

Abstract
Since the most significant ischemic sequelae occur within hours of stroke, it is necessary to understand how neuronal function changes during this time. While histologic and behavioral models show the extent of stroke-related damage, only in vivo recordings can illustrate changes in brain activity during stroke and validate effectiveness of neuroprotective compounds. Spontaneous and evoked field potentials (fEPs) were recorded in the deep layers of the cortex with a linear microelectrode array for 3 hours after focal stroke in anesthetized rats. Tat-NR2B9c peptide, which confers neuroprotection by uncoupling the PSD-95 protein from N-methyl-D-aspartate receptor (NMDAR), was administered 5 minutes before ischemia. Evoked field potentials were completely suppressed within 3 minutes of infarct in all ischemic groups. Evoked field potential recovery after stroke in rats treated with Tat-NR2B9c (83% of baseline) was greater compared with stroke-only (61% of baseline) or control peptide (Tat-NR2B-AA; 67% of baseline) groups (P<0.001). Electroencephalography (EEG) power was higher in Tat-NR2B9c-treated animals at both 20 minutes and 1 hour (50% and 73% of baseline, respectively) compared with stroke-only and Tat-NR2B-AA-treated rats (P<0.05). Tat-NR2B9c significantly reduces stroke-related cortical dysfunction as evidenced by greater recovery of fEPs and EEG power; illustrating the immediate effects of the compound on poststroke brain function.
Keywords: brain ischemia, electrophysiology, evoked potentials, excitotoxicity, neurophysiology, neuroprotection
Introduction
Cortical focal ischemia is most commonly caused by occlusion of a cerebral artery. The core of the infarct experiences greatest loss of blood supply and cell death occurs within minutes. Neurons surrounding the core, in an area known as the penumbra, experience only a moderate reduction in blood flow and thus retain structural integrity, but may die in the hours and days following ischemia.1 This delayed death presents an excellent opportunity for therapeutic intervention.
Considerable research has been directed toward finding effective treatments for acute ischemic injury. Slow progress in this area reflects the complex nature of the mammalian nervous system, a lack of understanding in how neurons communicate during ischemic injury, and the role of communication in neuronal survival.2 Excitotoxicity via N-methyl-D-aspartate (NMDA)-type glutamate receptor overactivation is thought to have an important role in neuronal death in the ischemic penumbra and thus, glutamate receptor blockade has been a focus of neuroprotective research.3 However, this approach has not proven clinically useful because of poor pharmacology, poor neuroprotection in long-term recovery, and severe side effects of blocking excitatory communication.4 Uncoupling NMDARs from specific intracellular signal transduction cascades can confer neuroprotection in vitro and in vivo without affecting the basal function of the channel.5, 6 To this end, Aarts et al1 discovered that selective uncoupling of the scaffold protein PSD-95 from NMDAR can prevent neuronal death without blocking normal NMDAR function. They accomplished this by using the cell-permeable 20-mer peptide Tat-NR2B9c, containing the nine carboxy-terminal amino acids of the NMDA receptor NR2B subunit fused to the 11-mer HIV-1 Tat protein transduction domain. The peptide disrupts the protein–protein interactions of PSD-95 with NMDAR,7 thus preventing toxic signaling. In short-term survival (24 hours) experiments, Tat-NR2B9c significantly reduced focal ischemic brain damage in rats. In long-term survival (60 days) from stroke, Tat-NR2B9c preserves motor, learning, and behavioral abilities and preserves the brain tissue so that little or no brain lesion is evident upon histologic assessment.8
Research into ischemia and ischemic neuroprotection has typically been performed using histologic or behavioral analyses. Histologic analysis provides only gross assessment of infarct size and location, whereas behavioral function has been used to assess the extent of deficit in poststroke motor control, learning, and memory. In vitro studies have focused on cell death pathways associated with stroke-related degeneration but many have not yet been translated into in vivo approaches for stroke. In order to fully understand how stroke affects the brain tissue, we need an in vivo neurophysiologic model that can assess real-time changes in cortical excitability during and after stroke. It has been shown in primates and proof-of-concept clinical studies that uncoupling the PSD-95 protein improves neurologic outcomes after strokes involving middle cerebral artery occlusions.9, 10 It is well known that a relatively short-time window exists between the first infarct and the progression of a more prolonged transient ischemic attack where reversible symptoms such as ionic fluxes exacerbate neurologic deficits.11 As such, we were interested to find out what effect Tat-NR2B9c has on synaptic function during this critical early phase of degeneration prevention. In order to accomplish this, we developed a linear 8-channel microelectrode in vivo model of stroke that can monitor extracellular field potentials before, during, and after stroke.12 Using this model, we show that uncoupling PSD-95 scaffolding protein from NMDAR using Tat-NR2B9c peptide causes rapid and significant recovery of cortical function after stroke. To our knowledge, this is the first study showing neurophysiologic recovery of cortical dysfunction after administration of a neuroprotective compound during the poststroke therapeutic window.
