminar shear activation of p38 MAPK (in arterial endothelium) might enhance junction assembly, while conversely, venous shear might disassemble junctions. It is possible that orally available p38 MAPK inhibitors, (for example, SB 239063), might stabilize venous junctions and limit vascular permeability.
MS and venous remodeling
In MS, ‘Dawson’s fingers’ are fine periventricular white matter venous lesions that appear early on in the course of MS, and are often arranged around the longitudinal axis of the central veins [8]. The venous association of this lesion has long been suspected to link venous system disturbances with the etiology of MS [61-65]. This phenomenon may represent inflammation, shear-mediated mechanical trauma, or disturbances in pressure. Anatomic reports by Schelling suggested that these lesions reflect ‘hemodynamic back jetting,’ which is theorized to be an important cause of venous injury [29]. Such lesions may be correlated with restricted outflow, which may caused by structural disturbances present in MS veins Coen et al., [66]. These structural alterations may involve switching from collagen type I to type III, which may provoke other structural abnormalities, including valve disturbances, which might alter venous hemodynamics [49]. This type of matrix remodeling might be adaptive in acute venous congestion to limit hemorrhages and iron deposition; such changes in matrix thickness or composition in ‘mature’ lesions could limit exchange or perfusion. Such non-inflammatory wall thickening is normal during aging. It is unclear whether venous structural or flow disturbances in MS might represent part of a spectrum of venous diseases seen outside of the CNS. The incidence of chronic venous disease outside of the CNS increases with age, although the age of onset for MS is between the ages of 20 and 30 years, with a female preponderance [4,67]. Like chronic venous disease, MS also shows greater prevalence in female and European populations. Interestingly, CVI, which is characterized by weak flow of venous blood, especially in the legs [68], is also characterized by collagen isoform remodeling, but shows elevation of collagen type I expression and diminished type III expression [46], increased fibrillin-1 and laminin, and overproduction of MMP1, MMP2, and MMP3 [69]. Interestingly, transforming growth factor (TGF)β1 induces endothelial apoptosis in a collagen-dependent manner, with matrix collagen type I maintaining endothelial viability despite exposure to TGF-β1 [70]. Conversely, endoglin appears to oppose TGF-β1 induced collagen synthesis by p38 MAPK activation [71], and was found to suppress TGF β1-induced collagen synthesis when ERK1/2 signaling was present. The use of p38 MAPK inhibitors, such as dilmapimod [45], might help to prevent TGF-β1-associated venous remodeling.
Both the elevation and suppression of TGF-β1 in venous structure suggest a role for TGF-β1 in CVI pathogenesis [72-75]. Active TGF-β1 increases inducible nitric oxide synthase, which dysregulates venous tone and blood flow [73]. CVI is associated with suppression of the proliferative responses of fibroblasts and smooth muscle cells to TGF-β1 [76]. TGF-β1 signaling in fibroblasts is mediated by ERK1/2 and SMAD activation [76-78]. It is unclear whether TGF modifiers, such as avotermin, might have clinical benefit in MS, as has been suggested in CVI [47]. Similarly, the angiotensin II receptor antagonist candesartan inhibits TGF-β1-induced MMP9 via Smad7 Yu et al., [79], therefore, angiotensin antagonists may also be able to suppress the vessel remodeling that can contribute to vascular abnormalities in MS.
Bevacizumab has been shown to diminish injury in the experimental autoimmune encephalomyelitis model of MS by suppressing angiogenesis, suggesting that VEGF may play some part in the development of MS [80], Argaw et al. suggested that astrocytes might represent an important source of VEGF-A, which leads to the activation of eNOS and plays a significant role in the loss of BBB that occurs in MS [42]. Although not yet tested, the effects of VEGF-A on venous structure could lead to a similar loss of BBB, leading to the extravasation of lymphocytes and plasma protein, which could provoke injury and vessel remodeling. Therefore, anti-angiogenic drugs such as bevacizumab or cavtratin may find clinical applications in MS treatment (Table 1). Immunochemical and MRI methods have confirmed erythrocyte penetration in a subset of MS lesions, and the accumulation of iron-laden macrophages occurs predominantly around venules, with venous vascular lesions regularly showing iron signatures [81-85]. Iron released by extravasated erythrocytes becomes susceptible to Fenton and Haber-Weiss oxidant-generating reactions in the parenchyma, mediated by reactive oxygen species, which leads to alterations in second messenger signaling and tissue injury (Figure 1). Iron chelators (for example, desferrioxamine) (Table 1) may be effective in lowering the overall iron (and oxidant) burden.
Figure 1.
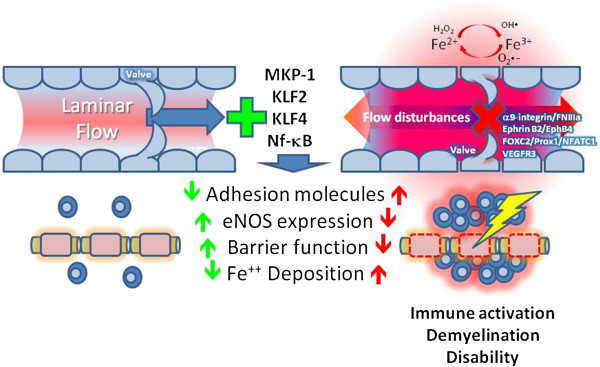
Venous endothelial injury in neuroinflammatory disease. Alterations in normal flow induced by changes in outflow resistance or valve failure lead to endothelial disturbances that provoke localized inflammatory responses, which may intensify immune activation, leading to demyelination and disability in MS. Flow sensors that may be dysregulated include MKP-1, KLF2, and KLF4, which control adhesion molecule, eNOS, and blood-brain barrier function and iron deposition. Venous valve structural and regulatory components that might be dysregulated in this schema include α9-integrin/fibronectin (FNIIIa), Ephrin B2/EphB4, FOXC2/Prox1/NFATC1, and VEGFR-3. Abbreviations: eNOS, endothelial nitric oxide synthase; FOXC2, Forkhead box protein C2; KLF, Krueppel-like factor; MKP, mitogen-activated protein kinase phosphatase; MS, multiple sclerosis; NFATC1, nuclear factor of activated T-cells, cytoplasmic 1; VEGFR, vascular endothelial growth factor receptor.
Vessel structure in CVI is correlated with vessel stiffening; a reversed collagen expression in MS might suggest a hypercompliant venous structure. CVI is also characterized by an higher TIMP-1/MMP-2 ratio, which might favor deposition of non-elastic matrix [86]. We reported previously that MS exhibits increased expression of MMP-8 and MMP-9, which was suppressed by IFNβ1b therapy and was correlated with reduced T2-weighted lesions [41]. We also reported that addition of doxycycline, an MMP inhibitor similar to IFNβ1b, significantly reduced contrast-enhancing lesion numbers and disability scores. Lower serum MMP-9 levels correlated with fewer contrast-enhancing lesions. Furthermore, transendothelial migration of monocytes, stimulated by MS serum, was reduced in patients undergoing combination therapy with doxycycline [43]. Like MS, CVI is also characterized by increased circulating levels of MMP-9, and MMP-1, MMP-2, and TIMP-1 were also reported to be increased in CVI [Saito et al., 2001]. During chronic venous disease, the venous valves and the vessel wall exhibit monocyte and macrophage infiltration [87], which is characterized by increased expression of ICAM-1 [88]. Importantly, Takase et al. found inflammation of the vasa vasorum, which could provoke wall remodeling. Individuals with CVI retain more leukocytes, in which may explain the greater quantities of circulating leukocytes in CVI Bergan et al. [89]. Patients with CVI also have higher leukocyte activation and oxidant production compared with controls [90]. Powell et al. found more platelet-monocyte aggregates in CVI (29% versus 8%; P < 0.0002), while CD11b expression on monocytes in CVI was approximately twice that of controls (7.5 vs. 3.7; P<0.01). The presence of CVI also led to greater generation of platelet leukocyte aggregates [91]. Therefore, low or retrograde flow states, as may exist in CVI, might lead to a perilous imbalance favoring vascular inflammatory programs.
Are there hemodynamic influences in venous vascular disturbances?
Other forms of venous restriction may also contribute to alterations in BBB. Early studies by Putnam using venous obstruction showed development of MS-like lesions [92]. More recently, Mayhan and Heistad [93] found that deliberate occlusion of the superior vena cava produced vascular solute leakage, primarily in venules. We also found recently that experimentally increased intra-abdominal hypertension (IAH) in mice (produced by abdominal volume), also caused a rapid and reversible failure of BBB (as shown by extravasation of Evans blue stain). Such changes are presumably hydrodynamic, because they resolved within 2 hours after relief of IAH [94]. Clinically, IAH above 20 mm Hg diminished venous return, and translated into increased intracranial pressure [95]. Interestingly, with respect to the potential influence of altered hemodynamics and cyclical pressure changes in the venous barrier, Shin et al. [96-99] showed that cyclical pressure modulates venous endothelial proliferative and barrier responses through mechanotransduction-regulated changes in fibroblast growth factor receptor/basic fibroblast growth factor and VEGF-C signaling. Interestingly, cyclical high (but not low) pressure disorganized tight (ZO-1) rather than adherens (VE-cadherin) junctional organization , and this was associated with diminished blood brain barrier.These studies provide mechanistic links between environmental pressure changes and an ‘inflammatory’ venous phenotype. It is not yet clear if such responses are unique to venous (and not arterial) endothelium.
