

Abstract
Intravenous immunoglobulin (IVIg) is currently evaluated in clinical trials for the treatment of various disorders of the central nervous system. To assess its capacity to reach central therapeutic targets, the brain bioavailability of IVIg must be determined. We thus quantified the passage of IVIg through the blood–brain barrier (BBB) of C57Bl/6 mice using complementary quantitative and qualitative methodologies. As determined by enzyme-linked immunosorbent assay, a small proportion of systemically injected IVIg was detected in the brain of mice (0.009±0.001% of injected dose in the cortex) whereas immunostaining revealed localization mainly within microvessels and less frequently in neurons. Pharmacokinetic analyses evidenced a low elimination rate constant (0.0053 per hour) in the cortex, consistent with accumulation within cerebral tissue. In situ cerebral perfusion experiments revealed that a fraction of IVIg crossed the BBB without causing leakage. A dose-dependent decrease of brain uptake was consistent with a saturable blood-to-brain transport mechanism. Finally, brain uptake of IVIg after a subchronic treatment was similar in the 3xTg-AD mouse model of Alzheimer disease compared with nontransgenic controls. In summary, our results provide evidence of BBB passage and bioavailability of IVIg into the brain in the absence of BBB leakage and in sufficient concentration to interact with the therapeutic targets.
Keywords: Alzheimer's disease, blood–brain barrier, in situ cerebral perfusion, intravenous immunoglobulin, 3xTg-AD
Introduction
The poor brain bioavailability of immunoglobulins has limited their use for treating diseases of the central nervous system (CNS), either for passive or active immunotherapy. To enter the brain, Ig must cross the blood–brain barrier (BBB), which regulates the exchange of compounds between blood and brain. Endothelial cells sheathing brain capillaries differ from those in the rest of the body by the absence of fenestrations, limited pinocytic vesicular transport, prevalent adherens, and tight junctions.1 The BBB normally permits water, ions, and small lipophilic substances to diffuse freely along their concentration gradient.1 Nutrients such as glucose and amino acids enter the brain via transporters whereas receptor-mediated transcytosis allows the uptake of larger molecules including insulin, leptin, and transferrin.1 Alternatively, epithelial cells of the choroid plexuses and of the arachnoid membrane indirectly allows the transport of plasma-borne molecules to the cerebrospinal fluid (CSF), which bathes the cerebral interior and exterior surfaces of the brain parenchyma.2 IgG concentration in the CSF is routinely measured clinically with a normal steady-state CSF/serum ratio of 0.0027.3 Although the protein movement across the blood–CSF barrier is limited, the tight junctions at the choroid plexus are considerably more permeable than those of capillaries of the BBB.3, 4 However, the presence of IgG in the CSF remains thus a poor assessment of its brain bioavailability,4 because exchange of large molecules between brain cells and the CSF is limited.4 For technical reasons, however, the passage of drugs into the brain is scarcely studied in humans. Because clinical efficacy of any drug mostly relies on interaction with its target, the need in determining brain bioavailability of therapeutic Ig is inescapable. Yet, few in vivo studies have sought to determine the brain bioavailability of polyclonal or monoclonal Ig with both qualitative and quantitative methodologies.
Intravenous Ig (IVIg) is a blood-derived therapeutic preparation composed of over 98% polyclonal human IgG (hIgG). This heterogeneous product is used for the treatment of immune deficiencies, acute infections, autoimmune diseases and for neurologic disorders.5, 6 Interestingly, IVIg treatment has been associated with a lower risk of developing Alzheimer's disease (AD)7 and cognitive improvement has been reported for AD patients treated with IVIg.8 Moreover, phase 2 and 3 clinical trials are undergoing in AD (http://www.ClinicalTrials.gov). However, treating a 70-kg individual with IVIg (0.4 g/kg every 2 weeks)8 would require the use of plasma fractionated from over 150 blood donations every year.6 As more than 24 million people are affected with AD worldwide,9 the utilization of IVIg for such a prevalent disease would most certainly lead to a shortage. Although indirect evidence of IVIg reaching the parenchyma exists,10, 11, 12 it is still unclear if the penetration of IVIg is a prerequisite for its clinical effects, because no quantification of the passage into the CNS has been performed. Therefore, to develop alternatives, it is essential to first determine the extent by which IVIg can access cerebral tissues. In this study, we investigated the concentrations of IVIg in the blood, peripheral tissues, and brain structures after systemic administration in mouse models. Our data provide a quantitative and qualitative assessment of the brain bioavailability and pharmacokinetic parameters, suggest that therapeutically relevant concentrations can be reached in cerebral tissues in the absence of BBB disruption, and support a role for brain IgG in the treatment of CNS diseases.
Materials and Methods
Animals and Intravenous Immunoglobulin Treatment
For pharmacokinetic experiments, C57Bl/6 mice (The Jackson Laboratory, Bar Harbor, ME, USA) were used. Triple-transgenic mice (3xTg-AD) and age- and sex-matching nontransgenic (NonTg) littermates (mixed C57Bl/6 × 129S genetic background) produced in our animal research facility as well as B6129SF1/J (C57Bl/6 × 129S, F1) mice (also designated NonTg) purchased from The Jackson Laboratory were used. The 3xTg-AD mouse harbors three mutant genes coding for the following: beta-amyloid precursor protein (APPswe), presenilin-1 (PS1M146V), and tau (tauP301L),13 reproduces the main pathologic features of AD patients and is used to model AD neuropathology and behavioral defects.14 The animal research committee of Université Laval approved all procedures according to the guidelines from the Canadian council on animal care in science.
The animals were subjected to three IVIg regimens. First, a subacute treatment was used for basic pharmacokinetic analyses. Mice (n=3–5 per time point) were injected intraperitoneally (i.p.) with 1.5 g/kg IVIg (25 mg per mouse, in 0.2 mol/L glycine buffer pH 4.25, Gamunex, Grifols Canada, Mississauga, ON, Canada) and were killed after 6, 24, 48, 96, 144 or 192 hours. This protocol allowed us to determine the half-life (T1/2), the elimination rate constant (Ke), the time (T-max) to reach maximum concentration (C-max), and the % of injected dose in plasma and tissues. The second injection regimen, the subchronic treatment, was selected to analyze, all at once at the time of killing, the presence of hIgG in the brain during the absorption phase (75 minutes), the pure elimination phase (96 hours) and at brain maximum concentration (Cmax, 24 hours). This subchronic treatment consisted in three injections are as follows: two intraperitoneal injections of 30 mg/mouse IVIg (96 and 24 hours before death) followed by one intravenous injection of 20 mg/mouse (75 minutes before death). The purpose of the last intravenous injection was to ensure that 100% of IVIg administered reached the blood stream (plasmatic C0, Table 1). In assays comparing 3xTg-AD and NonTg animals, 4- to 13-month-old mice were used to evaluate the impact of the neuropathology on BBB transport. Finally, to assess the effects of a chronic treatment on the passage of IVIg to the brain, 15-month-old 3xTg-AD and NonTg mice were injected twice weekly for 1 month with 1.5 g/kg IVIg (i.p.) or an equivalent volume of vehicle. Injections of an equivalent volume of 0.2 mol/L glycine pH 4.25 (vehicle in Gamunex preparation) were used as a negative control.