Materials and Methods
Animals
Experiments were conducted using a total of 34 adult male Sprague–Dawley rats (280 to 400 g) in compliance with the ethics protocol of the University Health Network Animal Care Committee (UHN ACC) and ARRIVE guidelines. The present study has been approved in its entirety by UHN ACC. Animals were placed into four groups — stroke-only (n=11), sham (n=6), Tat-NR2B9c+stroke (n=6), Tat-NR2B-AA+stroke (n=7), and Tat-NR2B9c+sham (n=4). Every effort was made to reduce the number of experimental animals used and minimize discomfort. Rats were housed in the animal care facility at the Toronto Western Hospital with food and water ad libitum. Experiments were performed in the light phase of a 12 to 12-hour light–dark cycle.
Anesthesia was induced using isoflurane (Pharmaceutical Partners of Canada) and maintained with an intraperitoneal injection of urethane (Sigma, Oakville, ON, Canada) at a dose of 1.2 g/kg. Urethane anesthesia allows continuous monitoring of cortical electrophysiology over a prolonged period with minimal cardiovascular and respiratory complications.13 Furthermore, spontaneous potentials measured in rats anesthetized with urethane have similar characteristics to those generated in the conscious animal.14 Adequate levels of anesthesia were confirmed every 15 minutes by absence of a withdrawal reflex to hindlimb pinch and maintained as required by supplemental doses (20% of initial dose) of urethane. Body temperature was maintained at 36°C to 37°C.
Pre-Experiment Surgery
After induction of anesthesia, the hair on the dorsal surface of the head was shaved and the head was mounted in a stereotaxic frame (Stoelting, Wood Dale, IL, USA), with blunt ear bars and a mouthpiece with an incisor bar. Lidocaine (2% solution; Astra Pharma, Mississauga, ON, Canada) was administered by intradermal injection before an incision was made along the anterior–posterior axis on the dorsal surface of the cranium. The skin was retracted and clamped, exposing the bregma and lambda. The anterior–posterior (AP) and medial–lateral (ML) coordinates for the region of recording were located on the dorsal surface of the exposed skull. A right rostrocaudal cranial window (AP −5.0 to +4.0, ML midline to ridge), for access to the somatosensory cortex, was made using a high-speed drill leaving the dura intact. Finally, to access the middle cerebral artery for ischemia induction, a 5 mm lateral extension of the craniotomy was made underneath the cranial ridge on the right hemisphere.
Electrophysiological Recordings
All experiments were performed using a parasagittal 8-channel microelectrode array, with 1 mm tip separation, as previously published by our group.12 Microelectrodes were assembled from Parylene-C-insulated tungsten wires (Micro Probe, Gaithersburg, MD, USA), with a 20 μmol/L tip length. To decrease the initial impedance of 1 MΩ for recording, the electrode tips were electroplated in 24 karat yellow gold electroplating solution (Krohn Technical Products, Carlstadt, NJ, USA) and followed by platinizing solution (VWR Scientific Products, Mississauga, ON, Canada) using a Stimulus Isolator (Sarasota, FL, USA; A360 World Precision Instruments) and 1 μA of cathodal direct current applied to the electrode for approximately 10 seconds, giving final impedances of 200 to 400 kΩ. Finally, the microelectrodes were insulated by a sleeve of polyimide Kapton (Micro ML Tubing, Midway, MA, USA). Recording electrodes were inserted into a 10-pin connector (Omnetics Connector) with eight channels, one ground and one reference connected to the neck skin. The array was arranged rostrocaudally across the frontoparietal hemicortex about the bregma (ML +3.0 to 3.5, DV 2.0 mm) (Figure 1). Electrode 1 was the most anterior and electrode 8 the most posterior electrode, with inner electrodes 4 and 5 centered on bregma. Evoked field potentials were generated by stimulating the ventral posterolateral nucleus of the thalamus (VPL; AP -2.3, ML +1.8, DV 6.3 mm), ipsilateral to the ischemic cortex. As the animal was fully anesthetized, direct electrical stimulation of the thalamus was chosen over peripheral nerve or whisker barrel stimulation.15
Figure 1.

Illustration of the parasagittal 8-channel microelectrode array in the rat cortex. The array was arranged rostrocaudally across the frontoparietal hemicortex. Ischemia was induced by coagulating the arteriolar branches of the middle cerebral artery (MCA) and one branch of the anterior cerebral artery (ACA) using a microcautery pen (denoted by X). Outline around array denotes size of the craniotomy.