Interestingly, Miyamoto et al. [100] and Yura et al. [101] showed that bilateral occlusion of the external jugular veins, like in mice subjected to middle cerebral artery occlusion, led to an increase in brain ischemia. Therefore, if resistance to venous outflow, either pressure-mediated or structurally mediated, provokes decreased cerebral blood flow as has been suggested [8-10], such disturbances could trigger tissue injury and demyelination (as seen in MS). An important question remains as to how downstream restriction of venous outflow might lead to a dysregulated vascular phenotype upstream of the point of insufficient venous drainage. Restriction to venous outflow would also be expected to impair normal flow-mediated vasodilatation. Impaired production of dilators such as prostacyclin, nitrous oxide, and endothelium-derived hyperpolarizing factor would lead to a retrograde volume/pressure transmission that might present as venous vascular injury. Restriction of venous outflow and congestion has been suggested to lead to distention and remodeling of venous capillaries into veins, which may have very differently structural and functional properties. Venous congestion may also provoke thrombus formation via both reduced flow and altered endothelial surface properties.
Whether intracranial venous pressure (IVP) is increased in MS remains highly controversial. McTaggart et al. described significant internal jugular vein (IJV) ‘flattening’ in MS and a trend toward more non-IJV collaterals [48]. Although increased intra-abdominal pressure may be produced by venous obstruction or jugular valve insufficiency, and might thenbe transmitted to the intracranial venous system, causing intracranial hypertension, [102], the significance of this mechanism in chronic cerebrospinal venous insufficiency (CCSVI) remains very controversial. Meyer-Schwickerath reported that venous pressures are normal in patients with MS [103], as measured by ophthalmodynamometry. Haacke et al. [2] pointed out that angioplasty in patients with MS Zamboni et al., [104] reduced venous pressure, consistent with relative pre-operative venous hypertension. Several recent reports have indicated that altered craniocervical venous outflow may also be detected in individuals diagnosed with chronic migraine [105,106], suggesting that cranial venous outflow disturbances may represent a ‘secondary’ rather than a primary phenomenon. Conversely, Lee et al. considered the ontogeny of several venous malformations, as they may contribute to flow disturbances in patients with MS, supporting the idea of cerebrospinal venous malformations as a primary event, which might lead to venous hypertension Lee et al., [107]. Although abnormal venous flow patterns in MS are being corrected through endovascular approaches, future studies to correlate and validate clinical outcomes and pathological mechanism are clearly needed.
Increased intracranial venous pressure without venous leakage or demyelination: pseudotumor cerebri
Venous vascular leakage attributed to MS might be explained intuitively as the result of increased IVP, although this explanation has not been fully accepted. Of relevance to this issue is the disorder pseudotumor cerebri (PC) (also known as idiopathic intracranial hypertension), in which prolonged and demonstrably high intracranial pressures are not associated with venous leakage or demyelination. PC belongs to a set of disorders that include hydrocephalic states and spontaneous (primary) intracranial hypotension, in which the CSF circulation interfaces with the blood circulatory system. CSF moves by bulk flow and pulsatile forces (transferred from the cerebral arteries) from the ventricles into the spinal and cortical subarachnoid spaces. CSF is then largely absorbed via the arachnoid villi into the superior sagittal sinus (SSS). The pressure of the CSF (intracranial pressure, ICP) must always exceed blood pressure in the SSS for this absorption to take place. With reversal of this gradient, such as in newborns with stenosis of the jugular foramina, hydrocephalus results, as the unfused cranial sutures allow for an expansion of the ventricles, which are accumulating CSF [108]. With sutures closed, a fully myelinated, healthy brain will resist ventricular expansion, although ICP will rise, a condition predisposing to PC.
PC is a disorder mainly of females aged 15 to 45 years, with the greatest incidence in the young adult years [109]. It is characterized by high ICP, papilledema, headache, visual blurring and loss, tinnitus, retrobulbar pain, and neck stiffness [110]. Ventricular size is normal or slightly reduced. In most cases, dural venous sinus outflow obstructions or increased right atrial pressures raise IVP to the point where it challenges the ICP [111].
The MRI diagnosis of PC is partly one of exclusion of other causes of increased ICP, such as choroid plexus papilloma, cerebral edema, tumor, and obstructive hydrocephalus. Positive signs of intracranial hypertension include empty sella, bilateral increased fluid in the optic sheath, mild flattening of the posterior sclera, enhancement of the prelaminar optic nerve, distension of the periotic subarachnoid space, vertical tortuosity of the optic nerve, and gadolinium enhancement of the prelaminar optic nerve [112].
Absent from these patients are the MRI hyperintensity signals indicating demyelination. In the study of Wall et al., microscopic examination of brain tissue from patients with PC at autopsy showed no neuronal necrosis, gliosis, or inflammation, and no prominence of perivascular spaces or pallor of myelin in neuropil or white matter [113]. Although these findings do not exclude endothelial injury, the absence of inflammation and demyelination under conditions of prolonged venous hypertension points to the existence of factors that may protect these patients from demyelinating disease. Experimental studies focusing on high ICP states, as found in PC, would help identify these factors.
Genes regulating venous valves
At the molecular level, if congenital or pathologic alterations in venous valve structure contribute to the etiology of CVI and other venous disturbances [114], identifying genes that control venous valve structure might provide important clues to the basis of venous pathology Bazigou et al. [115,116] described that venous valves are organized by interactions of several genes at different developmental stages and post-natally. The development of venous valves requires signaling from Prospero-related homeobox 1 (Prox1), vascular endothelial growth factor receptor (VEGFR)-3, and integrin α9. The binding of integrin α9 to fibronectin-IIIa is also an important structural motif necessary for venous/lymphatic valve assembly [115]. Lymphatic valve formation also involves Cnb1/NFATc1, connexin 37 and 43, and laminin-a5. Nuclear factor of activated T-cells, cytoplasmic 1 (NFATc1) also modulates cardiac valve formation [117]. The later maturation of these valves also requires Sema3A/Neuropilin-1/PlexinA1 signaling Bouvrée et al. [3]. Interestingly, Ephrin-B2 and integrin α9 were both shown to be necessary for maintenance of venous valves, as post-natal editing of these genes induced valve atresia [116]. Further, integrin α9, VEGFR3, and Prox1 were also found to be expressed in lymphatic vessel valves. Additional regulators of venous and lymphatic valve structure may include the Tie2 receptor tyrosine kinase and multiple angiopoietin ligands [116]. Tie2 signaling also appears to be responsive to altered patterns of fluid shear and is dysregulated by abnormal flow. Flow may also influence valve structure/function, as eNOS levels within valve endothelium are increased [118], and eNOS expression appears to modulate valve development, at least in aortic valves [119].
Forkhead box protein C2 (FOXC2) is another transcription factor expressed in venous and lymphatic valves, which controls their development Mellor et al., [120]. FOXC2 is dysregulated in lymphedema distichiasis [121]. Mellor et al. showed that individuals exhibiting mutations in FOXC2 uniformly exhibited valve disturbances in the saphenous and deep veins, contributing to venous reflux and lymphedema Mellor et al., [120]. Interestingly, although FOXC2 gene mutations were closely associated with venous valve failure and were seen in carriers, these individuals did not have lymphedema. Currently, it is still unclear whether ‘silent’ alterations in venous/lymphatic programming genes such as FOXC2 might contribute to venous valve failure in CNS pathology.
Among these regulators, Ephrin-B2 is usually considered to be an arterial and lymphatic specific family transmembrane ligand that binds the receptor tyrosine kinase EphB4, and participates in venous specification [122,123]. EphA2 and ephrinA1 are both persistently expressed by cultured brain endothelial cells, and treatment of brain endothelial cells with inflammatory cytokines caused the shedding of these markers into brain endothelial derived microparticles, which are small (<0.1 μm) plasma membrane vesicles [124]. Similarly, samples of control and MS serum showed increased levels of ephrin A1 and EphA2 expression in vessel structures in MS brain tissue [125]. Several vascular ephrins and Eph receptors may therefore be dysregulated in CNS inflammation, although not all of them have an influence on vascular remodeling. In the setting of CVI, IJV incompetence has been correlated with transient global amnesia [107,126-129], which may contribute to cognitive disturbances in several neurodegenerative conditions. Ephrin-B2 is strongly expressed in venous endothelium, and suppresses endothelial proliferative responses towards VEGF and Ang-2 Kim et al., [130]. By comparison, the receptors EphB2 and EphB3 are strongly expressed by arterial endothelium, and EphB/ephrin-B interactions have been suggested to modulate arteriovenous specification and separation. It is interesting to note that during inflammation, endothelial expression of EphA2 receptor and ephrin-B2 is increased [131].
Other genes modulating venous remodeling
We have previously examined genes that were modified in cerebrovascular endothelial cells in response to serum from patients with RRMS, and found several markers that were modulated by soluble factors present in MS serum and by IFN-β1b therapy, including 14-3-3, metavinculin, myosin-3, plasminogen, reticulocalbin-2 and eticulocalbin--3, ribonuclease/angiogenin inhibitor, annexin A1, tropomyosin, and Rap1A [5]. Ferlini et al. (performed a gene array on chromosome 6p21.32 (human leukocyte antigen (HLA) locus) in patients exhibiting venous malformations associated with MS, and found several candidate genes that were altered including heat shock protein (HSP)A1L, HSPA1A, metabotropic glutamate receptor (GRM)4, and growth factor receptor-bound protein 2, an adaptor involved in MAPK signaling Ferlini et al., [132]. Pirmohamed et al, showed that HSPA1L might be linked with HLA-associated drug hypersensitivity [133], and increased GRM4 has also been reported in MS lesions [134]. Several genes that are associated with MS and inflammatory disease progression (VEGF, endothelin-1, IL-6, VCAM-1, ICAM-1, MMP-2, MMP-9 and PAI-1) are also modulated by alterations in mechanical stretch on the vessel wall [135]. Therefore, genes that drive venous disturbances might reflect the coincident presence of both heritable and environmental (shear/stretch) risk factors.
