

Abstract
Fibrinogen is a circulating multifunctional plasma protein vital for hemostasis. Activation of the coagulation cascade converts soluble fibrinogen to insoluble polymerized fibrin, which, along with platelets, forms the hemostatic clot. However, inappropriate formation of fibrin clots may result in arterial and venous thrombotic disorders that may progress to life-threatening adverse events. Often thrombotic disorders are associated with inflammation and the production of oxidants. Fibrinogen represents a potential target for oxidants and several oxidative post-translational modifications that influence fibrinogen structure and function have been associated with disease pathogenesis. Here, we review various oxidative modifications of fibrinogen and the consequences of these modifications on protein structure, ability to form fibrin and the resulting alterations on fibrin architecture, viscoelastic and biochemical properties that may contribute to disease.
Keywords: fibrinogen, fibrin, coagulation, thrombosis, oxidation, nitrotyrosine
Introduction to fibrinogen and fibrin structure and biochemistry
Fibrinogen is synthesized in hepatocytes and secreted into the blood [1] where it circulates with a half-life of about 3 days [2]. Fibrinogen is a 340 kDa hexamer, composed of two pairs of three non-identical chains termed Aα, Bβ, and γ. Specific sites in each of these chains are more subject to oxidative modification (Figure 1) as elaborated below. Together the chains comprise a symmetrical molecule composed of one globular E region flanked on each side by globular D regions [3–6] that are connected by three-stranded α-helical coiled-coils [4–6]. The E region, which is composed of all three chains, contains fibrinopeptides A (FpA) and B (FpB) [4–6]. Cleavage of these peptides by thrombin exposes knobs ‘A’ and ‘B’, resulting in the formation of fibrin monomers [7–11]. Positively charged ‘A’ and ‘B’ knobs have complementary negatively charged binding sites within the γ and β nodules in the D regions of adjoining monomers termed holes ‘a’ and ‘b’ [12, 13]. Knob-hole associations each result in the formation of half-staggered, double-stranded protofibrils [14, 15], which associate laterally to form fibrin fibers [14, 16], and ultimately the branching network structure of the hemostatic or thrombotic clot [17]. The formation of fibrin clots using either isolated fibrinogen or platelet poor plasma (PPP) after the addition of thrombin and Ca2+ can be monitored spectrophotometrically by following turbidity changes over time (Figure 2A). This assay yields some generalized structural and kinetic information about fibrin clot formation, but does not provide specifics on properties such as rate of FpA and FpB release, which requires HPLC methodology [18], or fibrin clot structure, which can be obtained by scanning electron microscopy (Figure 2B) [19] or confocal microscopy [20].
Figure 1.
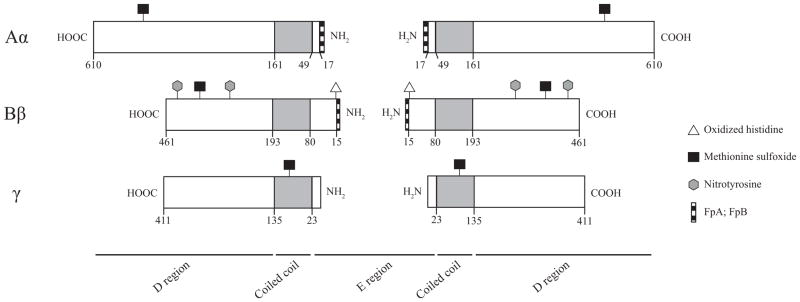
Sites of oxidative post-translational modifications on fibrinogen. Schematic representation of the Aα, Bβ, and γ chains of fibrinogen drawn to scale in length. Residue numbering follows the removal of the signal peptide. FpA and FpB (stripes) are localized towards the N-termini of the Aα and Bβ chains, respectively. Coiled-coil domains are represented in grey. His16 is located within the ‘B’ knob. Met476, Met367, and Met78 are localized to the αC region, the carboxy termini of the Bβ chains, and the coiled-coils of the γ chain, respectively. Both Tyr292 and Tyr422 are localized within the carboxy termini of the Bβ chains, however, Tyr292 is located near the ‘b’ holes.
Figure 2.