The parasagittal microelectrode array recorded field potentials in the ‘watershed' border region between the anterior and middle cerebral arteries. Signals were amplified using a 20-gain headstage and amplifier (Plexon, Dallas, TX, USA) (filter settings 3 Hz to 5 kHz, Gain 5000). Each output was digitized at a sampling rate of 12 kHz, displayed, and recorded using a MICRO 1401 mkII computer interface device, and Spike2 neurologic capture software (CED, Cambridge, UK) for offline analysis.
Ischemia and Sham Surgeries
At the beginning of each experiment, a stable baseline of 5 to 10 minutes of spontaneous cortical oscillations was obtained. Cerebral blood flow was measured for the duration of the experiment (3 hours) with a Doppler Flowmeter (Moor, Devon, UK; Instruments Limited). The laser probe was placed 2 to 3 mm above the surface of the cortex near in the vicinity of the 3rd and 4th electrode. Large veins and arteries were avoided to obtain regional blood flow supplying the affected cortical parenchyma.
The right middle cerebral artery was localized under a high-power microscope (Carl Zeiss, Toronto, ON, Canada; AG). Ischemia was induced by coagulating the visible arteriolar branches of the middle cerebral artery and one protruding branch of the anterior cerebral artery using a microcautery pen. This is a modified version of the previously published model of focal cortical stroke.8, 16 Recordings were maintained up to 3 hours after vessel occlusion. The sham surgeries were identical to those in the ischemia group; however, no arterial coagulation was performed. Test pulses (1 Hz) were administered continuously for the next 3 minutes. Subsequently, 10 pulses were delivered every 10 seconds for 12 minutes, then every 5 minutes for 45 minutes and lastly every 15 minutes for the next 2 hours (Figure 2).
Figure 2.
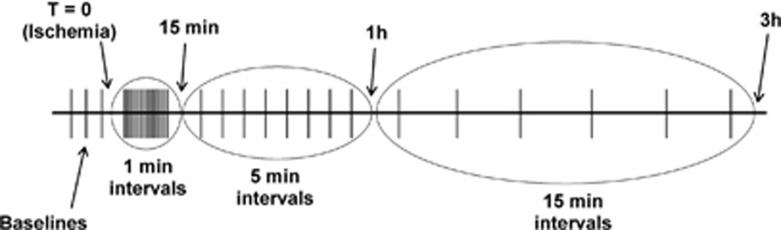
Diagram of the thalamic stimulation parameters before, during, and after ischemia. Each horizontal line is a cluster of 20-test pulses (1 Hz). Upon ischemia (T=0), a stimulation cluster was administered every minute for 15 minutes. Subsequently, the interval between clusters was increased to 5 minutes for the duration of 45 minutes (up to 1 hour post stroke). From 1 hour to 3 hours post stroke, each cluster was administered every 15 minutes.
Tat-NR2B9c Peptide Synthesis and Administration
Tat-NR2B9c and Tat-NR2B-AA peptides were custom synthesized at the Advanced Protein Technology Centre at the Hospital for Sick Children in Toronto using solid-phase peptide synthesis. Full-length peptides were cleaved from the solid substrate using trifluoroacetate, analyzed by mass spectrophotometry to confirm sequence, and lyophilized under nitrogen. Peptides were reconstituted at a concentration of 1 mol/L in water and dialyzed overnight at 4°C against 10 mmol/L ammonium bicarbonate buffer, pH 7.0, to remove TFA. The dialyzed peptide was aliquoted and stored at –80°C. Final peptide concentrations were determined by the OD at 280 nm. Tat-NR2B9c contains an 11 amino acid membrane transduction domain from the HIV-1 Tat protein, fused N-terminal to the last 9 C-terminal residues of the NMDAR NR2B subunit. The NR2B portion of the peptide contains the T/SxV motif critical for PSD-95 binding. Tat-NR2B-AA (control peptide) is identical to Tat-NR2B9c with the exception of a double-point alanine mutation in the T/SxV thus eliminating PSD-95 binding. In the present study, Tat-NR2B9c and Tat-NR2B-AA (3 nmol/g of body weight) were hand injected via tail vein in six animals each, 5 minutes before ischemia induction.
Data Analysis
After experiments, extracellular evoked potentials were smoothed and averaged using Spike2. Fast Fourier transforms were used to analyze acute loss of power spectra after stroke. Electroencephalograph (EEG) power was obtained from 2-minute time windows before and after ischemia without any overlap. The frequency spectrum was largely restricted to the 2 to 8 Hz range across all electrodes and animals. The delta slow wave activity (2to 4 Hz) was the most prevalent frequency band and was readily observed in ischemic and sham rats. In order to characterize the loss of Fast Fourier transform power across animals, we calculated the area under curve by taking 0 to 20 Hz range on the x-axis.