Optic neuritis and vascular endothelial injury
Optic neuritis (ON) an inflammatory demyelinating disease of the optic nerve, is a common early feature of MS, and often leads to some degree of visual loss in patients. Inflammatory demyelination of the optic nerve in ON can histopathologically resemble acute MS plaques in the brain. For example, ON shows nerve sheath edema, perivenous ‘cuffing’, destruction of myelin, and vascular fluorescein leakage. Retinal VE inflammation usually precedes demyelination, and is often detected as retinal vein ‘sheathing’ [136]. Papillitis (inflammation of the optic nerve head), with increased blood flow and retinal edema, blurring of disk margins, and swollen veins can be seen in up to 30% of patients presenting with ON. A significant number of patients with ON have retrobulbar neuritis, and present with abnormal funduscopic findings.
An interesting finding in patients with MS is focal sheathing of the retinal veins (periphlebitis retinae), which includes local perivenous infiltration of lymphocytes and plasma cells [137,138], post-inflammatory peri-venular gliosis [137], and evidence of focal extravasation of plasma proteins [137]. Although the human retina has limited myelin and myelin basic proteins (limited by the lamina cribrosa [139]), or myelinating oligodendrocytes, it is still unclear as to why some patients with MS patients periphlebitis retinae. One theory to explain such findings is that other myelin-associated antigens, such as, the human natural killer-1 carbohydrate epitope and myelin-associated glycoprotein, can be expressed by retinal Müller glial cells [140,141]. However, this hypothesis cannot sufficiently explain the retinal findings in MS. To consider this issue, Engell et al. [142] investigated retinal venous alterations in patients with acute ON. MS was found in 41 of 76 patients examined for ophthalmologic issues; 1 patient had periphlebitis retinae and two had venous ‘sheathing’. It was concluded that altered venous structure in the retina indicated an ultimate diagnosis of MS. Therefore, because retinal venous abnormalities in patients with MS occur outside the key areas of demyelination, perivenular inflammation may represent the early event contributing to new lesions. Perivenous sheathing (periphlebitis retinae) indicates some loss of normal blood-retinal barrier. Therefore, sheathing may most be often perivenular because the venous endothelial junctions are inherently less restrictive than those of the corresponding arterial endothelium. The increased venous tendency to express adhesive inflammation-associated ECAMs in response to inflammatory or hypoxic stimuli, along with immune cell retention, may initiate or sustain exaggerated responses. In retinal endothelial monolayers (which exhibit BBB properties,) we found that the junctional solute barrier required actin microfilament assembly, was positively regulated by β-adrenoreceptor signaling [143], and was dysregulated by increased glucose levels [144]. Therefore, the BBB may be dysregulated by changes in circulating autacoids or metabolic disturbances.
Developmental venous anomalies
Haacke et al. [2] suggested that that venous hypertension caused by congenital or pathologic changes could provoke the development of dural arteriovenous structural abnormalities in MS. It has even been suggested [145]that the presence of congenital venous anomalies may occur in some isolated populations (such as in Sardinia) that could contribute to more frequent or earlier-onset venous disturbances. It has been proposed that more profound vascular flow disturbances in these populations might provoke neurovascular forms of injury, which could include CCSVI or MS [145]. It is unclear whether additional risk factors are necessary to increase the penetrance of this phenotype and appearance of this condition.
Pathophysiology of ADEM, with emphasis on venous dysfunction
ADEM is a relatively rare CNS inflammatory demyelinating disease, which affects both adults and children. ADEM typically occurs as a single-stage syndrome. It is often seen after immunization (also described as ‘post-vaccination encephalomyelitis’), and may also occur after some systemic viral infections (for example, measles). Clinically, ADEM produces a variety of symptoms, including fever, headache, meningismus, seizures, loss of sensation/tingling, visual loss, weakness or paralysis, loss of coordination, involuntary spasms, and loss of sphincter control. Neuropathologically, ADEM exhibits scattered focal demyelination, which is usually limited to the perivenous areas. The underlying neuropathological defects in ADEM can affect both the brain and spinal cord, with MRI often revealing large and diffuse or multifocal lesions. This appearance differs from that of MS in that MS lesions are focal, smaller, and confluent [146]. The MRI lesions of ADEM involve both gray and white matter [147].
Neuropathological studies in ADEM have shown merged regions of perivenular demyelination throughout the cerebral hemispheres, brainstem, cerebellum, and spinal cord. Although these lesions are usually most numerous in the white matter, they can affect deeper layers of the cerebral cortex, thalamus, hypothalamus, and other gray-matter areas within the brain. Microscopically, ADEM affects small distended veins enclosed within parenchymal infiltrates of reactive microglia, lymphocytes, macrophages, and occasionally neutrophils, associated with demyelination [39].
Although the details of ADEM pathogenesis remain only partially understood, interactions between inflamed and activated underlying cerebral venous endothelium and activated leukocytes play major roles in its development. Following activation of the immune system, either because of molecular mimicry or sensitization against the self-antigens following a viral infection, myelin basic protein-reactive lymphocytes can interact with the venous endothelium [148]. Such interactions between the inflamed venous endothelium and the activated leukocytes can disrupt the normal functional and anatomical integrity of the cerebral venous endothelium, and eventually promote the transendothelial migration of leukocytes and release of neuroinflammatory mediators such as cytokines and chemokines. Further research into the immunopathogenesis of ADEM versus MS reveals that T helper (Th)1-related and Th2-related chemokines are generated during both ADEM and MS. ADEM shows upregulation of chemokines for neutrophils (CXCL1, CXCL7), monocytes/T cells (CCL3, CCL5), Th1 cells (CXCL10), and Th2 cells (CCL1, CCL22, and CCL17) [39]. Further, the involvement of MMP-9 [149] and increased serum levels of soluble ICAM-1 in the pathogenesis of ADEM has been shown [150], which places more emphasis on endothelial disturbances underlying ADEM pathology. Interestingly, the inflammatory demyelinating lesions of ADEM do not form near arterial vessels. This finding itself lends support to the concept that inherent venous (rather than arterial) endothelial anatomic or functional abnormalities drives ADEM.
Conclusions
The roles of anatomical and functional abnormalities of the cerebral venous endothelium in the pathogenesis of human CNS inflammatory diseases such as MS and ADEM often remain unrecognized, underinvestigated, and untreated. Rather than these diseases simply being the result of structural disturbances of veins, together with the combined hemodynamic (low/abnormal flow, pressure/congestion), programmatic (arterial, venous, valvular) and environmental (metabolic, hypoxic) stresses to which venous endothelial cells are exposed may render them particularly susceptible to inflammatory activation, contributing to several neurovascular pathologies. Presently, markers of arterial and venous endothelial specification and the role of each cell type in inflammation are now receiving more attention. A more thorough understanding of such mechanisms based on the developmental, cellular, and molecular mechanisms underlying the hemodynamic disturbances of these conditions will open many new therapeutic targets for debilitating diseases such as Alzheimer’s disease and MS.
Abbreviations
ADEM: Acute disseminated encephalomyelitis; BBB: Blood-brain barrier; CCSVI: Chronic cerebrospinal venous insufficiency; CNS: Central nervous system; CSF: Cerebrospinal fluid; CVI: Chronic venous insufficiency; ECAM: Endothelial cell adhesion molecule; eNOS: Endothelial nitric oxide synthase; FOXC2: Forkhead box protein C2; GRM: Metabotropic glutamate receptor; HDAC: Histone deacetylase; HLA: Human leukocyte antigen; HSP: Heat shock protein; IAH: Intra-abdominal hypertension; ICAM: Intercellular adhesion molecule; ICP: Intracranial pressure; IFN: Interferon; IJV: nternal jugular vein; IL: Interleukin; IVP: ntracranial venous pressure; KLF: Krueppel-like factor; LPS: Lipopolysaccharide; MAPK: Mitogen-activated protein kinase; MKP: mitogen-activated protein kinase phosphatase; MMP: Matrix metalloproteinase; MRI: Magnetic resonance imaging; MS: Multiple sclerosis; NAWM: Normal-appearing white matter; NMDA: N-methyl-D-aspartate; ON: Optic neuritis; PAI: Plasminogen activator inhibitor; PC: Pseudotumor cerebri; PPMS: Primary progressive multiple sclerosis; Prox1: Prospero-related homeobox 1; RRMS: Relapsing-remitting multiple sclerosis; SSS: Superior sagittal sinus; Th: T helper; TIMP: Tissue inhibitor of metalloproteinase; TM: Thrombomodulin; TNF: Tumor necrosis factor; VCAM: Vascular cell adhesion molecule; VE: Vascular endothelial; VEGF: Vascular endothelial growth factor.
Competing interests
The authors declare no competing interests.
Authors’ information
JJSA and CVG are members of the Molecular and Cellular Physiology Department, LSUHSC-Shreveport; LP is a member of the Pathology Department, LSUHSC-Shreveport, IT is a member of the Department of Microbiology and Immunology, LSUHSC-Shreveport, and AM is a member of Department of Neurology, LSUHSC-Shreveport.
Contributor Information
Jonathan S Alexander, Email: jalexa@lsuhsc.edu.
Leonard Prouty, Email: lprout@lsuhsc.edu.
