

Abstract
FSHD2 is a rare form of facioscapulohumeral muscular dystrophy (FSHD) characterized by the absence of a contraction in the D4Z4 macrosatellite repeat region on chromosome 4q35 that is the hallmark of FSHD1. However, hypomethylation of this region is common to both subtypes. Recently, mutations in SMCHD1 combined with a permissive 4q35 allele were reported to cause FSHD2. We identified a novel p.Lys275del SMCHD1 mutation in a family affected with FSHD2 using whole-exome sequencing and linkage analysis. This mutation alters a highly conserved amino acid in the ATPase domain of SMCHD1. Subject III-11 is a male who developed asymmetrical muscle weakness characteristic of FSHD at 13 years. Physical examination revealed marked bilateral atrophy at biceps brachii, bilateral scapular winging, some asymmetrical weakness at tibialis anterior and peroneal muscles, and mild lower facial weakness. Biopsy of biceps brachii in subject II-5, the father of III-11, demonstrated lobulated fibers and dystrophic changes. Endomysial and perivascular inflammation was found, which has been reported in FSHD1 but not FSHD2. Given the previous report of SMCHD1 mutations in FSHD2 and the clinical presentations consistent with the FSHD phenotype, we conclude that the SMCHD1 mutation is the likely cause of the disease in this family.
1. Introduction
Facioscapulohumeral muscular dystrophy (FSHD) is the third most common myopathy, characterized by autosomal dominant inheritance and progressive, often asymmetrical weakness of the facial, scapular and upper arm muscles, typically with onset in childhood or adolescence. There are two forms of FSHD, FSHD1 (95%) and FHSD2 (5%), with indistinguishable clinical presentations [1]. FSHD is caused by chromatin relaxation of the D4Z4 macrosatellite repeat on chromosome 4, resulting in expression of the retrotransposed gene DUX4 in skeletal muscle [2], causing a toxic effect, either with contraction of the repeat (FSHD1) or without contraction (FSHD2).
Recently, it was reported that mutations in SMCHD1, which has a role in epigenetic gene silencing [3], cause many cases of FSHD2 [4]. In that study, whole-exome and follow-up Sanger sequencing led to the identification of 12 different heterozygous out-of-frame deletions, splice site mutations, and missense mutations in SMCHD1 in 15 out of 19 FSHD2 pedigrees. These mutations co-segregated with D4Z4 hypomethylation in an autosomal dominant pattern. Mutations p.Ser868Asn, p.Cys492Arg, p. Asp537Ilefs*10 and p.Thr1522Thr caused decreased SMCHD1 protein expression in patient fibroblasts, and p.Phe1554Ser mutant SMCHD1 had decreased binding to D4Z4 repeats compared to control protein. Knockdown of SMCHD1 caused activation of DUX4 expression in normal skeletal muscle cells, and DUX4 was expressed when SMCHD1 transcripts and protein levels were reduced to less than 50% of normal levels. The authors concluded that haploinsufficiency of SMCHD1 causes the hypomethylation of D4Z4 repeats and subsequent pathogenic DUX4 expression in these FSHD2 pedigrees.
Here we report a novel SMCHD1 mutation, p.Lys275del, which was identified by whole-exome sequencing and linkage analysis in a family affected with FSHD2. This mutation is the second mutation in SMCHD1 that resides in the ATPase domain of the encoded protein, and it is located upstream of all previously reported mutations. Both the mutation and D4Z4 hypomethylation co-segregate with the phenotype in our family, replicating the observation of di-locus inheritance of FSHD2. In addition, a muscle sample obtained from an affected individual at biopsy showed reproducible DUX4 expression. This report of SMCHD1-associated FSHD2 with inflammation expands the allelic and clinical heterogeneity of SMCHD1 mutations in the pathogenesis of FSHD2.
2. Patients and Methods
2.1. Patient samples
This study was approved by the Institutional Review Board of Boston Children’s Hospital. Written informed consent was obtained from all subjects.