Table 1. PK values were measured after a single intraperitoneal injection of 25 mg IVIg (mean±s.e.m. of n=3–5 animals × 6 time points) as presented in Figure 1A.
| Plasma | Spleen | Liver | Cortex | Hippocampus | |
|---|---|---|---|---|---|
| Weight or volume of tissue | 1.02±0.04 mLa | 57.6±2.3 mgb | 791±31 mga | 158.5±9.1 mgb | 29.6±2.4 mgb |
| Tissue/body weight | 60.36 μl/ga | 3.34±0.33 mg/gb | 46.3±5.5 mg/ga | 9.4±0.1 mg/gb | 1.8±0.1 mg/gb |
| hIgG (C0)c | 15.2±1.3 g/L | 711±231 μg/g | 161.3±42.7 μg/g | 13.5±2.0 μg/g | 14.1±2.9 μg/g |
| hIgG (total)d | 15.5±2.0 mg | 41.0±15.5 μg/g | 127.6±40.2 μg/g | 2.14±0.46 μg/g | 0.42±0.13 μg/g |
| % injected dosed | 61.8±8.0% | 0.16±0.06% | 0.51±0.16% | 0.009±0.001% | 0.0017±0.0005% |
| T-max, hours | 6 | 6 | 6 | 24 | 24 |
| C-max | 15.2 mg/mL | 699 μg/g | 232 μg/g | 9.0 μg/g | 12.7 μg/g |
| Half-life, hourse | 90 | 46 | 60 | 117 | 140 |
| Ke, hour-1f | 0.0063 | 0.0137 | 0.009 | 0.0053 | 0.0042 |
C-max, experimental maximum concentration in plasma or tissue; hIgG, human IgG; IVIg, intravenous immunoglobulin; Ke, elimination rate constant in plasma or tissue (per hour); PK, pharmacokinetic; T-max, time to reach maximum concentration.
=measured experimentally.
=C0 was extrapolated (Y-intercept) from experimental linear regression of semi-log curve after absorption phase, including C-max values but excluding t=144 and 192 hours.
=calculated from tissue weight and C0.
=measured experimentally after absorption phase.
=measured experimentally (slope) from experimental linear regression of semi-log curves excluding absorption phase and C-max values.
In Situ Cerebral Perfusion
In situ cerebral perfusion (ISCP) technique was used as previously described15 for the measurement of the distribution volume and the brain uptake coefficient (Kin) of IVIg. This technique ensures that 100% of the injected dose runs through the cerebral vasculature, avoiding peripheral distribution and metabolism. Moreover, the perfusion flow rate during the ISCP can be fully controlled, bypassing the heart.16 Briefly, mice were deeply anesthetized using a mixture of ketamine/xylazine. To ensure that 100% of the perfusate reached the BBB, the right common carotid artery was catheterized after ligation of the external branch. The thorax was then opened, the heart removed, and perfusion with IVIg and [3H]-inulin was immediately initiated at a flow rate of 2.5 mL/minute for 60 seconds followed by a washing step of 120 seconds with [14C]-sucrose in the perfusion buffer. Intravenous immunoglobulin (200, 1,000, or 5,000 μg/mL), [3H]-inulin, as well as [14C]-sucrose were prepared in the following perfusion buffer: 128 mmol/L NaCl, 24 mmol/L NaH-CO3, 4.2 mmol/L KCl, 2.4 mmol/L NaH2PO4, 1.5 mmol/L CaCl2, 0.9 mmol/L MgCl2, and 9 mmol/L D-glucose, gassed with 95% O2 and 5% CO2 to obtain a pH of 7.4 and heated at 37°C. The procedure was terminated by the decapitation of the mouse. The right cerebral hemisphere (the perfused hemisphere) was collected and homogenized in 5 volumes/mg tissue of lysis buffer (50 mmol/L Tris-HCl, 150 mmol/L NaCl, 0.5% deoxycholate, 1% triton containing protease and phosphatase inhibitors) then sonicated three times for 5 × 1-second pulses.12 A volume of 300 μl was used for radioactive measures (WinSpectral 1414 Liquid Scintillation Counter, Wallac/Perkin Elmer, Waltham, MA, USA). The rest of the homogenate was spun 20 minutes at 100,000 g, the supernatant was removed and kept at −80°C for hIgG quantification.12
The brain transport coefficient (Kin) for the IVIg, the [3H]-inulin, and the [14C]-sucrose was calculated from the measured volume of distribution using the following equations:
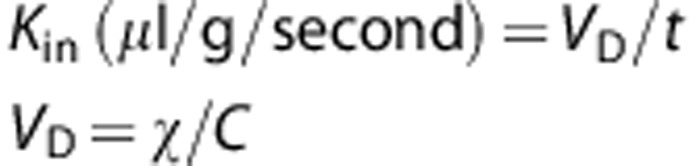 |
where VD (μl/g) represents the volume of distribution of the compound under study, t (s) is the time of perfusion, χ is the concentration in the brain (d.p.m./g or μg/g) and C is the concentration of the compound in the perfusion fluid. Under physiologic conditions, [14C]-sucrose and [3H]-inulin do not cross the BBB and were used here to confirm BBB integrity and washing efficiency throughout the perfusion experiment. The VD value of sucrose remained unchanged indicating that IVIg perfusion did not lead to BBB disruption. The [3H]-inulin counts were below the detection level in all animals, confirming that the postperfusion capillary washing was complete.
Tissue Preparation for Postmortem Analyses
For all analyses, mice were killed by intracardiac perfusion under deep anesthesia, with the exception of ISCP as described above. After transcardiac administration of 50 mL 0.1 mol/L phosphate-buffered saline (PBS) buffer (Bioshop Canada, Burlington, ON, Canada) containing protease and phosphatase inhibitors (SigmaFAST Protease Inhibitor Tablets, Sigma-Aldrich, St Louis, MO, USA with sodium fluoride and sodium pyrophosphate), the spleen, liver, and brain were collected (about 50 mg of tissue each). For IgG quantification, the hippocampus and the cortex were dissected, snap frozen on dry ice, and stored at −80°C until used. The cortex, hippocampus, spleen, and liver were homogenized in lysis buffer and processed as described for ISCP brain homogenates.
For brain cell dissociation, mice (n=3) were perfused without protease and phosphatase inhibitors. The whole brain was processed using the NeuroCult Enzymatic Dissociation Kit for adult CNS tissue (rat and mouse) from StemCell Technology (Vancouver, BC, Canada) according to the instructions from the manufacturer. Isolated cells were counted and frozen in 1 × 107 cell aliquots for further assays.