Two-way analysis of variances were used to test main effects and a P-value of 0.05 or less was taken as significant (Sigma Stat). Suppression of fEPs was evaluated using the biphasic W_FP script in Spike2. Amplitudes of the first positive (P1) and first negative (N1) components were calculated and summated to obtain a peak-to-trough (P1+N1) measurement. These measurements were then normalized to baseline for channel 3 in each animal and baselines were averaged across experiments.
Results
Evoked Field Potential
A depth profile of thalamically evoked cortical fEPs was performed to observe the changes in evoked components (Figure 3A). Deeper layers of the cortex were chosen as recording sites, as this region is directly activated by the thalamus.17 Note that the largest fEP is recorded from deep layers of the cortex (Figure 3A), which consisted of two main components—the positive component (P1) and negative component (N1) that together formed a biphasic wave (P1/N1) (Figure 3B), which has been previously reported.18 Thalamic stimulation evoked fEPs with largest amplitudes at ch1 and smallest at ch8, decreasing incrementally with distance (Figure 3B). Upon stroke, the amplitude of P1/N1 was completely suppressed in all stroke groups (Figure 4A, C, and D) Following either partial or complete P1/N1 recovery, amplitudes in all five groups were stable to the 3-hour end point (Figure 4A–E). This presented an ideal quantitative measurement of poststroke brain function.
Figure 3.

(A) Illustration of the depth profile of cortical evoked field potentials (fEPs) in ch1, as evoked by thalamic stimulation in the ventral posterolateral nucleus (VPL). Deep layers of the cortex are directly activated by the VPL, where fEPs with the largest amplitudes were recorded. This region was chosen for subsequent multielectrode recordings. (B) Illustration of thalamically evoked fEPs recorded from the deep cortex on all electrode contacts. The fEP consisted of two main components — positive (P1) and negative (N1) that together formed a biphasic wave (P1/N1). Note that largest P1/N1 amplitudes were recorded at the most anterior contacts, which are directly activated by the VPL.
Figure 4.

(A) P1/N1 recorded from ch1 in an untreated stroke animal. Note complete suppression of P1/N1 upon stroke and significant attenuation lasting up to the 3 hours time point. (B) P1/N1 recorded from ch1 in a sham animal. Note virtually no change in P1/N1 amplitude, which was stable for the duration of the experiment. (C) P1/N1 recorded from ch1 in an ischemic rat treated with Tat-NR2B9c. Note complete attenuation of the fEP, followed by significant recovery of P1/N1 amplitude, which was stable until the endpoint of the experiment. (D) P1/N1 recorded from ch1 in an ischemic rat treated with control peptide Tat-NR2B-AA. Note complete suppression of fEP, followed by partial recovery of P1/N1 amplitude, comparable to untreated stroke-only animals. (E) P1/N1 recorded from ch1 in a sham animal treated with Tat-NR2B9c. Note no change in P1/N1 amplitude, comparable to untreated sham animals. (F) In the stroke-only group (n=6), P1/N1 recovered significantly below shams (n=6, P<0.001). Significantly greater recovery was observed in animals treated with the Tat-NR2B9c peptide (n=6) when compared with stroke-only animals (P<0.001) and stroke animals treated with the inactive Tat-NR2B-AA peptide (n=6, P<0.001). No significant change was observed in sham animals that received the Tat-NR2B9c peptide (n=4).
In stroke animals, P1/N1 recovered only to 61% of baseline, which was significantly less than shams (97% of baseline, P<0.001) (Figure 4F). However, application of Tat-NR2B9c to stroke animals significantly recovered P1/N1 (83% of baseline) when compared with stroke-only animals (P<0.001) and stroke animals treated with inactive Tat-NR2B-AA peptide (67% of baseline, P<0.001). Tat-NR2B9c application without stroke did not lead to a significant increase of P1/N1 amplitude (108% of baseline) when compared with untreated shams.
Spontaneous Electroencephalograph Power
Measurements of EEG power were taken at baseline, 20 minutes and 1 hour poststroke (Figure 5A). To determine whether the infarct produced a neurophysiologic correlate of an ischemic core and penumbra, we compared EEG amplitude across electrodes in ischemic animals. In the stroke-only group, anterior contacts closest to the ischemic focus (ch1–4) displayed a greater drop in power compared with ch5–8 posterior of the affected tissue (Figure 5B) showing a spatial relationship between proximity of ischemia and extent of EEG suppression. At 20 minutes poststroke, EEG power on ch5–8 (47% of baseline) was significantly greater compared with ch1–4 (25% of baseline, P<0.05). This difference remained significant 1 hour after stroke (P<0.01). There was no significant difference in EEG power between ch1–4 and ch5–8 in sham animals (P=0.29). At 20 minutes poststroke in the ischemic group, ch1–4 had significantly greater loss of power compared with sham animals (107% of baseline, P<0.001). This difference between sham and ischemic groups was still significant 1 hour after stroke onset (stroke=36%, sham=102%, P<0.001).