Ikuo Tsunoda, Email: itsuno@lsuhsc.edu.
Chaitanya Vijay Ganta, Email: vganta@lsuhsc.edu.
Alireza Minagar, Email: aminag@lsuhsc.edu.
Acknowledgements
We thank acknowledge Courtney Parker and Christopher Monceaux for editing and bibliographic assistance with this review.
References
- Schaller B. Physiology of cerebral venous blood flow: from experimental data in animals to normal function in humans. Brain Res Brain Res Rev. 2004;46:243–260. doi: 10.1016/j.brainresrev.2004.04.005. [DOI] [PubMed] [Google Scholar]
- Haacke EM, Beggs CB, Habib C. The role of venous abnormalities in neurological disease. Rev Recent Clin Trials. 2012;7:100–116. doi: 10.2174/157488712800100305. [DOI] [PubMed] [Google Scholar]
- Bouvrée K, Brunet I, Del Toro R, Gordon E, Prahst C, Cristofaro B, Mathivet T, Xu Y, Soueid J, Fortuna V, Miura N, Aigrot MS, Maden CH, Ruhrberg C, Thomas JL, Eichmann A. Semaphorin3A, Neuropilin-1, and PlexinA1 are required for lymphatic valve formation. Circ Res. 2012;111:437–445. doi: 10.1161/CIRCRESAHA.112.269316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372:1502–1517. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- Alexander JS, Minagar A, Harper M, Robinson-Jackson S, Jennings M, Smith SJ. Proteomic analysis of human cerebral endothelial cells activated by multiple sclerosis serum and IFNbeta-1b. J Mol Neurosci. 2007;32:169–178. doi: 10.1007/s12031-007-0018-3. [DOI] [PubMed] [Google Scholar]
- Haghjooy Javanmard S, Saadatnia M, Homayouni V, Nikoogoftar M, Maghzi AH, Etemadifar M, Chaitanya VG, McGee JC, Minagar A, Alexander JS. Interferon-beta-1b protects against multiple sclerosis-induced endothelial cells apoptosis. Front Biosci (Elite Ed) 2012;4:1368–1374. doi: 10.2741/e466. [DOI] [PubMed] [Google Scholar]
- Minagar A, Ostanin D, Long AC, Jennings M, Kelley RE, Sasaki M, Alexander JS. Serum from patients with multiple sclerosis downregulates occludin and VE-cadherin expression in cultured endothelial cells. Mult Scler. 2003;9:235–238. doi: 10.1191/1352458503ms916oa. [DOI] [PubMed] [Google Scholar]
- Ge Y, Law M, Herbert J, Grossman RI. Prominent perivenular spaces in multiple sclerosis as a sign of perivascular inflammation in primary demyelination. AJNR Am J Neuroradiol. 2005;26:2316–2319. [PMC free article] [PubMed] [Google Scholar]
- Law M, Saindane AM, Ge Y, Babb JS, Johnson G, Mannon LJ, Herbert J, Grossman RI. Microvascular abnormality in relapsing-remitting multiple sclerosis: perfusion MR imaging findings in normal-appearing white matter. Radiology. 2004;231:645–652. doi: 10.1148/radiol.2313030996. [DOI] [PubMed] [Google Scholar]
- Saindane AM, Law M, Ge Y, Johnson G, Babb JS, Grossman RI. Correlation of diffusion tensor and dynamic perfusion MR imaging metrics in normal-appearing corpus callosum: support for primary hypoperfusion in multiple sclerosis. AJNR Am J Neuroradiol. 2007;28:767–772. [PMC free article] [PubMed] [Google Scholar]
- Swank RL, Roth JG, Woody DC Jr. Cerebral blood flow and red cell delivery in normal subjects and in multiple sclerosis. Neurol Res. 1983;5:37–59. doi: 10.1080/01616412.1983.11739631. [DOI] [PubMed] [Google Scholar]
- Simka M, Zaniewski M. Reinterpreting the magnetic resonance signs of hemodynamic impairment in the brains of multiple sclerosis patients from the perspective of a recent discovery of outflow block in the extracranial veins. J Neurosci Res. 2010;88:1841–1845. doi: 10.1002/jnr.22350. [DOI] [PubMed] [Google Scholar]
- Kalogeris TJ, Kevil CG, Laroux FS, Coe LL, Phifer TJ, Alexander JS. Differential monocyte adhesion and adhesion molecule expression in venous and arterial endothelial cells. Am J Physiol. 1999 ;276:L9–L19. doi: 10.1152/ajplung.1999.276.1.L9. [DOI] [PubMed] [Google Scholar]
- Amberger A, Maczek C, Jürgens G, Michaelis D, Schett G, Trieb K, Eberl T, Jindal S, Xu Q, Wick G. Co-expression of ICAM-1, VCAM-1, ELAM-1 and Hsp60 in human arterial and venous endothelial cells in response to cytokines and oxidized low-density lipoproteins. Cell Stress Chaperones. 1997;2:94–103. doi: 10.1379/1466-1268(1997)002<0094:ceoive>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Feuerstein GZ. Induced expression of adhesion molecules following focal brain ischemia. J Neurotrauma. 1995;12:825–832. doi: 10.1089/neu.1995.12.825. [DOI] [PubMed] [Google Scholar]
- Kevil CG, Payne DK, Mire E, Alexander JS. Vascular permeability factor/vascular endothelial cell growth factor-mediated permeability occurs through disorganization of endothelial junctional proteins. J Biol Chem. 1998;273:15099–15103. doi: 10.1074/jbc.273.24.15099. [DOI] [PubMed] [Google Scholar]
- Morita K, Sasaki H, Furuse M, Tsukita S. Endothelial claudin: claudin-5/TMVCF constitutes tight junction strands in endothelial cells. J Cell Biol. 1999;147:185–194. doi: 10.1083/jcb.147.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burek M, Arias-Loza PA, Roewer N, Förster CY. Claudin-5 as a novel estrogen target in vascular endothelium. Arterioscler Thromb Vasc Biol. 2010;30:298–304. doi: 10.1161/ATVBAHA.109.197582. [DOI] [PubMed] [Google Scholar]
- Volgina NE, Gurina OI, Grinenko NF, Baklaushev VP, Ivanova NV, Chekhonin VP. Expression of tight junction proteins by umbilical vein epithelial cells co-cultured with allogenic astrocytes. Bull Exp Biol Med. 2012;154:124–129. doi: 10.1007/s10517-012-1891-5. [DOI] [PubMed] [Google Scholar]
- Chaitanya GV, Cromer WE, Wells SR, Jennings MH, Couraud PO, Romero IA, Weksler B, Erdreich-Epstein A, Mathis JM, Minagar A, Alexander JS. Gliovascular and cytokine interactions modulate brain endothelial barrier in vitro. J Neuroinflammation. 2011;162:1–16. doi: 10.1186/1742-2094-8-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamson RH, Sarai RK, Altangerel A, Clark JF, Weinbaum S, Curry FE. Microvascular permeability to water is independent of shear stress, but dependent on flow direction. Am J Physiol Heart Circ Physiol. 2013;304:H1077–H1084. doi: 10.1152/ajpheart.00956.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakkar M, le Luong A, Chaudhury H, Ruud O, Punjabi PP, Anderson JR, Mullholand JW, Clements AT, Krams R, Foin N, Athanasiou T, Leen EL, Mason JC, Haskard DO, Evans PC. Dexamethasone arterializes venous endothelial cells by inducing mitogen-activated protein kinase phosphatase-1: a novel antiinflammatory treatment for vein grafts? Circulation. 2011;123:524–532. doi: 10.1161/CIRCULATIONAHA.110.979542. [DOI] [PubMed] [Google Scholar]
- Liu M, Kluger MS, D'Alessio A, García-Cardeña G, Pober JS. Regulation of arterial-venous differences in tumor necrosis factor responsiveness of endothelial cells by anatomic context. Am J Pathol. 2008;172:1088–1099. doi: 10.2353/ajpath.2008.070603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Xiao H, Laguna-Fernandez A, Villarreal G Jr, Wang KC, Geary GG, Zhang Y, Wang WC, Huang HD, Zhou J, Li YS, Chien S, Garcia-Cardena G, Shyy JY. Flow-Dependent Regulation of Kruppel-Like Factor 2 Is Mediated by MicroRNA-92a. Circulation. 2011;124:633–641. doi: 10.1161/CIRCULATIONAHA.110.005108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y, Davies PF. Site-specific microRNA-92a regulation of Kruppel-like factors 4 and 2 in atherosusceptible endothelium. Arterioscler Thromb Vasc Bio. 2012;32:979–987. doi: 10.1161/ATVBAHA.111.244053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee HJ, Kwon JS, Shin S, Ahn Y, Jeong MH, Trichostatin KH. A prevents neointimal hyperplasia via activation of Krüppel like factor 4. Vascul Pharmacol. 2011;55:127–134. doi: 10.1016/j.vph.2011.07.001. [DOI] [PubMed] [Google Scholar]
- SenBanerjee S. et al. KLF2 Is a novel transcriptional regulator of endothelial proinflammatory activation. J Exp Med. 2004;199:1305–1315. doi: 10.1084/jem.20031132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamik A, Lin Z, Kumar A, Balcells M, Sinha S, Katz J, Feinberg MW, Gerzsten RE, Edelman ER, Jain MK. Kruppel-like factor 4 regulates endothelial inflammation. J Biol Chem. 2007;282:13769–13779. doi: 10.1074/jbc.M700078200. [DOI] [PubMed] [Google Scholar]