For immunofluorescence and immunohistochemistry experiments, mice (n=4–8) from the subchronic treatment groups were perfused first with 50 mL 0.1 mol/L PBS buffer followed with 50 mL paraformaldehyde 4% in PBS buffer. The whole brain was recovered, fixed in paraformaldehyde 4% for 6 to 8 hours and incubated in 20% sucrose at 4°C for >72 hours. The brain was cut on a frozen microtome in 35-μm-thick sections and stained. For immunohistochemistry, free-floating sections were first incubated for 30 minutes in 3% H2O2 and blocked with 5% normal horse serum, 10 μg/mL 2.4G2 antibody (to block mouse Fc receptors) and 0.1% Triton X-100 in PBS. Sections were then incubated overnight at 4°C, with goat anti-hIgG antibody (heavy and light chain specific, Jackson ImmunoResearch Laboratories, West Grove, PA, USA). After the overnight incubation, sections were washed in PBS and incubated for 1 hour with biotin-conjugated horse anti-goat antibody (heavy and light chain specific; Vector Laboratories, Burlington, ON, Canada) at room temperature. The sections were further washed and placed in a solution containing an avidin/HRP complex (ABC Elite kit, Vector Laboratories) for 1 hour at room temperature. The bound peroxidase was revealed with 0.5 mg/mL 3,3′-diaminobenzidine (Sigma-Aldrich) and 0.01% hydrogen peroxide in 0.05M mol/L Tris (pH 7.6). Extensive washings in PBS stopped the reaction. The sections were counterstained with cresyl violet, dehydrated, and coverslipped with dibutyl phthalate xylene as mounting media. For immunofluorescence labeling, goat anti-hIgG, mouse anti-NeuN (Chemicon, EMD Millipore, Billerica, MA, USA), and rat anti-CD31 (BioLegend, San Diego, CA, USA), were used as primary antibodies for the staining of IVIg, neurons, and endothelial cells, respectively, followed by detection with the appropriate AF-labeled secondary donkey antibody (all from Life Technologies, Burlington, ON, Canada). The 2.4G2 antibody was added to the blocking buffer and labeling steps to block mouse Fc receptors. The nuclei were counterstained with 4′, 6-diamidino-2-phenylindole (ThermoFisher Scientific, Rockford, IL, USA). Finally, sections were mounted on SuperFrost Plus slides, treated with 0.5% Sudan black (in 70% methanol) for 5 minutes, and coverslipped with Mowiol mounting media. Confocal laser scanning microscopy was performed with an Olympus IX81 multispectral FV1000 confocal microscope, equipped with 405 nm, 488 nm, 546 nm, and 633 nm laser lines (Olympus America, Richmond Hill, ON, Canada). Images were acquired and processed using the Olympus FluoView ASW 3.1a and the Bitplane Imaris 7.5 Softwares (Bitplane, South Windsor, CT, USA). Maximum intensity projections were calculated using the surpass module. An analysis of each section was performed in a blinded fashion.
Enzyme-Linked Immunosorbent Assay and Immunoblot
The concentrations of human and mouse IgG (mIgG) were determined by species-specific enzyme-linked immunosorbent assay using IgG Fc-specific antibodies for capture and the corresponding HRP-conjugated antibodies for detection (Jackson ImmunoResearch Laboratories). For immunoblots, proteins from 5 × 106 dissociated brain cells were heated at 95°C for 5 minutes in Laemmli loading buffer, separated by SDS-Page on a 10% polyacrylamide gel, transferred on a polyvinylidene difluoride membrane (Immobilon-P, EMD Millipore, Billerica, MA, USA) and blocked in 5% non-fat dry milk, 0.5% bovine serum albumin, 0.1% tween 20 in PBS buffer as previously described.12 Proteins were detected using the following primary antibodies: anti-NeuN, anti-PDGFR (receptor for platelet-derived growth factor), anti-GFAP (Sigma-Aldrich), anti-oligodendrocyte specific antibody (Abcam, Toronto, ON, Canada), anti-collagen IV (Chemicon), and anti-actin (ABM, Richmond, BC, Canada) followed by HRP-labeled secondary antibodies and chemiluminescence reagents (Lumiglo Reserve, KPL, Gaithersburg, MD, USA). Immunoblots were analyzed with a KODAK Imaging Station 4000MM Digital Imaging System (Molecular Imaging Software version 4.0.5f7, Carestream Health, Rochester, NY, USA).
Pharmacokinetic and Statistical Analyses
Pharmacokinetic analyses were performed in accordance with the standard procedure17 and using Prism 5.0d software (GraphPad Software, La Jolla, CA, USA). Group means are presented with standard error of the mean (s.e.m.). A Bartlett test was executed on all data to test for equal variances. Statistical differences were determined using the appropriate one-way analysis of variance or Kruskal–Wallis nonparametric tests and post hoc tests for comparison between groups. The threshold for statistical significance was set to P<0.05. All statistical analyses were performed using the JMP software (version 9.0.2; SAS Institute, Cary, IL, USA) and Prism 5.0d.
Results
Pharmacokinetic Profile of Intravenous Immunoglobulin in Mouse Models
The use of IVIg in mouse models of human diseases has been widely reported.10, 11, 12 However, scarce pharmacokinetic data are available. In humans, the half-life of IVIg in plasma is evaluated to be 35 days6 but the quantification of tissue distribution, specifically in the brain, remains technically challenging. We first undertook to evaluate the time-related distribution of human IVIg in plasma and tissues from young C57Bl/6 mice after a single i.p. injection of 25 mg IVIg (Figure 1A, Table 1). In the plasma, a half-life of 90 hours was determined experimentally with a T-max of 6 hours. We also investigated the time course of hIgG distribution in mouse tissues after removal of contaminating blood by intracardiac perfusion. This procedure efficiently removes blood from cerebral tissue but is generally less complete with highly irrigated organs, such as the liver or spleen. Similar to plasma, the IVIg reached a maximum concentration at around 6 hours in the liver and the spleen, suggesting a high vascularization and fast distribution equilibrium (Figure 1A, Table 1). However, the blood-to-brain IVIg transport was delayed in comparison with peripheral tissues (Figure 1A, Table 1) with a maximum concentration (C-max) being reached 24 hours after IVIg administration. Accordingly, a low brain elimination rate constant (Ke) was measured from experimental data (Table 1). From these results, we also calculated the percentage of the injected dose retained in each type of tissue under analysis. In the brain, we evaluated that 0.009±0.001% and 0.0017±0.0005% of systemically administered IVIg were distributed to the cortex and hippocampus of injected animals (Table 1). These results provide a quantitative assessment of the amount of hIgG in the brain after a systemic injection of IVIg and suggest a limited but significant accumulation of IVIg in the brain.
Figure 1.