2.2. Parametric linkage analysis
Individuals II-5, II-6, III-8, III-10 and III-11 were genotyped at 10,204 single nucleotide polymorphisms (SNPs) using the GeneChip Human Mapping 10K 2.0 Xba Array (Affymetrix). Parametric linkage analysis was performed using MERLIN v1.1.2 software under the assumption of autosomal dominant inheritance, as described previously [5].
2.3. Whole-Exome Sequencing and Data analysis
DNA samples were sequenced at the Genomic Diagnostic Laboratory and analyzed by the Interpretive Genomic Services team at Boston Children’s Hospital. Whole blood DNA was subjected to solution capture (SureSelect Human All Exon V4, Agilent Technologies) to generate barcoded whole-exome sequencing libraries. Libraries were sequenced on an Illumina HiSeq, employing paired end reads (100 bp × 2) to a mean target coverage of 20X, 58X, and 24X, resulting in 78%, 94%, and 78% of the target covered by ≥10 reads for subjects II-5, II-6, and III-10, respectively. Alignment, variant calling, and annotation were performed with a custom informatics pipeline employing BWA [6], Picard (http://picard.sourceforge.net), GATK [7], and ANNOVAR [8]. The human genome reference used for these studies was hg19/GRCh37. Single nucleotide changes, microdeletions, and microinsertions were reported and annotated using the NCBI and UCSC reference sequences and online genome databases (NHLBI ESP with ~5400 exomes, 1000 Genomes Project, dbSNP135, Complete Genomics 52). Species conservation was determined using the LRT and PhyloP programs, and predicted pathogenicity was determined using the SIFT and PolyPhen-2 programs.
2.4. Bisulfite Sequencing (BSS)
DNA methylation of the 4qA region was analyzed in subjects II-5, II-6, III-8 and III-10 by bisulfite sequencing (BSS) assay. Converted DNA was amplified by nested PCR using primers and conditions that specifically amplify 4qA and not 4qB, 10qA, or 10qB; initial PCR was performed with primers BSS1438F: GTTTTGTTGGAGGAGTTTTAGGA and BSS3742R: AACATTCAACCAAAATTTCACRAAA and then followed by nested PCR with primers BSS1438F and BSS3626R: AACAAAAATATACTTTTAACCRCCAAAAA using 10% of the first PCR product. Polymorphic nucleotide changes that preferentially amplify 4qA subtelomeric region are underlined. All PCRs were performed using GoTaq Hot Start Polymerase (Promega) as follows: 94°C 2 min, 25 cycles of 94°C 15 sec, 58°C 20 sec, and 72°C 40 sec, and a final 72°C for 10 min. The 594-bp PCR product spans from the end of full length DUX4 exon 1 to the beginning of exon 3, therefore specifically analyzing the methylation status of the most distal 4qA D4Z4 repeat, which contains 57 CpGs. PCR products were cloned with pGEM-T Easy Vector system I (Promega) for sequencing analysis. At least 8 clones were sequenced for each subject and their methylation status was analyzed using web-base analysis software BISMA (http://biochem.jacobs-university.de/BDPC/BISMA/) [9] with the default parameters.
2.5. DUX4 expression
Total RNA was isolated from biopsied biceps brachii muscle from subject II-5. Polyadenylated full length DUX4 mRNA was then detected by reverse transcription polymerase chain reaction (RT-PCR) as previously described [10].
3. Results
3.1. Patients
The pedigree is shown in Fig. 1. Subject III-11 had no developmental or neurological problems in childhood. At age 13 years, he first noticed arm weakness when lifting up groceries and experienced difficulty rising from chairs. Physical examination revealed marked bilateral atrophy at biceps brachii, bilateral scapular winging, some asymmetrical weakness at tibialis anterior and peroneal muscles, and mild lower facial weakness. Clinical genetic testing on subject III-11 using EcoRI and EcoRI-BlnI restriction endonuclease digestion, pulsed-field gel electrophoresis, and Southern blot analysis demonstrated a D4Z4 allele size > 48 kilobases for both alleles, with a normal range ≥ 42 kilobases, indicating that he does not have FSHD1. Serum creatine kinase (CK) level was 652 IU/L (normal 20–220). At age 20, he remained ambulatory but could not navigate stairs. Cardiac evaluation was normal. Subject II-5, the father of III-11, was diagnosed with FSHD at the age of 40 years. He had marked bilateral atrophy of the biceps brachii and scapular winging. A muscle biopsy at age 53 years revealed chronic dystrophic changes (Fig. 2a) with many lobulated fibers (Fig. 2b), some small angulated fibers (Supplemental fig. 1), and perivascular and endomysial inflammatory cell infiltration (Figs. 2c–d). Subject III-10, a brother of III-11, began to experience right shoulder weakness at the age of 23 years. He had right pectoral muscle atrophy and right scapular winging, but no facial muscle weakness. His serum CK levels were between 800–900 IU/L. At the age of 24, he developed intermittent blurry vision in his right eye.