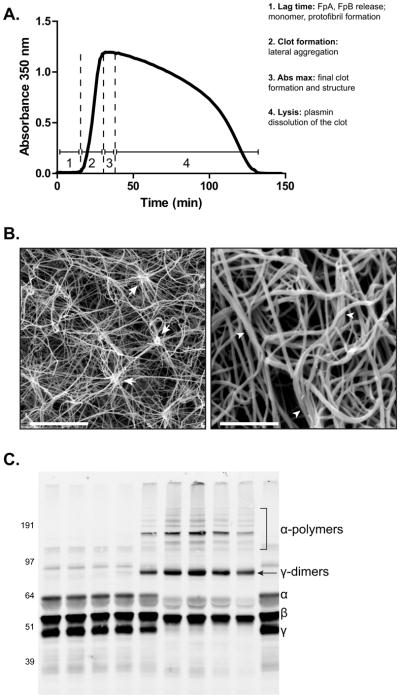
Techniques for measuring functional properties of fibrinogen. A) Turbidity assay showing both clot formation and lysis over time as a function of changes in absorbance. Thrombin, CaCl2, and either tPA alone or tPA and plasminogen are added simultaneously to PPP or isolated fibrinogen, respectively and clot formation and lysis time are monitored by changes in absorbance at 350 nm as a function of time. 1. The lag phase represents the formation of fibrin monomers, oligomers, and protofibrils. 2. Protofibrils that have reached a sufficient length laterally aggregate to generate fibers, causing a rapid increase in absorbance. 3. Eventually the clot is fully formed, which is represented by the maximum absorbance. 4. Subsequently, with binding of both plasminogen and t-PA to fibrin, plasmin lyses the clot, indicated by a decrease in absorbance. B) (Left) Scanning electron micrograph of a clot generated from isolated fibrinogen containing high levels of nitrated fibrinogen molecules. Magnification bar represents 10 μm. Arrows indicate clusters, defined as 8 single fibers crossing at a single point. (Right) Scanning electron micrograph of a clot generated from the PPP of a PE subject. Magnification bar represents 2 μm. Arrowheads designate fiber bundles, defined as 3 or more fibers aligning together for greater than 1 μm. Fiber density is commonly measured manually by counting the number of fibers in a small, designated area repeated over the entirety of the micrograph. Fiber diameter is also measured manually, often using Image J (Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, http://imagej.nih.gov/ij/, 1997–2011) or other software that is capable of determining distance. Here, the widths of many fibers are measured within the micrograph either in designated areas or at random. C) Western blot of FXIIIa crosslinking fibrin in PPP. Addition of thrombin and CaCl2 in PPP initiates both fibrin polymerization and FXIII activation. In isolated fibrinogen, FXIII must also be added to induce crosslinking. FXIIIa crosslinking of fibrin creates both γ-dimers and α-polymers. The reaction is allowed to proceed for the desired time point(s) followed by quenching with concentrated sample buffer containing a reducing agent and boiling. The products of the reaction are then run on an SDS PAGE gel followed by Western blot analysis with an anti-fibrinogen antibody. Lane 1) 0 hr 2) 5 min 3) 15 min 4) 30 min 5) 1 hr 6) 2 hr 7) 4 hr 8) 8 hr 9) 24 hr 10) No thrombin or CaCl2.
Fibrin is covalently crosslinked by the transglutaminase Factor XIII (FXIII), which provides stability and elasticity to the clot [20–23]. FXIII is activated (FXIIIa) by thrombin through cleavage of the N-terminal activation peptide [24]. FXIIIa crosslinks fibrin via the formation of ε-(γ-glutamyl) lysine bonds within and between α and γ chains, creating γ-dimers, α-polymers, and α-γ-heteropolymers [25, 26]. The rate and extent of FXIIIa crosslinking of fibrin, made by addition of thrombin plus FXIII and Ca2+ to either isolated fibrinogen or PPP, can be determined by quenching the reaction and running the products on an SDS PAGE gel with crosslinked fibrin products identified and quantified by western blot analysis and densitometry (Figure 2C).
Physiological lysis of the fibrin clot proceeds through the enzyme plasmin, which is converted from its inactive zymogen precursor, plasminogen, by the enzyme, tissue plasminogen activator (tPA) [27]. This reaction requires fibrin as a cofactor [28] and the kinetics of lysis can be followed by changes in turbidity (Figure 2A). Plasmin binds and cleaves fibrin at several high-affinity lysine residues, one of which is located in the flexible region of the carboxy termini of the Aα chains termed αC regions [29, 30] subsequently exposing additional lysine residues that plasmin further cleaves into smaller fragments [29]. Endogenous inhibitors of fibrinolysis target both tPA and plasmin, and include plasminogen activator inhibitor-1 (PAI-1), α2-antiplasmin (α2-PI), and thrombin-activatable fibrinolysis inhibitor (TAFI).