Biodistribution of human and mouse immunoglobulin G (IgG) in the plasma and tissue homogenates. (A) Pharmacokinetic evaluation of human IgG (hIgG) distribution in homogenates of brain and peripheral tissues after a single intraperitoneal (i.p.) injection of 25 mg intravenous immunoglobulin (IVIg) in 4- to 6-week-old C57Bl/6 females (n=3–5 per time point). (B) A single i.p. injection also leads to a reduction of brain endogenous mouse IgG (mIgG). (C) Subchronic treatment: 9- to 13-month-old nontransgenic (nontg) and 3 × Tg-AD mice (n=6–8) received three injections of IVIg (two i.p. injections of 30 mg (96 and 24 hours before killing) and one intravenous injection ((75 minutes before killing) of 20 mg) or equivalent volumes of vehicle (control) and were perfused intracardially with 50 mL phosphate-buffered saline (PBS) buffer under deep anesthesia. (D) Chronic treatment: nontg and 3xTg-AD mice (n=11–13) were injected twice a week for 1 month (nine i.p. injections) with 1.5 g/kg IVIg and killed at 16 months of age, using intracardiac perfusion. Results are presented as means +/− s.e.m. Statistical analyses: one-way analysis of variance followed by Dunnett's multiple comparison test. mIgG: ∞P<0.05, ∞∞P<0.01; hIgG: *P<0.05, **P<0.01 versus controls. (A–C) Spleen, liver, cortex, and hippocampus homogenates were prepared using five volumes of radio immunoprecipitation assay buffer/mg tissue. The quantification of hIgG and mIgG was performed using specific enzyme-linked immunosorbent assays.
Apparent Competition Between Human Immunoglobulin G and Endogenous Mouse Immunoglobulin G to Enter the Brain
Interestingly, subsequent to the injection of IVIg, mIgG was reduced by 47% in the cortex and 42% in the hippocampus after 96 hours, suggesting that IVIg competes with mIgG to reach the brain (Figure 1B). To characterize the accumulation of IVIg in the brain and peripheral tissues after repeated injections in 3xTg-AD mice, hIgG concentration was evaluated and compared with endogenous mIgG after a subchronic treatment. The expression of AD-related transgenes in the 3xTg-AD mouse did not influence the entry of IVIg in the brain and peripheral tissues. The concentration of hIgG in the brain rose to 11.6±0.4 and 10.8±1.0 μg hIgG/g tissue in the cortex of NonTg and 3xTg-AD animals, 75 minutes after the last of a series of subchronic injections (Figure 1C). A similar mean concentration of 10.3±1.7 μg mIgG/g tissue was observed in the cortex of controls at the same time point (Figure 1C). In parallel with our pharmacokinetic data, endogenous mIgG concentrations were decreased by 40.4% and 41.2% in the cortex and hippocampus of IVIg mice, suggesting a regulation of mouse and human Ig entry into the CNS under normal- and AD-like conditions. To reproduce the chronic treatment conditions normally used in human trials, we also quantified the IVIg in the cortex after 1-month treatment. Interestingly, the chronic regimen led to a further increase in hIgG concentrations up to 18.3±1.2 and 20.2±1.7 μg hIgG/g tissue in 16-month-old NonTg and 3xTg-AD animals, respectively (Figure 1D).
Brain Uptake of Intravenous Immunoglobulin is a Saturable Process
Furthermore, we quantified the IVIg brain uptake using an ISCP method (Figure 2A).15 This technique was performed by injecting IVIg directly into the carotid artery, allowing almost 100% of the substance under study to quickly reach the cerebrovasculature. As expected, we measured a dose-dependent increase of IVIg concentration in the brain (Figure 2B). However, increasing the concentration was associated with a decreased percentage of hIgG reaching the brain (Figure 2C), evidencing a saturation of IVIg passage through the BBB. The Kin of IVIg was also reduced from 0.007±0.001 (500 μg IVIg) to 0.002±0.0002 μl/g per second (12500 μg IVIg) (Figure 2D). We further evaluated the distribution volume of sucrose to control the BBB integrity after the administration of IVIg. Indeed, as sucrose does not cross the BBB, enhanced permeability would result in an increased distribution volume of sucrose, which was not the case in this study (Figure 2E). Therefore, ISCP data strongly suggest that a saturable transport mechanism underlies IVIg uptake through the BBB.
Figure 2.

Quantification of intravenous immunoglobulin (IVIg) brain uptake: saturation of blood–brain barrier (BBB) transport. Mice were deeply anesthetized and (A) in situ cerebral perfusion was performed through the carotid artery, at a flow rate of 2.5 mL/minute for 60 seconds with the perfusate (3H-inulin with IVIg or vehicle) followed with a washing step of 120 seconds. 3H-inulin served as a control for the washing step and was below the detection limit in all mice. (B) The brain concentration of IVIg increased while (C) brain IVIg uptake and (D) brain transport coefficient (Kin) decreased with increasing doses of IVIg, suggesting a saturation of blood-to-brain transport. (B–D) Intravenous immunoglobulin was detected using an anti-human immunoglobulin G (IgG)-specific enzyme-linked immunosorbent assay. (E) 14C-sucrose was added to the washing buffer to measure the effects of IVIg on the BBB integrity. Intravenous immunoglobulin did not increase the vascular volume (VD) of 14C-sucrose, suggesting no modification in BBB permeability. Each data point represents a single individual (n=4–7 per group). Statistical analysis: one-way analysis of variance with Tukey's multiple comparison test *P<0.05, **P<0.01 versus 500 μg IVIg.
Localization of Intravenous Immunoglobulin in Brain Microvessels and Cells after Systemic Administration
Immunohistochemical staining was used to localize IVIg in the CNS. We observed significant immunoreactivity for hIgG throughout the brain parenchyma (Figure 3A) after a subchronic treatment compared with controls, despite the fact that it was largely excluded from the white matter in 3xTg-AD and NonTg animals. Interestingly, an important proportion of hIgG-immunopositive cells were noticeable in the cortex of treated mice. To confirm hIgG localization in the brain, confocal microscopy was also performed. Intravenous immunoglobulin staining (Figure 3B) was strongly colocalized with the endothelial cell marker CD31. Remarkably, hIgG immunostaining also colocalized with NeuN, a neuronal marker. Results obtained in 4- to 9-month-old 3xTg-AD and age-matched NonTg mice were similar, despite the fact that amyloid plaques were clearly detectable at that age (Figure 3C).13 Taken together, our findings indicate that IVIg crosses the BBB reaching the brain parenchyma, and hIgG-immunopositive endothelial cells and neurons were identified in 3xTg-AD and NonTg mice. As non-specific immunostaining was detected in the vehicle animals, mostly in the circumventricular organs, these regions were excluded from all subsequent analyses and photomicrographs (Figure 3A).
Figure 3.