Figure 1.
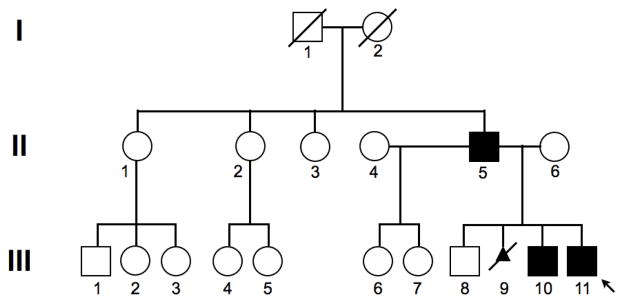
Pedigree of the family demonstrating the autosomal dominant pattern of inheritance. The arrow identifies the proband (subject III-11).
Figure 2.

A biopsy was obtained from the biceps brachii of an affected family member (II-5). Panels A to C represent images from frozen sections and panel D represents an image from a paraffin block section. All scale bars = 50 μm. (A) Hematoxylin and eosin (H&E) staining shows marked variation in fiber size including some hypertrophic fibers. Scattered regenerating fibers are seen (black arrow head). There is mild to moderate endomysial fibrosis. (B) NADH staining shows many lobulated fibers (white arrow head). (C) H&E staining demonstrates mild perivascular inflammatory cell infiltration (arrow). (D) H&E staining of a paraffin section shows moderate endomysial inflammatory cellular infiltration in some areas.
3.2. Linkage analysis and Whole-Exome sequencing
Linkage analysis demonstrated positive LOD scores at 23 genomic loci, all of which approached the maximum possible LOD score of 0.602. The 4q35 region was excluded by linkage analysis. Exome sequencing revealed more than 20,000 variants in each individual tested. There were no mutations detected in other genes that have been associated with FSHD-like phenotypes, including CAPN3, VCP, FHL1, and GAA. We excluded benign polymorphisms and filtered possible mutations (Fig. 3). Eighteen variants within the linkage peaks co-segregated with the phenotype among the three subjects whose exomes were sequenced (Supplemental Table 1).
Figure 3.

Exome sequencing revealed 23,952, 26,466, and 24,686 variants in individuals II-5 (affected), II-6 (unaffected) and III-10 (affected), respectively. After excluding reported SNPs in public databases (dbSNP 135 (http://www.ncbi.nlm.nih.gov/projects/SNP/), NHLBI Ground Opportunity Exome Sequencing Project (https://esp.gs.washington.edu/drupal/) and the 1000 Genomes Project (http://www.1000genomes.org/) and filtering for nonsynonymous, nonsense, or splice variants, or coding insertion/deletions, the numbers of candidate variants were reduced to 156, 183, and 161 in subjects II-5, II-6 and III-10, respectively. Among 60 variants that co-segregated with the phenotype of these three members of the family, there were 18 variants that were within the linkage peaks. Sanger sequencing of those 18 variants in II-5, II-6, and III-10, as well as in other family members (II-1, II-2, III-8 and III-11) confirmed the whole exome sequencing results and demonstrated that 3 of the variants co-segregated with the phenotype in the broader family (Supplemental Table 1).