- Schelling F. Damaging venous reflux into the skull or spine: relevance to multiple sclerosis. Med Hypotheses. 1986;21:141–148. doi: 10.1016/0306-9877(86)90003-4. [DOI] [PubMed] [Google Scholar]
- Lin Z. et al. Kruppel-like factor 2 (KLF2) regulates endothelial thrombotic function. Circ Res. 2005;96:48–57. doi: 10.1161/01.RES.0000159707.05637.a1. [DOI] [PubMed] [Google Scholar]
- Parmar KM, Larman HB, Dai G, Zhang Y, Wang ET, Moorthy SN, Kratz JR, Lin Z, Jain MK, Gimbrone MA Jr, García-Cardeña G. Integration of flow-dependent endothelial phenotypes by Kruppel-like factor 2. J Clin Invest. 2006;116:49–58. doi: 10.1172/JCI24787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das H, Kumar A, Lin Z, Patino WD, Hwang PM, Feinberg MW, Majumder PK, Jain MK. Kruppel-like factor 2 (KLF2) regulates proinflammatory activation of monocytes. Proc Natl Acad Sci U S A. 2006;103:6653–6658. doi: 10.1073/pnas.0508235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fledderus JO, van Thienen JV, Boon RA, Dekker RJ, Rohlena J, Volger OL, Bijnens AP, Daemen MJ, Kuiper J, van Berkel TJ, Pannekoek H, Horrevoets AJ. Prolonged shear stress and KLF2 suppress constitutive proinflammatory transcription through inhibition of ATF2. Blood. 2007;109:4249–4257. doi: 10.1182/blood-2006-07-036020. [DOI] [PubMed] [Google Scholar]
- van Agtmaal EL, Bierings R, Dragt BS, Leyen TA, Fernandez-Borja M, Horrevoets AJ, Voorberg J. The shear stress-induced transcription factor KLF2 affects dynamics and angiopoietin-2 content of Weibel-Palade bodies. PLoS One. 2012;7:e38399. doi: 10.1371/journal.pone.0038399. 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Methe H, Balcells M, Alegret Mdel C, Santacana M, Molins B, Hamik A, Jain MK, Edelman ER. Vascular bed origin dictates flow pattern regulation of endothelial adhesion molecule expression. Am J Physiol Heart Circ Physiol. 2007;292:H2167–H2175. doi: 10.1152/ajpheart.00403.2006. [DOI] [PubMed] [Google Scholar]
- Sen-Banerjee S, Mir S, Lin Z, Hamik A, Atkins GB, Das H, Banerjee P, Kumar A, Jain MK. Kruppel-like factor 2 as a novel mediator of statin effects in endothelial cells. Circulation. 2005;112:720–726. doi: 10.1161/CIRCULATIONAHA.104.525774. [DOI] [PubMed] [Google Scholar]
- Ohnesorge N, Viemann D, Schmidt N, Czymai T, Spiering D, Schmolke M, Ludwig S, Roth J, Goebeler M, Schmidt M. Erk5 activation elicits a vasoprotective endothelial phenotype via induction of Kruppel-like factor 4 (KLF4) J Biol Chem. 2010;285:26199–26210. doi: 10.1074/jbc.M110.103127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali F, Hamdulay SS, Kinderlerer AR, Boyle JJ, Lidington EA, Yamaguchi T, Soares MP, Haskard DO, Randi AM, Mason JC. Statin-mediated cytoprotection of human vascular endothelial cells: a role for Kruppel-like factor 2-dependent induction of heme oxygenase-1. J Thromb Haemost. 2007;5:2537–2546. doi: 10.1111/j.1538-7836.2007.02787.x. [DOI] [PubMed] [Google Scholar]
- Franciotta D, Zardini E, Ravaglia S, Piccolo G, Andreoni L, Bergamaschi R, Romani A, Tavazzi E, Naldi P, Ceroni M, Marchioni E. Cytokines and chemokines in cerebrospinal fluid and serum of adult patients with acute disseminated encephalomyelitis. J Neurol Sci. 2006;247:202–207. doi: 10.1016/j.jns.2006.05.049. [DOI] [PubMed] [Google Scholar]
- Huddleson JP, Ahmad N, Srinivasan S, Lingrel JB. Induction of KLF2 by fluid shear stress requires a novel promoter element activated by a phosphatidylinositol 3-kinase-dependent chromatin-remodeling pathway. J Biol Chem. 2005 Jun 17;280:23371–23379. doi: 10.1074/jbc.M413839200. [DOI] [PubMed] [Google Scholar]
- Alexander JS, Harris MK, Wells SR, Mills G, Chalamidas K, Ganta VC, McGee J, Jennings MH, Gonzalez-Toledo E, Minagar A. Alterations in serum MMP-8, MMP-9, IL-12p40 and IL-23 in multiple sclerosis patients treated with interferon-beta1b. Mult Scler. 2010;16:801–809. doi: 10.1177/1352458510370791. [DOI] [PubMed] [Google Scholar]
- Argaw AT, Asp L, Zhang J, Navrazhina K, Pham T, Mariani JN, Mahase S, Dutta DJ, Seto J, Kramer EG, Ferrara N, Sofroniew MV, John GR. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J Clin Invest. 2012;122:2454–2468. doi: 10.1172/JCI60842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minagar A, Alexander JS, Schwendimann RN, Kelley RE, Gonzalez-Toledo E, Jimenez JJ, Mauro L, Jy W, Smith SJ. Combination therapy with interferon beta-1a and doxycycline in multiple sclerosis: an open-label trial. Arch Neurol. 2008;65:199–204. doi: 10.1001/archneurol.2007.41. [DOI] [PubMed] [Google Scholar]
- Sharp CD, Houghton J, Elrod JW, Warren A, Jackson TH 4th, Jawahar A, Nanda A, Minagar A, Alexander JS. N-methyl-D-aspartate receptor activation in human cerebral endothelium promotes intracellular oxidant stress. Am J Physiol Heart Circ Physiol. 2005;288:H1893–H1899. doi: 10.1152/ajpheart.01110.2003. [DOI] [PubMed] [Google Scholar]
- Anand P, Shenoy R, Palmer JE, Baines AJ, Lai RY, Robertson J, Bird N, Ostenfeld T, Chizh BA. Clinical trial of the p38 MAP kinase inhibitor dilmapimod in neuropathic pain following nerve injury. Eur J Pain. 2011;15:1040–1048. doi: 10.1016/j.ejpain.2011.04.005. [DOI] [PubMed] [Google Scholar]
- Sansilvestri-Morel P, Rupin A, Badier-Commander C, Fabiani JN, Verbeuren TJ. Chronic venous insufficiency: dysregulation of collagen synthesis. Angiology. 2003;54:S13–S18. doi: 10.1177/0003319703054001S03. [DOI] [PubMed] [Google Scholar]
- McCollum PT, Bush JA, James G. et al. Randomized phase II clinical trial of Avotermin versus placebo for scar improvement. Br J Surg. 2011;98:925–934. doi: 10.1002/bjs.7438. [DOI] [PubMed] [Google Scholar]
- McTaggart RA, Fischbein NJ, Elkins CJ, Hsiao A, Cutalo MJ, Rosenberg J, Dake MD, Zaharchuk G. Extracranial venous drainage patterns in patients with multiple sclerosis and healthy controls. AJNR Am J Neuroradiol. 2012;33:1615–1620. doi: 10.3174/ajnr.A3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radak D, Kolar J, Tanaskovic S, Sagic D, Antonic Z, Mitrasinovic A, Babic S, Nenezic D, Ilijevski N. Morphological and haemodynamic abnormalities in the jugular veins of patients with multiple sclerosis. Phlebology. 2012;27:168–172. doi: 10.1258/phleb.2011.011004. [DOI] [PubMed] [Google Scholar]
- Sarchielli P, Greco L, Floridi A, Floridi A, Gallai V. "Excitatory amino acids and multiple sclerosis: evidence from cerebrospinal fluid. Arch Immunol. 2003;60:1082–1088. doi: 10.1001/archneur.60.8.1082. [DOI] [PubMed] [Google Scholar]
- Sharp CD, Hines I, Houghton J, Warren A, Jackson TH 4th, Jawahar A, Nanda A, Elrod JW, Long A, Chi A, Minagar A, Alexander JS. Glutamate causes a loss in human cerebral endothelial barrier integrity through activation of NMDA receptor. Am J Physiol Heart Circ Physiol. 2003;285:H2592–H2598. doi: 10.1152/ajpheart.00520.2003. [DOI] [PubMed] [Google Scholar]
- Wachtel M, Frei K, Ehler E, Fontana A, Winterhalter K, Gloor SM. Occludin proteolysis and increased permeability in endothelial cells through tyrosine phosphatase inhibition. J Cell Sci. 1999;112:4347–4356. doi: 10.1242/jcs.112.23.4347. [DOI] [PubMed] [Google Scholar]
- Yang Y, Rosenberg GA. MMP-mediated disruption of claudin-5 in the blood-brain barrier of rat brain after cerebral ischemia. Methods Mol Biol. 2011;762:333–345. doi: 10.1007/978-1-61779-185-7_24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magid R, Murphy TJ, Galis ZS. Expression of matrix metalloproteinase-9 in endothelial cells is differentially regulated by shear stress. Role of c-Myc. J Biol Chem. 2003;278:32994–32999. doi: 10.1074/jbc.M304799200. [DOI] [PubMed] [Google Scholar]