Intravenous immunoglobulin (IVIg) was detected in the capillary endothelial cells and neurons after systemic administration in mice. (A and B) Nontransgenic (NonTg) and 3xTg-AD mice received three injections of IVIg (subchronic treatment) or equivalent volumes of vehicle (Ctrl) and were perfused with 50 mL phosphate-buffered saline (PBS) buffer followed with 50 mL 4% paraformaldehyde. Representative (A) immunohistochemistry and (B) immunofluorescent labeling of IVIg on 35-μm-thick brain sections of 4- to 9-month-old 3xTg-AD and NonTg mice (4–8 animals per group). Intravenous immunoglobulin was detected throughout the brain of injected mice, in capillaries, brain parenchyma, and brain cells. Immunohistochemistry: sections from the brains of IVIg-treated and control mice were mounted in alternation on the same slide. The three slides represent six independent animals. Photomicrographs were taken with a MicroFire 1.0 camera (Optronics, Goleta, CA, USA) linked to an E800 Nikon 274 microscope using the imaging software Picture Frame. Immunofluorescent staining: IVIg-treated and control 3xTg-AD animals are presented. Secondary antibodies conjugated with AF-488 for the detection of CD31 (endothelial cells in green), AF-565 for NeuN (neurons in red), AF-647 for human IgG (white) and 4′,6-diamidino-2-phenylindole (nucleus in blue). Confocal laser scanning microscopy was performed at room temperature with an Olympus IX81 multispectral FV1000 confocal microscope, equipped with 405 nm, 488 nm, 546 nm and 633 nm laser lines (Olympus America Inc., Richmond Hill, ON, Canada). Images were acquired sequentially with 2 lines Kalman integration using a × 60 OSC NA1.4 objective lens, and some regions of interest were acquired at zoom × 2.5. Image stacks were then imported in Bitplane Imaris 7.5 software (Bitplane, South Windsor, CT, USA). Maximum intensity projections were calculated using the surpass module and were exported for each channel as TIF files. (C) Age-dependent progression of the amyloid neuropathology in the 3xTg-AD mice. Four, 12, and 19-month-old 3xTg-AD mice were perfused with PBS buffer. Representative immunofluorescence labeling of amyloid plaques was detected on 12-μm-thick brain sections using 6E10 antibody (specific to amino acid 1 to 16 of APP) with a i90 Nikon fluorescence microscope (Nikon, Québec, QC, Canada) coupled to a Hamamatsu 1394 ORCA-285 monochrome camera, exploited by Simple PCI software version 5.3.0.1102 (Compix Imaging Systems, Cranberry, PA, USA) (n=3–5 animals per group).
To confirm the immunolabeling results, we next dissociated brain cells from cerebral tissue of IVIg-treated mice and verified the isolation of astrocytes, oligodendrocytes, endothelial cells, pericytes, and neurons by immunoblot, thus validating the efficiency of the dissociation process (Figure 4A). To quantify the number of IVIg molecules per cell, we prepared cell lysates from 1 × 107 cells and performed a hIgG-specific enzyme-linked immunosorbent assay. In these conditions, 5842±2021 and 4236±803 molecules/cell were detected in NonTg and 3xTg-AD animals after a subchronic treatment (Figure 4B). Again, no statistical differences were observed between 3xTg-AD and NonTg mice. These data are supportive of a direct interaction between IVIg and brain cells.
Figure 4.

Quantification of intravenous immunoglobulin (IVIg) binding to brain cells. (A) Brain cells were isolated from 3xTg-AD and NonTg mice using NeuroCult Enzymatic Dissociation Kit for Adult CNS Tissue (StemCell Technologies, Vancouver, BC, Canada). Immunoblot was used to confirm the presence of a variety of cell types after dissociation with specific cell markers: PDGFR for pericytes, NeuN for neurons, GFAP for astrocytes, oligodendrocyte specific antigen (OligoD) for oligodendrocytes, Collagen IV (Col IV) for endothelial cells and actin as a control for the homogeneity of the loading (each lane represents an individual animal). (B) To determine the number of IVIg molecules per cell, cell lysates from 1 × 107 cells of n=3 mouse brains were prepared in radio immunoprecipitation assay buffer and human IgG-specific enzyme-linked immunoprecipitation assay was performed. Statistical analysis is as follows: unpaired t-test P=0.5012. NS=not significant.
Discussion
To unravel the mechanisms of action and identify potential pharmacologic targets for IVIg in AD and other neurological disorders, it is crucial to determine whether IVIg is limited to the periphery or is able to cross the BBB and reach therapeutic targets directly in the brain. Published evidence of systemically administered IVIg exerting CNS effects in animal models hints toward central bioavailability.10, 11, 12 However, BBB passage of IVIg remains poorly defined. In this work, we quantified the migration of systemically administered hIgG to the brain, in the absence of BBB-enhanced permeability.
Our results are consistent with an accumulation of IVIg in the brain, albeit in a small percentage of the injected dose. Previous experiments with monoclonal antibodies injected directly into the parenchyma in rats indicate a half-life of elimination in the brain of approximately 45 minutes.18 In contrast, our findings rather show a low elimination rate constant in the hippocampus and cortex of IVIg-treated animals and, thus, a long half-life in the brain. Our experiments also demonstrate that the absorption rate of IVIg is slower in the brain than in peripheral tissues. In a previous study in the mouse, Bard et al19reported half-lives of 6 days for mouse monoclonal IgG in the serum and 50 days in the brain compared with 90 hours (3.8 days) and 117 to 140 hours (4.9–5.8 days) as calculated here for IVIg. In addition, a blood clearance half-life of 20 to 30 hours was documented for polyclonal anti-Aβ antibodies purified from human IVIg when assayed in the APP23 mouse model of AD.20 Differences in the experimental design such as the use of purified polyclonal or monoclonal IgG, time points chosen, and antibody doses could explain these dissimilarities.
We determined the percentage of injected IVIg migrating to the CNS to provide a quantitative assessment of the capacity of IVIg to interact with therapeutic targets within the brain. We report that 0.009±0.001% and 0.0017±0.0005% of systemically administered IVIg reached the cortex and hippocampus of animals after a single injection. Surprisingly, although IgG are commonly considered for the treatment of CNS diseases, very few quantitative data on their brain bioavailability can be found in the literature. Statements of ∼0.1% of injected IgG reaching the brain in the mouse are often made,21, 22 referring to results mentioned as data not shown in a report published in 200023 or measured from relative % of injected dose per gram of brain from a single time point (1 hour post injection).24 In most of these analyses, the concentration was obtained from biotinylated or radiolabeled injected Ig. As stated by Bard et al,19 it is unknown how much of the radioactive (or biotin) counts were associated with intact antibody and how much antibodies were engaged in the process of degradation. In contrast, here we performed human-specific enzyme-linked immunosorbent assay that requires intact Fc fragments. Additionally, a brain uptake of 0.06% injected dose per gram of tissue is reported using a radiolabeled rat mAb administered through the jugular vein, but with blood radioactivity included in the evaluation of brain IgG content.25 Thus, although the present percentage of injected IgG found in the brain after a systemic injection appears lower than traditionally accepted numbers, it is important to underscore that our assessment was calculated from the total dose of injected IVIg instead of plasma IgG at a specific time point. Our data also represent an absolute percentage of IgG (not per gram of brain tissue) and was measured after intracardiac perfusion to eliminate the blood from the brain vasculature. Nevertheless, extrapolating from these percentages and assuming comparable transport dynamics in humans, one can estimate that, from a 28 g injected dose,8 roughly 250 mg and 50 mg reach the cortex and hippocampus, respectively. This would correspond to concentrations approximating 10−7 to 10−5 mol/L of cerebral interstitial fluids. The interaction between monoclonal antibodies and antigens display high specificity and affinity, with equilibrium dissociation ranging from 10−11 to 10−4 mol/L.26, 27 Therefore, if we assume similar affinity of IVIg for CNS targets, the brain concentrations measured here after systemic injections are therapeutically relevant.