Figure 5.

(A) Total electroencephalograph (EEG) power in sham (n=6), stroke (n=9), Tat-NR2B9c+stroke (n=6), control peptide Tat-NR2B-AA+stroke (n=6), and Tat-NR2B9c+sham (n=4) groups averaged across anterior electrodes ch1–4. Ischemic animals treated with Tat-NR2B9c showed significant recovery of EEG power when compared with stroke-only rats (P<0.01,**) and stroke rats treated with Tat-NR2B-AA (P<0.01,**). No significant change was observed in sham animals that received the Tat-NR2B9c peptide (n=4). (B) Change in EEG power: each bar corresponds to one channel on the multielectrode array. Ch1 is the most anterior, Ch8 the most posterior, and Bregma is located between channels 4 and 5. Sham animals (green, n=6) did not lose EEG power at 20 minutes and 1 hour after baseline. Stroke-only (yellow, n=9) rats showed a decrease in EEG power 20 minutes after infarct with greater suppression in the anterior of the array (P<0.05). Partial recovery was observed in the stroke-only group at 1 hour after ischemia. Anterior array still showed increased loss of EEG power (P<0.01). Ischemic rats treated with Tat-NR2B9c (orange, n=6) showed no difference in power loss between anterior and posterior contacts.
Administration of Tat-NR2B9c resulted in significantly improved recovery of EEG power when compared with stroke-only and stroke animals treated with the control peptide. Tat-NR2B9c+stroke group showed significantly greater EEG power at 20 minutes after stroke on ch1–4 (50% of baseline) when compared with stroke-only rats (P<0.001) and stroke rats treated with Tat-NR2B-AA (26% of baseline, P<0.001). These differences were still significant at 1 hour (P<0.001). Electroencephalograph levels in Tat-NR2B9c-treated animals were not significantly different than shams (P=0.052). Sham animals treated with Tat-NR2B9c did not exhibit significant change in EEG power when compared with untreated shams at either 20 minutes (107% of baseline) or 1 hour (132% of baseline).
Confirmation of Stroke
Cerebral blood flow was monitored to verify that stroke occurred in real time and that it coincided with the occlusion of arteries and suppression of electrophysiological recordings. Cerebral blood flow immediately dropped after stroke to 37% of baseline and stayed below baseline for the duration of the experiment (Supplementary Figure 1). This drop in blood flow is indicative of moderate-to-severe middle cerebral artery occlusion.19 Craniotomy and electrode insertion without arteriolar occlusion in sham animals caused no changes in cerebral blood flow. Cerebral blood flow levels were significantly suppressed in all stroke animals when compared with shams (P<0.001), thus making the region ischemic. The reason for a non-complete reduction in cerebral blood flow levels observed in stroke animals is possibly because of the focal nature of the pial artery occlusion. Even after stroke, there is still supplementary blood flow that perfuses the affected area from small, unobstructed arterioles branching from the anterior cerebral artery near bregma.20
As our group and others have published extensive histologic analyses showing significantly reduced infarct size after Tat-NR2B9c administration,1, 8, 21 the present study focused on neurophysiologic function rather than histology.
Discussion
Tat-NR2B9c confers significant histologic neuroprotection and functional recovery in multiple stroke models, and has demonstrated significant protection in a human clinical stroke trial1, 8, 9, 10 The present study aims to understand the mechanisms by which Tat-NR2B9c confers neuroprotection by using neurophysiologic correlates characterizing deficit and recovery of brain function after stroke induction. We hypothesized that uncoupling the PSD-95 scaffolding protein from NMDAR would restore cortical function at an early time point after stroke onset. Indeed, administration of Tat-NR2B9c 5 minutes before a focal permanent pial arteriolar occlusion resulted in a significant recovery of evoked and spontaneous cortical electrical activity within 1 hour of stroke. Synaptic recovery after stroke-related neurophysiologic dysfunction illustrates the immediate therapeutic effects of Tat-NR2B9c during ischemia.