- Cao L, Wang H, Wang F. Amyloid-β-induced matrix metalloproteinase-9 secretion is associated with retinal pigment epithelial barrier disruption. Int J Mol Med. 2013;31:1105–1112. doi: 10.3892/ijmm.2013.1310. [DOI] [PubMed] [Google Scholar]
- Carden D, Xiao F, Moak C, Willis BH, Robinson-Jackson S, Alexander S. Neutrophil elastase promotes lung microvascular injury and proteolysis of endothelial cadherins. Am J Physiol. 1998;275:H385–H392. doi: 10.1152/ajpheart.1998.275.2.H385. [DOI] [PubMed] [Google Scholar]
- Hubbard RC, Ogushi F, Fells GA, Cantin AM, Jallat S, Courtney M, Crystal RG. Oxidants spontaneously released by alveolar macrophages of cigarette smokers can inactivate the active site of alpha 1-antitrypsin, rendering it ineffective as an inhibitor of neutrophil elastase. J Clin Invest. 1987;80:1289–1295. doi: 10.1172/JCI113204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Radisky ES, Das P, Batra J, Hata T, Hori T, Baine AM, Gardner L, Yue MY, Bu G, Del Zoppo G, Patel TC, Nguyen JH. TIMP-1 attenuates blood-brain barrier permeability in mice with acute liver failure. J Cereb Blood Flow Metab. 2013;33:1041–1049. doi: 10.1038/jcbfm.2013.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kevil CG, Oshima T, Alexander JS. The role of p38 MAP kinase in hydrogen peroxide mediated endothelial solute permeability. Endothelium. 2001;8:107–116. doi: 10.3109/10623320109165320. [DOI] [PubMed] [Google Scholar]
- Wong V, Ching D, McCrea PD, Firestone GL. Glucocorticoid down-regulation of fascin protein expression is required for the steroid-induced formation of tight junctions and cell-cell interactions in rat mammary epithelial tumor cells. J Biol Chem. 1999;274:5443–5453. doi: 10.1074/jbc.274.9.5443. [DOI] [PubMed] [Google Scholar]
- Adams CW. Perivascular iron deposition and other vascular damage in multiple sclerosis. J Neurol Neurosurg Psychiatry. 1988;51:260–265. doi: 10.1136/jnnp.51.2.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams CW, Abdulla YH, Torres EM, Poston RN. Periventricular lesions in multiple sclerosis: their perivenous origin and relationship to granular ependymitis. Neuropathol Appl Neurobiol. 1987;13:141–152. doi: 10.1111/j.1365-2990.1987.tb00177.x. [DOI] [PubMed] [Google Scholar]
- Fog T. On the vessel–plaque relations in the brain in multiple sclerosis. Acta Neurol Scand Suppl. 1963;39:258–262. doi: 10.1111/j.1600-0404.1963.tb01841.x. [DOI] [PubMed] [Google Scholar]
- Putnam TJ. Evidences of vascular occlusion in multiple sclerosis and encephalomyelitis. Arch Neurol Psychiatry. 1937;6:1298–1321. [Google Scholar]
- Putnam TJ, Adler A. Vascular architecture of the lesions of multiple sclerosis. Arch Neurol Psychiatry. 1937;38:1–15. [Google Scholar]
- Coen M, Menegatti E, Salvi F, Mascoli F, Zamboni P, Gabbiani G, Bochaton-Piallat ML. Altered collagen expression in jugular veins in multiple sclerosis. Cardiovasc Pathol. 2013;22:33–38. doi: 10.1016/j.carpath.2012.05.005. [DOI] [PubMed] [Google Scholar]
- Oklu R, Habito R, Mayr M, Deipolyi AR, Albadawi H, Hesketh R, Walker TG, Linskey KR, Long CA, Wicky S, Stoughton J, Watkins MT. Watkins MT Pathogenesis of varicose veins. J Vasc Interv Radiol. 2012;23:33–39. doi: 10.1016/j.jvir.2011.09.010. [DOI] [PubMed] [Google Scholar]
- Rathbun SW, Kirkpatrick AC. Treatment of chronic venous insufficiency. Curr Treat Options Cardiovasc Med. 2007;9:115–126. doi: 10.1007/s11936-007-0005-6. [DOI] [PubMed] [Google Scholar]
- Sansilvestri-Morel P, Fioretti F, Rupin A, Senni K, Fabiani JN, Godeau G, Verbeuren TJ. Comparison of extracellular matrix in skin and saphenous veins from patients with varicose veins: does the skin reflect venous matrix changes? Clin Sci (Lond) 2007;112:229–239. doi: 10.1042/CS20060170. [DOI] [PubMed] [Google Scholar]
- Pollman MJ, Naumovski L, Gibbons GH. Vascular cell apoptosis: cell type-specific modulation by transforming growth factor-beta1 in endothelial cells versus smooth muscle cells. Circulation. 1999;99:2019–20126. doi: 10.1161/01.cir.99.15.2019. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Barbero A, Obreo J, Alvarez-Munoz P, Pandiella A, Bernabéu C, López-Novoa JM. Endoglin modulation of TGF-beta1-induced collagen synthesis is dependent on ERK1/2 MAPK activation. Cell Physiol Biochem. 2006;18:135–142. doi: 10.1159/000095181. [DOI] [PubMed] [Google Scholar]
- Bujan J, Gimeno MJ, Jimenez JA. et al. Expression of elastic components in healthy and varicose veins. World J Surg. 2003;27:901–905. doi: 10.1007/s00268-003-6897-8. [DOI] [PubMed] [Google Scholar]
- Jacob T, Hingorani A, Ascher E. Overexpression of transforming growth factor-beta1 correlates with increased synthesis of nitric oxide synthase in varicose veins. J Vasc Surg. 2005;41:523–530. doi: 10.1016/j.jvs.2004.12.044. [DOI] [PubMed] [Google Scholar]
- Kowalewski R, Malkowski A, Sobolewski K, Gacko M. Evaluation of transforming growth factor-beta signaling pathway in the wall of normal and varicose veins. Pathobiology. 2010;77:1–6. doi: 10.1159/000272948. [DOI] [PubMed] [Google Scholar]
- Pascual G, Mendieta C, Garcia-Honduvilla N. et al. TGF-beta1 upregulation in the aging varicose vein. J Vasc Res. 2007;44:192–201. doi: 10.1159/000100375. [DOI] [PubMed] [Google Scholar]
- Lal BK, Saito S, Pappas PJ. et al. Altered proliferative responses of dermal fibroblasts to TGF-beta1 may contribute to chronic venous stasis ulcer. J Vasc Surg. 2003;37:1285–1293. doi: 10.1016/s0741-5214(02)75295-6. [DOI] [PubMed] [Google Scholar]
- Oklu R, Walker TG, Wicky S, Hesketh R. Angiogenesis and current antiangiogenic strategies for the treatment of cancer. J Vasc Interv Radiol. 2010;21:1791–1805. doi: 10.1016/j.jvir.2010.08.009. [DOI] [PubMed] [Google Scholar]
- Pappas PJ, Lal BK, Ohara N. et al. Regulation of matrix contraction in chronic venous disease. Eur J Vasc Endovasc Surg. 2009;38:518–529. doi: 10.1016/j.ejvs.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Zhao G, Li H, Liu X, Wang S. Candesartan antagonizes pressure overload-evoked cardiac remodeling through Smad7 gene-dependent MMP-9 suppression. Gene. 2012;497:301–306. doi: 10.1016/j.gene.2012.01.081. [DOI] [PubMed] [Google Scholar]
- MacMillan CJ, Furlong SJ, Doucette CD, Chen PL, Hoskin DW, Easton AS. Bevacizumab diminishes experimental autoimmune encephalomyelitis by inhibiting spinal cord angiogenesis and reducing peripheral T-cell responses. J Neuropathol Exp Neurol. 2012;71:983–999. doi: 10.1097/NEN.0b013e3182724831. [DOI] [PubMed] [Google Scholar]
- Craelius W, Migdal MW, Luessenhop CP, Sugar A, Mihalakis I. Iron deposits surrounding multiple sclerosis plaques. Arch Pathol Laboratory Med. 1982;106:397. [PubMed] [Google Scholar]
- Haacke EM, Garbern J, Miao Y, Habib C, Liu M. Iron stores and cerebral veins in MS studied by susceptibility weighted imaging. Int Angiol. 2010;29:149–157. [PubMed] [Google Scholar]
- LeVine SM. Iron deposits in multiple sclerosis and Alzheimer’s disease brains. Brain Res. 1997;760:298–303. doi: 10.1016/s0006-8993(97)00470-8. [DOI] [PubMed] [Google Scholar]
- Young NP, Weinshenker BG, Parisi JE, Scheithauer B, Giannini C, Roemer SF, Thomsen KM, Mandrekar JN, Erickson BJ, Lucchinetti CF. Perivenous demyelination: association with clinically defined acute disseminated encephalomyelitis and comparison with pathologically confirmed multiple sclerosis. Brain. 2010;133:333–348. doi: 10.1093/brain/awp321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivadinov R, Weinstock-Guttman B, Pirko I. Iron deposition and inflammation in multiple sclerosis. Which one comes first? BMC Neurosci. 2011;12:60. doi: 10.1186/1471-2202-12-60. 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bawdier-Commander C, Verbeuren T, Lebard C, Michel JB, Jacob MP. Increased TIMP/MMP ratio in varicose veins: a possible explanation for extracellular matrix accumulation. J Pathos. 2000;192:105–112. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH670>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Ono T, Bergan JJ, Schmid-Schönbein GW, Takase S. Monocyte infiltration into venous valves. J Vasc Surg. 1998;27:158–166. doi: 10.1016/s0741-5214(98)70303-9. [DOI] [PubMed] [Google Scholar]