3.3. Sanger sequencing and mutation analysis
Sanger sequencing of the 18 loci in those three subjects as well as in additional family members (II-1, II-2, III-8 and III-11) confirmed the 18 variants and also demonstrated that three of the variants co-segregated with the phenotype in the broader family (Fig. 3, Supplemental Table 1). Among these variants was the novel mutation c.823_825del, encoding the in-frame single amino acid deletion p.Lys275del, in SMCHD1 (Fig. 4a). This deleted amino acid localizes to the ATPase domain of SMCHD1 [4] and is conserved in multiple species (Fig. 4b). The amino acids affected by variants found in the two other genes, FAM105B and SETMAR, were not conserved (Supplemental Fig. 2) and these variants are predicted to be benign by in silico analysis (Supplemental Table 2).
Figure 4.

The novel SMCHD1 mutation was present in all affected family members. (A) Electropherogram of the SMCHD1 mutation loci of an unaffected (III-8) and affected (III-11) subject. Forward and reverse reads confirmed a three base pair deletion, c.823_825del, which results in the deletion of a single amino acid, p.Lys275del. (B) Lys275 in SMCHD1 is highly conserved through multiple species.
3.5. Bisulfite sequencing and DUX4 expression
Bisulfite sequencing analysis of DNA samples from subjects II-5, II-6, III-8, and III-10 showed decreased DNA methylation of the 4qA allele specifically in affected individuals, consistent with the diagnosis of FSHD2 (Table 1). Polyadenylated DUX4-full length mRNA expression was detected in biopsied muscle tissue obtained from affected subject II-5 (Supplemental fig. 3), whereas it is generally undetectable in muscle tissue from unaffected subjects[11, 12]. Therefore, the epigenetically altered 4qA allele for subject II-5 is permissive for FSHD as it contains a functional DUX4 polyadenylation signal in exon 3[2]. These data suggest that the FSHD2 in this family is likely to be caused by the novel SMCHD1 mutation p.Lys275del.
Table 1. Methylation of D4Z4 repeats in 4qA allele.
Methylation of D4Z4 repeats in 4qA allele in affected (II-5 and III-10) and unaffected (II-6 and II-8) subjects in the family. Bisulfite sequencing analysis shows DNA methylation of D4Z4 repeats in the 4qA allele is decreased in affected individuals compared to the unaffected ones.
| Patient | II-5 (affected) | II-6 (unaffected) | III-8 (unaffected) | III-10 (affected) |
|---|---|---|---|---|
| Methylation (%) | 20.4 | 48.6 | 49.7 | 11.1 |
4. Discussion
We report a novel mutation in SMCHD1 in a family with FSHD2. This second report of SMCHD1-associated FSHD2 independently replicates the previous report [4] using whole-exome sequencing and linkage analysis. In light of the di-locus inheritance pattern of FSHD, the exclusion of the 4q35 locus by linkage analysis can be explained by the inclusion of an unaffected child of an affected individual, i.e., III-8, in the linkage scan. This individual does not have the SMCHD1 mutation, but most likely has a permissive allele. In our family, other candidate mutations did not co-segregate with the phenotype or did not alter the sequence of well-conserved amino acids. Consistent with the previous report, our patients showed decreased DNA methylation of D4Z4 repeats at 4q35. We additionally showed that the DUX4 gene was expressed as its FSHD-associated polyadenylated full-length mRNA in biopsied muscle tissue obtained from an affected individual.
The novel p.Lys275del mutation deletes a lysine residue in exon 7, in the well-conserved ATPase domain of SMCHD1. The function of the ATPase domain in SMCHD1 is not understood but may have a role in chromatin interaction, as reported in studies of other SMC proteins [13]. In support of this hypothesis, the previous report demonstrated binding of SMCHD1 to D4Z4 by chromatin immunoprecipitation analysis [4]. They also showed that SMCHD1 protein levels are reduced in 7 cases with 4 different mutations outside the ATPase domain, indicating that haploinsufficiency contributes to the disease mechanism [4]. Among the previously reported mutations, only p.Tyr353Cys in exon 9 lies in the ATPase domain [4]. It is possible that both the p.Lys275del and p.Tyr353Cys mutations result in decreased ATPase activity in the protein due to haploinsufficiency of SMCHD1. This would be an interesting direction for future studies.