It is well known that electrical depression as a result of elevated potassium and glutamate after stroke is directly linked to the loss of neuronal excitability.22, 23, 24 Furthermore, preservation of neuronal excitability and recovery from synaptic depression has been shown to be critical for functional recovery after stroke.25, 26, 27 The prolonged disruption of ionic gradients may initiate a cascade of damaging events28 that lead to prolonged inhibition of evoked responses, as evidenced by decreased fEP amplitude in our study. The dependence of evoked responses on a normal membrane potential is consistent with its strong inhibition during ischemia.24, 29
It has been shown that EEG power and fEP amplitude are suppressed after stroke.29, 30 Here we show greater and more rapid recovery of EEG power and fEP amplitude in ischemic rats treated with Tat-NR2B9c when compared with stroke-only animals or ischemic rats treated with the inactive peptide Tat-NR2B-AA. To our knowledge, these are novel findings not reported in stroke literature. Previously studied anti-stroke compounds such as MK-801 reduce glutamatergic signaling through strong, non-competitive blockade of NMDARs.3 MK-801 can improve synaptic performance by decreasing poststroke excitotoxicity, but confers poor neuroprotection and leads to serious side effects in human administration.4, 31 By contrast, our research shows that Tat-NR2B9c does not alter glutamatergic calcium signaling via NMDARs.1 Disrupting PSD-95 with antisense oligonucleotides or PSD-95 inhibitors does not affect normal excitatory transmission.1, 5, 32 In the hippocampal slices of mutant mice lacking PSD-95, NMDAR-dependent long-term potentiation (LTP) is facilitated, whereas NMDAR-dependent long-term depression is absent.32 On the other hand, overexpression of PSD-95 suppresses LTP33 and decreases the threshold for long-term depression induction.34 At the molecular level, PSD-95 functions as a critical scaffold protein coupling the NMDAR-mediated rise in calcium to the intracellular signaling cascades necessary for initiating long-term depression.35 Together, these results suggest that PSD-95 acts as an inhibitor of LTP or promoter of long-term depression. At the same time, NMDAR currents, normal subunit expression, and synaptic morphology are unaffected by the selective uncoupling of PSD-95 from NMDAR. If PSD-95 suppresses LTP in normal NMDAR function, it is possible that uncoupling the two proteins in the present stroke model promotes LTP, as seen by recovery of EEG power and fEP amplitude.
Further evidence of involvement of LTP in Tat-NR2B9c-mediated recovery has been shown by selectively blocking the NR2B subunit from NMDARs, which leads to a blockade of activity-dependent LTP.36 However, an emerging concept states that LTP is not solely dependent on the expression of the NR2B subunit, but that LTP induction is facilitated by the correct balance of NR2A/NR2B subunit composition at the synapse.36, 37 A recent study by Costa et al38 examined hippocampal LTP in Parkinsonian rats and showed that blocking NMDAR–PSD-95 interactions with Tat-NR2B9c promoted LTP. Costa et al38 showed that the NR2A/2B ratio is decreased in Parkinsonian rats, resulting in LTP impairment. While Tat-NR2B9c decreased LTP in control animals with a normal NR2A/2B ratio, in Parkinsonian rats, the peptide restored this form of synaptic plasticity to control levels. The findings reported by Costa et al38 are similar to our observations for Tat-NR2B9c-treated stroke rats, where increased signaling of NR2B subunit may be ameliorated by restoring the normal NR2A/2B ratio, as evidenced in real time by the early recovery of synaptic function. Lastly, our group has recently shown that Tat-NR2B9c neuroprotection in vitro and in vivo requires CaMKIV and that Tat-NR2B9c enhances CaMKIV phosphorylation after ischemia.21 It is also known that CaMKIV has a role in mediating early synaptic plasticity in the rodent cortex.39 These findings support the results of our present study showing greater recovery of synaptic potentials after Tat-NR2B9c administration immediately preceding ischemia when compared with stroke-only rats. It could be that this potentiation is evidenced as early synaptic plasticity facilitated by enhancing CaMKIV phosphorylation with Tat-NR2B9c.
Conclusion
To date, there is no viable and approved treatment targeting ischemic neuroprotection. We present an acute focal stroke model, which enables us to study localized changes in spontaneous and evoked potentials in the rat cortex. We showed that uncoupling the PSD-95 scaffolding protein from NMDAR NR2B subunit recovers stroke function during a critical window of potential neuronal degeneration.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies the paper on the Journal of Cerebral Blood Flow & Metabolism website (http://www.nature.com/jcbfm)
This study was supported by the Canadian Stroke Network (CSN).