Data generated from ISCP experiments evidenced a saturable brain uptake of IVIg, strongly suggesting that transporters are involved in its transfer across the BBB. Such observations are in agreement with previous investigation using polyclonal IgG in guinea pigs, in which the brain transport of Ig isolated from guinea pigs is saturated at blood physiologic concentrations28 but contradicts other data showing absence of saturation when small doses (up to 30 mg/kg) of monoclonal mIgG were injected.19 In our study, higher concentrations of IVIg in the cortex and hippocampus were also associated with a reduction of mIgG, further suggesting that IgG are transported through the BBB by a saturable mechanism, in a species-independent manner. In light of the determination of a resident half-life of 45 minutes for monoclonal IgG18 and the observed reduction of brain mIgG after systemic injections of IVIg, our data suggest a regulated influx/efflux of IgG at the BBB.
Little is known about the transporters, receptors, or adhesion molecules implicated in IVIg entry into the CNS. In vitro and in vivo studies have involved FcRn and LRP-1,29, 30 both expressed at the BBB, as possible candidates for specific transport of antibodies. Recent studies indicate that IgG binding to transferrin or insulin receptors can access brain cells by mimicking their normal substrates.31 This strategy was used to engineer chimeric antibodies to promote blood-to-brain transport of therapeutic molecules.32 It is possible that some of the IgG molecules in the IVIg preparation might access the brain because of their antigen specificity to such transcytosis systems. Indeed, IVIg is a heterogeneous product representative of the hIgG repertoire of thousands of healthy donors5 with highly variable amino-acid composition.33 Thus, a fraction of these IgG molecules may cross the BBB because of characteristics such as their isoelectric point,34 degree of hydrophobicity, amino-acid composition, or antigen specificity,31 whereas the majority might be excluded from the brain. Thus, our data underscore the possibility that deciphering the characteristics of IVIg molecules that allows their migration into the CNS as well as the transporters involved could lead to the identification of brain-penetrant IgG and to the optimization of IVIg use in prevalent CNS disorders.
Differences between mouse and human antibodies, IgG subtypes for example, have been reported and should be taken into account when interpreting the present data. Indeed, in the mouse, the isotypes are IgG1, IgG2a (or IgG2c, depending on the strain used), IgG2b, and IgG3 whereas in humans, the four subtypes are as follows: IgG1, IgG2, IgG3, and IgG4. Although subtypes differ in their interactions with Fcγ receptors, in mouse models of AD and autoimmune diseases, treatments with hIgG or mIgG resulted in similar therapeutic action.35, 36, 37 Variations in glycosylation are also another important species-specific distinction. IgG is a glycoprotein with oligosaccharides present in the Fc and Fab regions. Species-related microheterogeneity has been reported and can affect the binding abilities.38, 39 In both humans and mice, the only known Fcγ receptors expressed by endothelial cells is FcRn.35 Nevertheless, other unknown receptors/transporters, for which the impact of species-related differences is still unexplored, could be implicated in the observed blood-to-brain transport. Because of these species-specific differences, our study presents some limitations. On the one hand, it is possible that hIgG incapable of targeting human BBB receptors could nevertheless specifically recognize their murine counterparts via their Fab region, thus artificially increasing their apparent transfer to the brain. However, the species-related affinity for the Fcγ receptors might rather decrease hIgG transport in the mouse CNS. Therefore, the present data on IVIg transport to the brain can be extrapolated to human only with caution.
Although our in situ brain perfusion data confirmed a direct transport of IVIg through the BBB, it should be noted that in theory, a subset of systemically administered IVIg can access the brain through the blood–CSF and the CSF–brain barriers.2 However, the surface of the CSF–brain barrier at plexus choroid is very small (in humans, 0.021 m2) compared with the BBB (in humans, ∼20 m2).4 Moreover, the diffusion of large molecules such as hIgG from the CSF to the brain is extremely slow with a range limited to a few millimeters.40 In contrast, the brain microvasculature is dense with an inter-capillary distance of ∼40 μm, only enough space for two neurons. This suggests that every neuron in the brain is perfused by its own capillary.4 The homogeneous and generalized staining pattern observed in our IHC analyses further argues against blood–CSF–brain recirculation, as the sole mechanism of brain transport of IVIg. Nevertheless, it should not be ruled out that part of the IVIg found in the brain after systemic injection may have traveled through the CSF.
To analyze the distribution of IVIg in the mouse brain, we used a subchronic treatment, to visualize IgG molecules that enter the brain during the absorption (75 minutes time point) and elimination phases (96 hour) as well as those present in the brain at C-max (24 hours), in the same experiment. Using these conditions, IVIg was detected in the cerebral microvessels and in the brain parenchyma. With dissociated brains, we further demonstrated that a small proportion of IVIg was bound to brain cells further suggesting that IVIg can interact with therapeutic targets in the CNS. Interestingly, increased hIgG concentrations were observed after administration of a chronic regimen, consistent with an accumulation of IVIg in the brain.
The brain uptake of IVIg did not differ between 3xTg-AD and NonTg mice, consistent with previous data with purified anti-Aβ administered to APP23 transgenic mice.20 This is also in agreement with the fact that no changes in BBB functional integrity and permeability were detected in the 3xTg-AD mouse.15 In contrast, other studies suggest that antibodies against Aβ accumulate preferentially in amyloid-rich brain of PDAPP and Tg2576 mice modeling AD, likely due to binding to the massive amount of Aβ plaques in these animals.19, 41 Such difference might reflect a low content in antibodies reactive against brain-derived Aβ in the present IVIg preparation, although significant concentrations of Aβ-specific autoantibodies have been reported in most IVIg formulations including Gamunex.42 Other explanations may derive from differences between IVIg preparations and monoclonal and polyclonal anti-Aβ antibodies or because we used 3xTg-AD mice at an age where Aβ plaques are not yet massively formed (Figure 3C).13, 14, 15 Although neurovascular dysfunctions have been described in AD and in a few animal models,43, 44 we have previously confirmed that BBB function and permeability are not significantly altered in the 3xTg-AD mice at 10 to 11 months.15 Altogether, our data suggest that AD-like neuropathology did not alter the passage of IVIg across the BBB.