FSHD1 and FSHD2 have been reported to be clinically indistinguishable [1], consistent with our observations. In addition, the patient muscle pathology reported herein is consistent with FSHD, including the presence of lobulated fibers and small angulated fibers. Perivascular as well as endomysial inflammatory cell infiltration have been reported in FSHD1 patients [14] but not previously in SMCHD1-associated FSHD2; this is therefore the first observation of such inflammation in FSHD2. These data suggest that both FSHD1 and FSHD2 result from an epigenetic misregulation of chromosome 4q35 and share similar downstream pathological mechanisms.
Supplementary Material
Acknowledgments
The authors thank Catherine Brownstein, David Margulies, the staff of the Genetic Diagnostic Laboratory and the entire Interpretive Genomic Services team at Boston Children’s Hospital for facilitating sample analysis. This study was supported by the William Randolph Hearst Fund at Harvard Medical School (SM), Muscular Dystrophy Association (MDA) Research Grant 186796 (PBK), NIH R01 NS080929 (PBK), the Association Française contre les Myopathies grant 15700 (TIJ, PLJ), Muscular Dystrophy Association (MDA) Development Grant 202863 (FR), and the Bernard F. and Alva B. Gimbel Foundation (LMK). Microarray genotyping and Sanger DNA sequencing experiments were performed in the Molecular Genetics Core Facility at Boston Children’s Hospital, supported by NIH P30 HD18655 through the Intellectual and Developmental Disabilities Research Center and NIH P50NS40828 through the Neuromuscular Disease Project.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.de Greef JC, Lemmers RJ, Camano P, et al. Clinical features of facioscapulohumeral muscular dystrophy 2. Neurology. 2010;75:1548–54. doi: 10.1212/WNL.0b013e3181f96175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lemmers RJ, van der Vliet PJ, Klooster R, et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science. 2010;329:1650–3. doi: 10.1126/science.1189044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blewitt ME, Gendrel AV, Pang Z, et al. SmcHD1, containing a structural-maintenance-of-chromosomes hinge domain, has a critical role in X inactivation. Nat Genet. 2008;40:663–9. doi: 10.1038/ng.142. [DOI] [PubMed] [Google Scholar]
- 4.Lemmers RJ, Tawil R, Petek LM, et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat Genet. 2012;44:1370–4. doi: 10.1038/ng.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boyden SE, Salih MA, Duncan AR, et al. Efficient identification of novel mutations in patients with limb girdle muscular dystrophy. Neurogenetics. 2010;11:449–55. doi: 10.1007/s10048-010-0250-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rohde C, Zhang Y, Reinhardt R, Jeltsch A. BISMA--fast and accurate bisulfite sequencing data analysis of individual clones from unique and repetitive sequences. BMC Bioinformatics. 2010;11:230. doi: 10.1186/1471-2105-11-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones TI, Chen JC, Rahimov F, et al. Facioscapulohumeral muscular dystrophy family studies of DUX4 expression: evidence for disease modifiers and a quantitative model of pathogenesis. Hum Mol Genet. 2012;21:4419–30. doi: 10.1093/hmg/dds284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dixit M, Ansseau E, Tassin A, et al. DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1. 2007;104:18157–62. doi: 10.1073/pnas.0708659104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Snider L, Geng LN, Lemmers RJ, et al. Facioscapulohumeral dystrophy: incomplete suppression of a retrotransposed gene. PLoS Genet. 2010;6:e1001181. doi: 10.1371/journal.pgen.1001181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hirano T. At the heart of the chromosome: SMC proteins in action. Nat Rev Mol Cell Biol. 2006;7:311–22. doi: 10.1038/nrm1909. [DOI] [PubMed] [Google Scholar]
- 14.Arahata K, Ishihara T, Fukunaga H, et al. Inflammatory response in facioscapulohumeral muscular dystrophy (FSHD): immunocytochemical and genetic analyses. Muscle Nerve. 1995;2:S56–66. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.