Supplementary Material
References
- Aarts M, Liu Y, Liu L, Besshoh S, Arundine M, Gurd JW, et al. Treatment of ischemic brain damage by perturbing NMDA receptor- PSD-95 protein interactions. Science. 2002;298:846–850. doi: 10.1126/science.1072873. [DOI] [PubMed] [Google Scholar]
- Tymianski M. Emerging mechanisms of disrupted cellular signaling in brain ischemia. Nat Neurosci. 2011;14:1369–1373. doi: 10.1038/nn.2951. [DOI] [PubMed] [Google Scholar]
- Ginsberg MD. Neuroprotection for ischemic stroke: past, present and future. Neuropharmacology. 2008;55:363–389. doi: 10.1016/j.neuropharm.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albers GW, Atkinson RP, Kelley RE, Rosenbaum DM. Safety, tolerability, and pharmacokinetics of the N-methyl-D-aspartate antagonist dextrorphan in patients with acute stroke. Dextrorphan Study Group. Stroke. 1995;26:254–258. doi: 10.1161/01.str.26.2.254. [DOI] [PubMed] [Google Scholar]
- Sattler R, Xiong Z, Lu WY, Hafner M, MacDonald JF, Tymianski M. Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science. 1999;284:1845–1848. doi: 10.1126/science.284.5421.1845. [DOI] [PubMed] [Google Scholar]
- Carmichael ST. Plasticity of cortical projections after stroke. Neuroscientist. 2003;9:64–75. doi: 10.1177/1073858402239592. [DOI] [PubMed] [Google Scholar]
- Kornau HC, Schenker LT, Kennedy MB, Seeburg PH. Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science. 1995;269:1737–1740. doi: 10.1126/science.7569905. [DOI] [PubMed] [Google Scholar]
- Sun H-S, Doucette Ta, Liu Y, Fang Y, Teves L, Aarts M, et al. Effectiveness of PSD95 inhibitors in permanent and transient focal ischemia in the rat. Stroke. 2008;39:2544–2553. doi: 10.1161/STROKEAHA.107.506048. [DOI] [PubMed] [Google Scholar]
- Cook DJ, Teves L, Tymianski M. Treatment of stroke with a PSD-95 inhibitor in the gyrencephalic primate brain. Nature. 2012;483:213–217. doi: 10.1038/nature10841. [DOI] [PubMed] [Google Scholar]
- Hill MD, Martin RH, Mikulis D, Wong JH, Silver FL, Terbrugge KG, et al. Safety and efficacy of NA-1 in patients with iatrogenic stroke after endovascular aneurysm repair (ENACT): a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2012;11:942–950. doi: 10.1016/S1474-4422(12)70225-9. [DOI] [PubMed] [Google Scholar]
- Hakim AM. Ischemic penumbra: the therapeutic window. Neurology. 1998;51:S44–S46. doi: 10.1212/wnl.51.3_suppl_3.s44. [DOI] [PubMed] [Google Scholar]
- Srejic LR, Valiante TA, Aarts MM, Hutchison WD. High-frequency cortical activity associated with postischemic epileptiform discharges in an in vivo rat focal stroke model. J Neurosurg. 2013;118:1098–1106. doi: 10.3171/2013.1.JNS121059. [DOI] [PubMed] [Google Scholar]
- De Wildt DJ, Hillen FC, Rauws AG, Sangster B. Etomidate-anaesthesia, with and without fentanyl, compared with urethane-anaesthesia in the rat. Br J Pharmacol. 1983;79:461–469. doi: 10.1111/j.1476-5381.1983.tb11019.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebenezer IS. The generation of cortical slow potentials in the rat anaesthetised with urethane and their modification by nicotine. Neuropharmacology. 1986;25:639–643. doi: 10.1016/0028-3908(86)90217-0. [DOI] [PubMed] [Google Scholar]
- Eckert MJ, Racine RJ. Long-term depression and associativity in rat primary motor cortex following thalamic stimulation. Eur J Neurosci. 2006;24:3553–3560. doi: 10.1111/j.1460-9568.2006.05220.x. [DOI] [PubMed] [Google Scholar]
- Forder JP, Munzenmaier DH, Greene AS. Angiogenic protection from focal ischemia with angiotensin II type 1 receptor blockade in the rat. Am J Physiol Heart Circ Physiol. 2005;288:H1989–H1996. doi: 10.1152/ajpheart.00839.2004. [DOI] [PubMed] [Google Scholar]
- Constantinople CM, Bruno RM. Deep cortical layers are activated directly by thalamus. Science. 2013;340:1591–1594. doi: 10.1126/science.1236425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth D, Shi. D. Three-dimensional in rat neocortex analysis of auditory-evoked potentials. JNeurophysiol. 1990;64:1527–1536. doi: 10.1152/jn.1990.64.5.1527. [DOI] [PubMed] [Google Scholar]
- Palomares SM, Gardner-Morse I, Sweet JG, Cipolla MJ. Peroxynitrite decomposition with FeTMPyP improves plasma-induced vascular dysfunction and infarction during mild but not severe hyperglycemic stroke. J Cereb Blood Flow Metab. 2012;32:1035–1045. doi: 10.1038/jcbfm.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle P, Jokelainen PT. Dorsal cerebral arterial collaterals of the rat. Anat Rec. 1982;203:397–404. doi: 10.1002/ar.1092030309. [DOI] [PubMed] [Google Scholar]