Fibrin is a highly extensible, viscoelastic polymer [22], allowing it to deform appropriately in response to shear stress. Changes in clot structure due to post-translational modifications (PTMs), elevated fibrinogen concentration, or interactions with other proteins have been shown to alter the viscoelastic properties of fibrin clots. Specifically, increased clot stiffness was significantly associated with coronary artery disease [31], as well as those at risk for thrombotic events such as smokers [32] and diabetics [33]. Traditionally, rheometry has been used to measure the elastic (stiffness) and inelastic (viscosity) properties of fibrin clots in vitro, although some studies have also utilized thromboelastography. Defined by the storage modulus (G′) and loss modulus (G″), respectively, these properties dictate the ability of the clot to store or dissipate energy due to the application of applied shear stress. The ratio of the loss modulus to the storage modulus, tan δ, is also used as a measure of the relative proportion of inelastic to elastic components. The relationships between viscoelastic properties, fibrin clot structure, and pathology remain unclear. Most studies have observed that increased fiber density or decreased porosity, coupled with decreased individual fiber diameter results in stiffer clots with delayed lysis, fostering a prothrombotic state [20, 31, 33]. In other cases, clot structural heterogeneities, including fiber clusters or bundles, have been observed as a consequence of PTMs and have varying effects on viscoelastic properties [32].
In addition to forming the fibrous network structure of a thrombus, fibrinogen also plays an important role in platelet aggregation. Individual platelets bind residues (400–411) located within the carboxy termini of the γ chains of fibrin(ogen) via their integrin receptor, αIIbβ3 [34]. These linkages tether platelets together resulting in platelet aggregation.
Fibrinogen is a critical protein in the formation of the clot, both in the fibrin network and platelet aggregation, which are ultimately required for the generation of the hemostatic thrombus. Perturbations in these functions may influence the formation and properties of the fibrin network and promote pathological states, including thrombosis and thromboembolism. Indeed, epidemiological studies have indicated that increased levels of circulating fibrinogen are an independent predictor of coronary heart disease and in some cases, of premature death from cardiovascular disease [35–38]. Despite these sound correlations, a causative association between high levels of fibrinogen and cardiovascular disease has not been firmly established. Evidence suggests that oxidative post-translational modifications may contribute to alterations in fibrinogen function and affect pathology. Here we review the impact of oxidative modifications on the structure and function of fibrinogen and discuss their relationship with various disease states.
Oxidative modifications of fibrinogen
Oxidative stress has been widely implicated in physiological processes such as aging, in disease pathogenesis ranging from carcinogenesis to atherogenesis, and the etiology of arterial and venous thrombosis. Proteins are major targets for oxidants and fibrinogen, which comprises a large percentage of plasma proteins (~4 %) is a likely target for oxidative post-translational modifications.
Fibrinogen oxidation
Fibrinogen was identified as the primary oxidized protein in the plasma of smokers, an at-risk group for lung cancer and thrombosis, and levels of oxidized fibrinogen were significantly increased in smokers as compared with controls [32, 39]. In thromboelastogram studies, clots made from whole blood collected from smokers shortly after smoking 2 cigarettes demonstrated decreased R value (lag phase), increased α angle (rate of fibrin polymerization), and increased G (stiffness) as compared with non-smokers and smokers before cigarette use [40]. In addition, clots formed from PPP of smokers post-cigarette use demonstrated increased fiber density and decreased fiber diameter compared with the control groups. Elevated plasma fibrinogen levels increased the risk for Alzheimer’s disease [41], increased brain atrophy in Alzheimer’s disease patients [42], and were associated with cognitive decline [43]. Previous studies have shown that oxidized proteins localize to sites of neurodegeneration [44]. Oxidation of the fibrinogen γ chain precursor protein quantified as carbonylation was increased in the plasma of subjects with Alzheimer’s disease [45]. Together these data suggest that inflammation and the production of oxidants may play overlapping roles in thrombosis and neurodegenerative diseases.
The potential effects of oxidation on fibrinogen and fibrin function have been explored in several studies that exposed fibrinogen to oxidants in vitro. Early studies using hematoporphyrins, photosensitive dyes, and chloramines to oxidize fibrinogen reported a dose-dependent inhibition of thrombin-induced fibrin polymerization [46, 47]. Photo-oxidized fibrin monomer did not bind fibrinogen as well as nascent fibrin monomer despite similar kinetics and amount of FpA and FpB released between oxidized and nascent fibrinogen [47]. Oxidation of fibrinogen by exposure to methylene blue photosensitive dye and light impaired polymerization of des A fibrin and rendered des AB NDSK (N-terminal disulphide knot (NDSK) region of fibrinogen lacking both FpA and FpB) incapable of binding to fibrinogen, with no effect on binding to D-dimer [48].