- Takase S, Bergan JJ, Schmid-Schönbein G. Expression of adhesion molecules and cytokines on saphenous veins in chronic venous insufficiency. Ann Vasc Surg. 2000;14:427–435. doi: 10.1007/s100169910092. [DOI] [PubMed] [Google Scholar]
- Bergan JJ, Schmid-Schönbein GW, Smith PD, Nicolaides AN, Boisseau MR, Eklof B. Chronic venous disease. N Engl J Med. 2006;355:488–498. doi: 10.1056/NEJMra055289. [DOI] [PubMed] [Google Scholar]
- Takase S, Schmid-Schönbein G, Bergan JJ. Leukocyte activation in patients with venous insufficiency. J Vasc Surg. 1999;30:148–156. doi: 10.1016/s0741-5214(99)70187-4. [DOI] [PubMed] [Google Scholar]
- Powell CC, Rohrer MJ, Barnard MR, Peyton BD, Furman MI, Michelson AD. Chronic venous insufficiency is associated with increased platelet and monocyte activation and aggregation. J Vasc Surg. 1999;30:844–851. doi: 10.1016/s0741-5214(99)70009-1. [DOI] [PubMed] [Google Scholar]
- Putnam T. Studies in multiple sclerosis: encephalitis and sclerotic plaques produced by venular obstruction. Arch of Neurol and Psych. 1935;33:929–940. [Google Scholar]
- Mayhan WG, Heistad DD. Role of veins and cerebral venous pressure in disruption of the blood-brain barrier. Circ Res. 1986;59:216–220. doi: 10.1161/01.res.59.2.216. [DOI] [PubMed] [Google Scholar]
- Youssef AM, Hamidian Jahromi A, Vijay CG, Granger DN, Alexander JS. Intra-abdominal hypertension causes reversible blood-brain barrier disruption. J Trauma Acute Care Surg. 2012;72:183–188. doi: 10.1097/TA.0b013e31822a3254. [DOI] [PubMed] [Google Scholar]
- Kovac N, Siranovic M, Mazul-Sunko B. Clinical significance of intra-abdominal pressure and abdominal perfusion pressure in patients with acute abdominal syndrome. SIGNA VITAE. 2007;2:14–17. [Google Scholar]
- Shin HY, Smith ML, Toy KJ, Williams PM, Bizios R, Gerritsen ME. VEGF-C mediates cyclic pressure-induced endothelial cell proliferation. Physiol Genomics. 2002;11:245–251. doi: 10.1152/physiolgenomics.00068.2002. [DOI] [PubMed] [Google Scholar]
- Shin HY, Gerritsen ME, Bizios R. Regulation of endothelial cell proliferation and apoptosis by cyclic pressure. Ann Biomed Eng. 2002;30:297–304. doi: 10.1114/1.1458595. [DOI] [PubMed] [Google Scholar]
- Shin HY, Bizios R, Gerritsen ME. Cyclic pressure modulates endothelial barrier function. Endothelium. 2003;10:179–187. doi: 10.1080/10623320390237883. [DOI] [PubMed] [Google Scholar]
- Shin HY, Schwartz EA, Bizios R, Gerritsen ME. Receptor-mediated basic fibroblast growth factor signaling regulates cyclic pressure-induced human endothelial cell proliferation. Endothelium. 2004;11:285–291. doi: 10.1080/10623320490904205. [DOI] [PubMed] [Google Scholar]
- Miyamoto K, Tsujimoto S, Tominaga M, Takeshima T, Morimoto T, Tsunoda S, Sakaki T. The focal brain ischemia with disturbance of venous drainage] No To Shinkei. 1992;44:621–627. [PubMed] [Google Scholar]
- Yura S, Sako K, Yonemasu Y. [The effects of disturbance of cerebral venous drainage on focal cerebral blood flow and ischemic cerebral edema] No To Shinkei. 1990;42:269–275. [PubMed] [Google Scholar]
- Nedelmann M, Kaps M, Mueller-Forell W. Venous obstruction and jugular valve insufficiency in idiopathic intracranial hypertension. Journal of Neurology. 2009;256:964–969. doi: 10.1007/s00415-009-5056-z. [DOI] [PubMed] [Google Scholar]
- Meyer-Schwickerath R, Haug C, Hacker A, Fink F, Seidel D, Hartung HP, Haupts MR. Intracranial venous pressure is normal in patients with multiple sclerosis. Mult Scler. 2011;17:637–638. doi: 10.1177/1352458510395982. [DOI] [PubMed] [Google Scholar]
- Zamboni P, Bertolotto A, Boldrini P, Cenni P, D'Alessandro R, D'Amico R, Del Sette M, Galeotti R, Galimberti S, Liberati A, Massacesi L, Papini D, Salvi F, Simi S, Stella A, Tesio L, Valsecchi MG, Filippini G. Chair of the Steering Committee. Efficacy and safety of venous angioplasty of the extracranial veins for multiple sclerosis. Brave dreams study (brain venous drainage exploited against multiple sclerosis): study protocol for a randomized controlled trial. Trials. 2012;13:183. doi: 10.1186/1745-6215-13-183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertl-Wagner B, Koerte I, Kümpfel T, Blaschek A, Laubender RP, Schick M, Steffinger D, Kaufmann D, Heinen F, Reiser M, Alperin N, Hohlfeld R. Non-specific alterations of craniocervical venous drainage in multiple sclerosis revealed by cardiac-gated phase-contrast MRI. Mult Scler. 2012;18:1000–1007. doi: 10.1177/1352458511432742. [DOI] [PubMed] [Google Scholar]
- Koerte IK, Schankin CJ, Immler S, Lee S, Laubender RP, Grosse C, Eftimov L, Milde-Busch A, Reiser M, Straube A, Heinen F, Alperin N, Ertl-Wagner B. Altered cerebrovenous drainage in patients with migraine as assessed by phase-contrast magnetic resonance imaging. Invest Radiol. 2011;46:434–440. doi: 10.1097/RLI.0b013e318210ecf5. [DOI] [PubMed] [Google Scholar]
- Lee AB, Laredo J, Neville R. Embryological background of truncular venous malformation in the extracranial venous pathways as the cause of chronic cerebro spinal venous insufficiency. Int Angiol. 2010;29:95–108. [PubMed] [Google Scholar]
- Sainte-Rose C, Lacombe J, Pierre-Kahn A, Renier D, Hirsch J-F. Intracranial venous sinus hypertension: cause or consequence of hydrocephalus in infants? J Neurosurg. 1984;60:727–736. doi: 10.3171/jns.1984.60.4.0727. [DOI] [PubMed] [Google Scholar]
- Johnston I, Owler B, Pickard J. The Pseudotumor Cerebri Syndrome. New York: Cambridge University Press; 2007. [Google Scholar]
- Wall M, George D. Idiopathic intracranial hypertension. A prospective study of 50 patients. Brain. 1991;114:155–180. [PubMed] [Google Scholar]
- Karahalios DG, Rekate HL, Khayata MH, Apostolides PJ. Elevated intracranial venous pressure as a universal mechanism in pseudotumor cerebri of varying etiologies. Neurology. 1996;46:198–202. doi: 10.1212/wnl.46.1.198. [DOI] [PubMed] [Google Scholar]
- Brodsky MC, Vaphiades MV. Magnetic resonance imaging in pseudotumor cerebri. Ophthalmology. 1998;105:1686–1693. doi: 10.1016/S0161-6420(98)99039-X. [DOI] [PubMed] [Google Scholar]
- Wall M, Dollar JD, Sadun AA, Kardon R. Idiopathic intracranial hypertension. Lack of histological evidence for cerebral edema. Arch Neurol. 1995. pp. 141–145. [DOI] [PubMed]
- Karmon Y, Ramanathan M, Minagar A, Zivadinov R, Weinstock-Guttman B. Arterial, venous and other vascular risk factors in multiple sclerosis. Neurol Res. 2012;34:754–760. doi: 10.1179/1743132812Y.0000000077. [DOI] [PubMed] [Google Scholar]
- Bazigou E, Xie S, Chen C, Weston A, Miura N, Sorokin L, Adams R, Muro AF, Sheppard D, Makinen T. Integrin-alpha9 is required for fibronectin matrix assembly during lymphatic valve morphogenesis Dev. Cell. 2009;17:175–186. doi: 10.1016/j.devcel.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazigou E, Lyons OT, Smith A, Venn GE, Cope C, Brown NA, Machine T. Genes regulating lymphangiogenesis control venous valve formation and maintenance in mice. J Clin Invest. 2011;121:2984–2992. doi: 10.1172/JCI58050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CP. et al. A field of myocardial-endocardial NFAT signaling underlies heart valve morphogenesis. Cell. 2004;118:649–663. doi: 10.1016/j.cell.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Bohlen HG, Gasheva OY, Zawieja DC. Nitric oxide formation by lymphatic bulb and valves is a major regulatory component of lymphatic pumping. Am J Physiol Heart Circ Physiol. 2011;301:H1897–H1906. doi: 10.1152/ajpheart.00260.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TC, Zhao YD, Courtman DW, Stewart DJ. Abnormal aortic valve development in mice lacking endothelial nitric oxide synthase. Circulation. 2000;101:2345–2348. doi: 10.1161/01.cir.101.20.2345. [DOI] [PubMed] [Google Scholar]