- Bell KFS, Bent RJ, Meese-Tamuri S, Ali A, Forder JP, Aarts MM. Calmodulin kinase IV-dependent CREB activation is required for neuroprotection via NMDA receptor-PSD95 disruption. JNeurochem. 2013;126:274–287. doi: 10.1111/jnc.12176. [DOI] [PubMed] [Google Scholar]
- Astrup J, Siesjö BK, Symon L. Thresholds in cerebral ischemia—the ischemic penumbra. Stroke. 1981;12:723–725. doi: 10.1161/01.str.12.6.723. [DOI] [PubMed] [Google Scholar]
- Nedergaard M.Spreading depression as a contributor to ischemic brain damage Adv Neurol 19967175–83.discussion 83–84. [PubMed] [Google Scholar]
- Chen S, Mohajerani MH, Xie Y, Murphy TH. Optogenetic analysis of neuronal excitability during global ischemia reveals selective deficits in sensory processing following reperfusion in mouse cortex. J Neurosci. 2012;32:13510–13519. doi: 10.1523/JNEUROSCI.1439-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bütefisch CM, Netz J, Wessling M, Seitz RJ, Hömberg V. Remote changes in cortical excitability after stroke. Brain. 2003;126:470–481. doi: 10.1093/brain/awg044. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Corbett D. Plasticity during stroke recovery: from synapse to behaviour. Nat Rev Neurosci. 2009;10:861–872. doi: 10.1038/nrn2735. [DOI] [PubMed] [Google Scholar]
- Clarkson AN, Huang BS, Macisaac SE, Mody I, Carmichael ST. Reducing excessive GABA-mediated tonic inhibition promotes functional recovery after stroke. Nature. 2010;468:305–309. doi: 10.1038/nature09511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirnagl U, Iadecola C, Moskowitz M. a. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- Sakatani K, Iizuka H, Young W. Somatosensory evoked potentials in rat cerebral cortex before and after middle cerebral artery occlusion. Stroke. 1990;21:124–132. doi: 10.1161/01.str.21.1.124. [DOI] [PubMed] [Google Scholar]
- Astrup J, Symon L, Branston NM, Lassen N. a. Cortical evoked potential and extracellular K+ and H+ at critical levels of brain ischemia. Stroke. 1977;8:51–57. doi: 10.1161/01.str.8.1.51. [DOI] [PubMed] [Google Scholar]
- Olney JW. Neurotoxicity of NMDA receptor antagonists: an overview. Psychopharmacol Bull. 1994;30:533–540. [PubMed] [Google Scholar]
- Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe a M, Makhinson M, et al. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature. 1998;396:433–439. doi: 10.1038/24790. [DOI] [PubMed] [Google Scholar]
- Ehrlich I, Klein M, Rumpel S, Malinow R. PSD-95 is required for activity-driven synapse stabilization. Proc Natl Acad Sci USA. 2007;104:4176–4181. doi: 10.1073/pnas.0609307104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beique J-C, Andrade R. PSD-95 regulates synaptic transmission and plasticity in rat cerebral cortex. J Physiol. 2002;546:859–867. doi: 10.1113/jphysiol.2002.031369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Schlüter OM, Steiner P, Czervionke BL, Sabatini B, Malenka RC. Molecular dissociation of the role of PSD-95 in regulating synaptic strength and LTD. Neuron. 2008;57:248–262. doi: 10.1016/j.neuron.2007.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett TE, Bannister NJ, Collett VJ, Dargan SL, Massey PV, Bortolotto ZA, et al. Differential roles of NR2A and NR2B-containing NMDA receptors in LTP and LTD in the CA1 region of two-week old rat hippocampus. Neuropharmacol. 2007;52:60–70. doi: 10.1016/j.neuropharm.2006.07.013. [DOI] [PubMed] [Google Scholar]
- Morishita W, Lu W, Smith GB, Nicoll RA, Bear MF, Malenka RC. Activation of NR2B-containing NMDA receptors is not required for NMDA receptor-dependent long-term depression. Neuropharmacol. 2007;52:71–76. doi: 10.1016/j.neuropharm.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Costa C, Sgobio C, Siliquini S, Tozzi A, Tantucci M, Ghiglieri V, et al. Mechanisms underlying the impairment of hippocampal long-term potentiation and memory in experimental Parkinson's disease. Brain. 2012;135:1884–1899. doi: 10.1093/brain/aws101. [DOI] [PubMed] [Google Scholar]
- Toyoda H, Zhao M-G, Mercaldo V, Chen T, Descalzi G, Kida S, et al. Calcium/calmodulin-dependent kinase IV contributes to translation-dependent early synaptic potentiation in the anterior cingulate cortex of adult mice. Mol Brain. 2010;3:27. doi: 10.1186/1756-6606-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.