In summary, our data demonstrate that a significant fraction of systemically administered IVIg reaches the cerebral microvessels and enters the brain through a saturable transport across the BBB. To our knowledge, this study is the first to provide quantitative measurement of IVIg brain entry and suggests that IVIg may interact with therapeutic targets within the CNS, in the absence of BBB leakage. Finally, further analysis is warranted to identify specific subpopulations of purified hIgG molecules with the ability to reach the brain and to decipher the underlying transport mechanisms.
Acknowledgments
The authors are grateful to Catherine Routhier for preliminary work on IVIg quantification in the brain, to Mélissa Ouellet for providing the sketch for ISCP description and to Dr Vincent Emond and Dr Charlotte Delay for their valuable editing of this manuscript.
DISCLAIMER
The authors have not received any royalties from Grifols or any other IVIg-producing companies and declare that they have no competing financial interests.
The authors declare no conflict of interest.
Footnotes
The work was supported in part by an unrestricted peer-reviewed grant from Grifols. This work was supported by grants to FC and RB from the Canadian Institutes of Health Research (grant ISO-102447), the Héma-Québec Foundation and Grifols. IS was supported by an Industrial Innovation PhD scholarship from CRSNG/FQRNT/Héma-Québec. FC is supported by a salary award from the Fonds de la recherche en santé du Québec (FRQ-S).
References
- Ballabh P, Braun A, Nedergaard M. The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis. 2004;16:1–13. doi: 10.1016/j.nbd.2003.12.016. [DOI] [PubMed] [Google Scholar]
- Johanson CE, Duncan JA, Stopa EG, Baird A. Enhanced prospects for drug delivery and brain targeting by the choroid plexus-CSF route. Pharm Res. 2005;22:1011–1037. doi: 10.1007/s11095-005-6039-0. [DOI] [PubMed] [Google Scholar]
- Johanson CE, Stopa EG, McMillan PN. The blood-cerebrospinal fluid barrier: structure and functional significance. Methods Mol Biol. 2011;686:101–131. doi: 10.1007/978-1-60761-938-3_4. [DOI] [PubMed] [Google Scholar]
- Pardridge WM. Drug transport in brain via the cerebrospinal fluid. Fluids Barriers CNS. 2011;8:7. doi: 10.1186/2045-8118-8-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelfand EW. Intravenous immune globulin in autoimmune and inflammatory diseases. N Engl J Med. 2012;367:2015–2025. doi: 10.1056/NEJMra1009433. [DOI] [PubMed] [Google Scholar]
- Ballow M, Berger M, Bonilla FA, Buckley RH, Cunningham-Rundles CH, Fireman P, et al. Pharmacokinetics and tolerability of a new intravenous immunoglobulin preparation, IGIV-C, 10% (Gamunex, 10%) Vox Sang. 2003;84:202–210. doi: 10.1046/j.1423-0410.2003.00286.x. [DOI] [PubMed] [Google Scholar]
- Fillit H, Hess G, Hill J, Bonnet P, Toso C. IV immunoglobulin is associated with a reduced risk of Alzheimer disease and related disorders. Neurology. 2009;73:180–185. doi: 10.1212/WNL.0b013e3181ae7aaf. [DOI] [PubMed] [Google Scholar]
- Relkin NR, Szabo P, Adamiak B, Burgut T, Monthe C, Lent RW, et al. 18-Month study of intravenous immunoglobulin for treatment of mild Alzheimer disease. Neurobiol Aging. 2008;30:1728–1736. doi: 10.1016/j.neurobiolaging.2007.12.021. [DOI] [PubMed] [Google Scholar]
- Mayeux R, Stern Y. Epidemiology of Alzheimer disease. Cold Spring Harbor Perspect Med. 2012;2:pii: a006239. doi: 10.1101/cshperspect.a006239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magga J, Puli L, Pihlaja R, Kanninen K, Neulamaa S, Malm T, et al. Human intravenous immunoglobulin provides protection against Abeta toxicity by multiple mechanisms in a mouse model of Alzheimer's disease. J Neuroinflammation. 2010;7:90. doi: 10.1186/1742-2094-7-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen SH, Storm N, Jensen PEH, Laursen H, Sorensen PS. IVIg enters the central nervous system during treatment of experimental autoimmune encephalomyelitis and is localised to inflammatory lesions. Exp Brain Res. 2006;178:462–469. doi: 10.1007/s00221-006-0752-8. [DOI] [PubMed] [Google Scholar]
- St-Amour I, Bousquet M, Pare I, Drouin-Ouellet J, Cicchetti F, Bazin R, et al. Impact of intravenous immunoglobulin on the dopaminergic system and immune response in the acute MPTP mouse model of Parkinson's disease. J Neuroinflammation. 2012;9:234. doi: 10.1186/1742-2094-9-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, et al. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM. Intraneuronal Abeta causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron. 2005;45:675–688. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- Bourasset F, Ouellet M, Tremblay C, Julien C, Do TM, Oddo S, et al. Reduction of the cerebrovascular volume in a transgenic mouse model of Alzheimer's disease. Neuropharmacology. 2009;56:808–813. doi: 10.1016/j.neuropharm.2009.01.006. [DOI] [PubMed] [Google Scholar]
- Hawkins BT, Egleton RD. Pathophysiology of the blood-brain barrier: animal models and methods. Curr Top Dev Biol. 2008;80:277–309. doi: 10.1016/S0070-2153(07)80007-X. [DOI] [PubMed] [Google Scholar]
- Leblanc P-P, Aiache J-M, Besner J-G, Buri P, Lesne M.Traite de Biopharmacie et Pharmacocinetique3rd edn.Les Presses de L'Universite de Montreal: Montreal; 1997. p 396. [Google Scholar]
- Zhang Y, Pardridge WM. Mediated efflux of IgG molecules from brain to blood across the blood-brain barrier. J Neuroimmunol. 2001;114:168–172. doi: 10.1016/s0165-5728(01)00242-9. [DOI] [PubMed] [Google Scholar]
- Bard F, Fox M, Friedrich S, Seubert P, Schenk D, Kinney GG, et al. Sustained levels of antibodies against Abeta in amyloid-rich regions of the CNS following intravenous dosing in human APP transgenic mice. Exp Neurol. 2012;238:38–43. doi: 10.1016/j.expneurol.2012.07.022. [DOI] [PubMed] [Google Scholar]