- Mellor RH, Brice G, Stanton AW, French J, Smith A, Jeffery S, Levick JR, Burnand KG. Mortimer PS; Mutations in FOXC2 are strongly associated with primary valve failure in veins of the lower limb. Lymphoedema Research Consortium. 2007;115:1912–1920. doi: 10.1161/CIRCULATIONAHA.106.675348. [DOI] [PubMed] [Google Scholar]
- Traboulsi EI, Al-Khayer K, Matsumoto M, Kimak MA, Crowe S, Wilson SE, Finegold DN, Ferrell RE, Meisler DM. Lymphedema-distichiasis syndrome and FOXC2 gene mutation. Am J Ophthalmol. 2002;134:592–596. doi: 10.1016/s0002-9394(02)01642-2. [DOI] [PubMed] [Google Scholar]
- Adams RH, Wilkinson GA, Weiss C, Diella F, Gale NW, Deutsch U, Risau W, Klein R. Roles of ephrinB ligands and EphB receptors in cardiovascular development: demarcation of arterial/venous domains, vascular morphogenesis, and sprouting angiogenesis. Genes Dev. 1999;13:295–306. doi: 10.1101/gad.13.3.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dela Paz NG, D'Amore PA. Arterial versus venous endothelial cells. Cell Tissue Res. 2009;335:5–16. doi: 10.1007/s00441-008-0706-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minagar A, Jy W, Jimenez JJ, Sheremata WA, Mauro LM, Mao WW, Horstman LL, Ahn YS. Elevated plasma endothelial microparticles in multiple sclerosis. Neurology. 2001;56:1319–1324. doi: 10.1212/wnl.56.10.1319. [DOI] [PubMed] [Google Scholar]
- Alexander JS, Ganta CV, Orr AW, Funk S, Yurdagul A, Cromer W, Mathis JM, Erdreich-Epstein A, Nagra RM, Minagar A, Tsunoda I, Sato F, Martinez N, Omura S. Ephrin/Eph receptor expression on brain microvascular endothelial cells and microparticles: regulation by inflammatory cytokines. BrainPET2011. Barcelona, Spain: XXVth International Symposium on Cerebral Blood Flow, Metabolism and Function and the Xth International Conference on Quantification of Brain Function with PET; 2011. [Google Scholar]
- Akkawi NM, Agosti C, Rozzini L, Anzola GP, Padovani A. Transient global amnesia and disturbance of venous flow patterns. Lancet. 2001;357:957. doi: 10.1016/S0140-6736(05)71655-X. [DOI] [PubMed] [Google Scholar]
- Hodges JR, Warlow CP. Syndromes of transient amnesia : towards a classification. A study of 153 cases. J Neurol Neurosurg Psychiatry. 1990;53:834–843. doi: 10.1136/jnnp.53.10.834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis SL. Aetiology of transient global amnesia. Lancet. 1998;352:397–399. doi: 10.1016/S0140-6736(98)01442-1. [DOI] [PubMed] [Google Scholar]
- Sander D, Winbeck K, Etgen T, Knapp R, Klingelhöfer J, Conrad B. Disturbance of venous flow patterns in patients with transient global amnesia. Lancet. 2000;356:1982–1984. doi: 10.1016/S0140-6736(00)03313-4. [DOI] [PubMed] [Google Scholar]
- Kim I, Ryu YS, Kwak HJ, Ahn SY, Oh JL, Yancopoulos GD, Gale NW, Koh GY. EphB ligand, ephrinB2, suppresses the VEGF- and angiopoietin 1-induced Ras/mitogen-activated protein kinase pathway in venous endothelial cells. FASEB J. 2002;16:1126–1128. doi: 10.1096/fj.01-0805fje. [DOI] [PubMed] [Google Scholar]
- Ivanov AI, Romanovsky AA. Putative dual role of ephrin-Eph receptor interactions in inflammation. IUBMB Life. 2006;58:389–394. doi: 10.1080/15216540600756004. [DOI] [PubMed] [Google Scholar]
- Ferlini A, Bovolenta M, Neri M, Gualandi F, Balboni A, Yuryev A, Salvi F, Gemmati D, Liboni A, Zamboni P. Custom CGH array profiling of copy number variations (CNVs) on chromosome 6p21.32 (HLA locus) in patients with venous malformations associated with multiple sclerosis. BMC Med Genet. 2010;11:64. doi: 10.1186/1471-2350-11-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirmohamed M. Genetic factors in the predisposition to drug-induced hypersensitivity reactions. AAPS Jl. 2006;8:E20–E26. doi: 10.1208/aapsj080103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geurts JJ, Wolswijk G, Bö L, Redeker S, Ramkema M, Troost D, Aronica E. Expression patterns of Group III metabotropic glutamate receptors mGluR4 and mGluR8 in multiple sclerosis lesions. J Neuroimmunol. 2005;158:182–190. doi: 10.1016/j.jneuroim.2004.08.012. [DOI] [PubMed] [Google Scholar]
- Anwar MA, Shalhoub J, Lim CS, Gohel MS, Davies AH. The effect of pressure-induced mechanical stretch on vascular wall differential gene expression. J Vasc Res. 2012;49:463–478. doi: 10.1159/000339151. [DOI] [PubMed] [Google Scholar]
- Lightman S, McDonald WI, Bird AC, Francis DA, Hoskins A, Batchelor JR, Halliday AM. Retinal venous sheathing in optic neuritis. Its significance for the pathogenesis of multiple sclerosis. Brain. 1987 Apr;110:405–414. doi: 10.1093/brain/110.2.405. [DOI] [PubMed] [Google Scholar]
- Arnold AC, Pepose JS, Hepler RS, Foos RY. Retinal periphlebitis and retinitis in multiple sclerosis. I. Pathologic characteristics. Ophthalmology. 1984;91:255–262. doi: 10.1016/s0161-6420(84)34296-8. [DOI] [PubMed] [Google Scholar]
- Engell T, Jensen OA, Klinken L. Periphlebitis retinae in multiple sclerosis. A histopathological study of two cases. Acta Ophthalmol (Copenh) 1985;63:83–88. doi: 10.1111/j.1755-3768.1985.tb05221.x. [DOI] [PubMed] [Google Scholar]
- Perry VH, Lund RD. Evidence that the lamina cribrosa prevents intraretinal myelination of retinal ganglion cell axons. J Neurocytol. 1990;19:265–272. doi: 10.1007/BF01217304. [DOI] [PubMed] [Google Scholar]
- Lucarelli MJ, Pepose JS, Arnold AC, Foos RY. Immunopathologic features of retinal lesions in multiple sclerosis. Ophthalmology. 1991;98:1652–1656. doi: 10.1016/s0161-6420(91)32080-3. [DOI] [PubMed] [Google Scholar]
- Stefansson K, Molnar ML, Marton LS, Molnar GK, Mihovilovic M, Tripathi RC, Richman DP. Myelin-associated glycoprotein in human retina. Nature. 1984;307:548–550. doi: 10.1038/307548a0. [DOI] [PubMed] [Google Scholar]
- Engell T, Sellebjerg F, Jensen C. Changes in the retinal veins in acute optic neuritis. Acta Neurol Scand. 1999;100:81–83. doi: 10.1111/j.1600-0404.1999.tb01041.x. [DOI] [PubMed] [Google Scholar]
- Haselton FR, Dworska E, Evans SS, Hoffman LH, Alexander JS. Modulation of retinal endothelial barrier in an in vitro model of the retinal microvasculature. Exp Eye Res. 1996;63:211–222. doi: 10.1006/exer.1996.0110. [DOI] [PubMed] [Google Scholar]
- Haselton FR, Dworska EJ, Hoffman LH. Glucose-induced increase in paracellular permeability and disruption of beta-receptor signaling in retinal endothelium. Invest Ophthalmol Vis Sci. 1998;39:1676–1684. [PubMed] [Google Scholar]
- Zamboni P, Cossu A, Carpanese L, Simonetti G, Massarelli G, Liboni A. The so-called venous aneurysms. Phlebology. 1990;5:45–50. [Google Scholar]
- Callen DJ, Shroff MM, Branson HM, Li DK, Lotze T, Stephens D, Banwell BL. Role of MRI in the differentiation of ADEM from MS in children. Neurology. 2009;72:968–973. doi: 10.1212/01.wnl.0000338630.20412.45. [DOI] [PubMed] [Google Scholar]
- Wingerchuk DM, Lucchinetti CF. Comparative immunopathogenesis of acute disseminated encephalomyelitis, neuromyelitis optica, and multiple sclerosis. Curr Opin Neurol. 2007;20:343–350. doi: 10.1097/WCO.0b013e3280be58d8. [DOI] [PubMed] [Google Scholar]
- Hafler DA, Benjamin DS, Burks J, Weiner HL. Myelin basic protein and proteolipid protein reactivity of brain- and cerebrospinal fluid-derived T cell clones in multiple sclerosis and post infectious encephalomyelitis. J Immune. 1987;139:68–72. [PubMed] [Google Scholar]
- Ichiyama T, Kajimoto M, Suenaga N, Maeba S, Matsubara T. Furukawa S Serum levels of matrix metalloproteinase-9 and its tissue inhibitor (TIMP-1) in acute disseminated encephalomyelitis. J Neuroimmunol. 2006;172:182–186. doi: 10.1016/j.jneuroim.2005.10.010. [DOI] [PubMed] [Google Scholar]
- Martino D, Branson JA, Church AJ, Candler PM, Livrea P, Giovannoni G, Dale RC. Soluble adhesion molecules in acute disseminated encephalomyelitis. Pediatr Neurol. 2005;33:255–258. doi: 10.1016/j.pediatrneurol.2005.05.006. [DOI] [PubMed] [Google Scholar]