- Bacher M, Depboylu C, Du Y, Noelker C, Oertel WH, Behr T, et al. Peripheral and central biodistribution of (111)In-labeled anti-beta-amyloid autoantibodies in a transgenic mouse model of Alzheimer's disease. Neurosci Lett. 2009;449:240–245. doi: 10.1016/j.neulet.2008.08.083. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, Rojiani A, Rosenthal A, Levkowitz G, Subbarao S, Alamed J, et al. Passive amyloid immunotherapy clears amyloid and transiently activates microglia in a transgenic mouse model of amyloid deposition. J Neurosci. 2004;24:6144–6151. doi: 10.1523/JNEUROSCI.1090-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levites Y, Smithson LA, Price RW, Dakin RS, Yuan B, Sierks MR, et al. Insights into the mechanisms of action of anti-Abeta antibodies in Alzheimer's disease mouse models. FASEB J. 2006;20:2576–2578. doi: 10.1096/fj.06-6463fje. [DOI] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- Banks WA, Terrell B, Farr SA, Robinson SM, Nonaka N, Morley JE. Passage of amyloid beta protein antibody across the blood-brain barrier in a mouse model of Alzheimer's disease. Peptides. 2002;23:2223–2226. doi: 10.1016/s0196-9781(02)00261-9. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Engelhardt B, Lesley J, Bickel U, Pardridge WM. Targeting rat anti-mouse transferrin receptor monoclonal antibodies through blood-brain barrier in mouse. J Pharmacol Exp Ther. 2000;292:1048–1052. [PubMed] [Google Scholar]
- Rose RJ, Labrijn AF, van den Bremer ET, Loverix S, Lasters I, van Berkel PH, et al. Quantitative analysis of the interaction strength and dynamics of human IgG4 half molecules by native mass spectrometry. Structure. 2011;19:1274–1282. doi: 10.1016/j.str.2011.06.016. [DOI] [PubMed] [Google Scholar]
- Zhao Q, Feng Y, Zhu Z, Dimitrov DS. Human monoclonal antibody fragments binding to insulin-like growth factors I and II with picomolar affinity. Mol Cancer Ther. 2011;10:1677–1685. doi: 10.1158/1535-7163.MCT-11-0281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV, Skundric DS, Segal MB, Lipovac MN, Mackic JB, Davson H. A saturable mechanism for transport of immunoglobulin G across the blood-brain barrier of the guinea pig. Exp Neurol. 1990;107:263–270. doi: 10.1016/0014-4886(90)90144-h. [DOI] [PubMed] [Google Scholar]
- Proulx DP, Rouleau P, Pare I, Vallieres-Noel MM, Bazin R. Interaction between intravenous immunoglobulin (IVIg) and the low-density lipoprotein receptor-related protein 1: a role for transcytosis across the blood brain barrier. J Neuroimmunol. 2012;251:39–44. doi: 10.1016/j.jneuroim.2012.06.009. [DOI] [PubMed] [Google Scholar]
- Deane R, Sagare A, Hamm K, Parisi M, LaRue B, Guo H, et al. IgG-assisted age-dependent clearance of Alzheimer's amyloid beta peptide by the blood-brain barrier neonatal Fc receptor. J Neurosci. 2005;25:11495–11503. doi: 10.1523/JNEUROSCI.3697-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paris-Robidas S, Emond V, Tremblay C, Soulet D, Calon F. In vivo labeling of brain capillary endothelial cells after intravenous injection of monoclonal antibodies targeting the transferrin receptor. Mol Pharmacol. 2011;80:32–39. doi: 10.1124/mol.111.071027. [DOI] [PubMed] [Google Scholar]
- Atwal JK, Chen Y, Chiu C, Mortensen DL, Meilandt WJ, Liu Y, et al. A therapeutic antibody targeting BACE1 inhibits amyloid-beta production in Vivo. Sci Transl Med. 2011;3:84ra43. doi: 10.1126/scitranslmed.3002254. [DOI] [PubMed] [Google Scholar]
- Schroeder HWJ. Similarity and divergence in the development and expression of the mouse and human antibody repertoires. Dev Comp Immunol. 2006;30:119–135. doi: 10.1016/j.dci.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Triguero D, Buciak JL, Pardridge WM. Cationization of immunoglobulin G results in enhanced organ uptake of the protein after intravenous administration in rats and primate. J Pharmacol Exp Ther. 1991;258:186–192. [PubMed] [Google Scholar]
- Bruhns P. Properties of mouse and human IgG receptors and their contribution to disease models. Blood Blood. 2012;119:5640–5649. doi: 10.1182/blood-2012-01-380121. [DOI] [PubMed] [Google Scholar]
- Sudduth TL, Greenstein A, Wilcock DM. Intracranial injection of gammagard, a human ivig, modulates the inflammatory response of the brain and lowers abeta in APP/PS1 mice along a different time course than anti-abeta antibodies. J Neurosci. 2013;33:9684–9692. doi: 10.1523/JNEUROSCI.1220-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsman Y, Foo AH, Leontyev D, Branch DR. Improved mouse models for the study of treatment modalities for immune-mediated platelet destruction. Transfusion. 2010;50:1285–1294. doi: 10.1111/j.1537-2995.2009.02558.x. [DOI] [PubMed] [Google Scholar]
- Raju TS, Briggs JB, Borge SM, Jones AJ. Species-specific variation in glycosylation of IgG: evidence for the species-specific sialylation and branch-specific galactosylation and importance for engineering recombinant glycoprotein therapeutics. Glycobiology. 2000;10:477–486. doi: 10.1093/glycob/10.5.477. [DOI] [PubMed] [Google Scholar]
- Jefferis R. Glycosylation as a strategy to improve antibody-based therapeutics. Nat Rev Drug Discov. 2009;8:226–234. doi: 10.1038/nrd2804. [DOI] [PubMed] [Google Scholar]
- Wolak DJ, Thorne RG. Diffusion of macromolecules in the brain: implications for drug delivery. Mol Pharm. 2013;10:1492–1504. doi: 10.1021/mp300495e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock D, Rojiani A, Rosenthal A, Subbarao S, Freeman M, Gordon M, et al. Passive immunotherapy against Abeta in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J Neuroinflammation. 2004;1:24. doi: 10.1186/1742-2094-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishnan K, Andrei-Selmer L-C, Selmer T, Bacher M, Dodel R. Comparison of intravenous immunoglobulins for naturally occurring autoantibodies against amyloid-beta. J Alzheimers Dis. 2010;20:135–143. doi: 10.3233/JAD-2010-1353. [DOI] [PubMed] [Google Scholar]
- Bell RD, Zlokovic BV. Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer's disease. Acta Neuropathol. 2009;118:103–113. doi: 10.1007/s00401-009-0522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrall AJ, Wardlaw JM. Blood-brain barrier: ageing and microvascular disease—systematic review and meta-analysis. Neurobiol Aging. 2009;30:337–352. doi: 10.1016/j.neurobiolaging.2007.07.015. [DOI] [PubMed] [Google Scholar]
- Riches AC, Sharp JG, Thomas DB, Smith SV. Blood volume determination in the mouse. J Physiol. 1973;228:279–284. doi: 10.1113/jphysiol.1973.sp010086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushida M, Kamendulis LM, Peat TJ, Klaunig JE. Dose-related induction of hepatic preneoplastic lesions by diethylnitrosamine in C57BL/6 mice. Toxicol Pathol. 2011;39:776–786. doi: 10.1177/0192623311409596. [DOI] [PubMed] [Google Scholar]