These results suggest that knob ‘A’-hole ‘a’ interactions are affected by oxidation. His16 in the β chain of fibrinogen was found to be modified in these oxidation studies, leading these authors to conclude that this amino acid was part of knob ‘A’ (Figure 1; Table 1) [48]. However, we now recognize [49] that His16 is part of knob ‘B’, suggesting that photo-oxidation of fibrinogen may also modify other previously unidentified sites, specifically those that affect A:a interactions. Oxidized fibrinogen also affects lysis. Pro-urokinase was activated faster in the presence of oxidized fibrin, and plasmin lysed oxidized fibrin at a faster rate implying a pro-fibrinolytic function for oxidized fibrinogen [50].
Table 1.
Site-specific oxidative modifications of fibrinogen and effects on fibrin function and clot structure.
| In vitro photo-oxidized fibrinogen [47, 48] | In vitro HOCl-oxidized fibrinogen [56] | Ex-vivo plasma of smokers and non-smokers [32] | |
|---|---|---|---|
| Type of modification | Oxidized histidine | Methionine sulphoxide | 3-Nitrotyrosine |
| Site of modification | Bβ His16; unknown Aα | * Aα Met476; Bβ Met367; γ Met78 | ** Bβ Tyr292; Bβ Tyr422 |
| Monomer association | Increased | NA | NA |
| Fibrin polymerization | Decreased | Decreased | Increased |
| Final turbidity | Decreased | Decreased | Increased |
| Fibrin clot lysis | NA | Slower | Slower |
| Viscoelastic properties | NA | Decreased stiffness and viscosity | Increased stiffness and viscosity |
| Fibrin clot structure | NA | Increased fiber density; decreased fiber diameter; decreased pore size | Increased fiber clusters |
Most abundant modification;
Most frequent modification.
Metal catalyzed oxidation of fibrinogen results in the formation of dityrosine crosslinks [51] and a dose-dependent increase in protein carbonyl levels [51, 52], which may affect several amino acid residues such as histidine, proline, arginine, and lysine [53]. Oxidation of fibrinogen with iron-ascorbate delayed release of FpA and FpB, concomitant with delayed fibrin polymerization and a lower maximum absorbance during turbidity assays [52]. Western blot analysis showed that the Aα and Bβ chains are susceptible to carbonylation following metal-catalyzed oxidation, while the γ chain remained unaffected [54]. ADP-induced platelet aggregation and adhesion were diminished with oxidized compared with nascent fibrinogen [51]. However, another study found that iron-ascorbate oxidation of fibrinogen increased platelet aggregation [55]. This study also demonstrated decreased catalytic efficiency of tPA conversion of plasminogen to plasmin, which requires fibrin as a cofactor. The authors proposed that decreased fibrinogen α-helical content caused by carbonylation of lysine residues may explain these findings. Moreover, aspirin-induced lysine acetylation prevented the effects of oxidation supporting a role for fibrinogen lysine residues in platelet aggregation. Both studies utilized similar oxidation systems and observed similar carbonyl levels per mg of protein. However, the shorter exposure time (0–1 hr [55] vs. 0–20 hr [51]) may have accounted for these differences. Although further experimentation is required, it is reasonable to assume that the number of amino acid residues, which amino acids, and the magnitude of oxidation per residue are significantly less during short exposures. Moreover, formation of reactive carbonyls indicated only one type of oxidative modification and it is possible that other oxidative modifications during prolonged exposures may account for the observed differences. These findings reinforce a common deficit in studies of protein oxidation that requires attention. The correlation between functional outcomes and protein oxidation should include precise identification of the modified amino acids, the chemical nature of the modification, as well as the extent of modification.
Fibrinogen oxidation with hypochlorite (HOCl) decreased the rate of fibrin polymerization and maximum absorbance by turbidity assays [56]. Treatment with hypochlorite dose dependently delayed lysis time, which may be due to the observed increase in fiber density, decrease in fiber diameter, and decrease in pore size, but not changes in plasminogen activation. Treatment of fibrinogen with hypochlorite also weakened clot stiffness, i.e. decreased G′ and G″. The maximum strain and maximum modulus in strain hardening experiments were lower for oxidized compared with nascent fibrin clots. The authors identified three oxidized methionine residues, Met476, Met367, and Met78 located on the Aα, Bβ, and γ chains, respectively (Figure 1, Table 1). Quantification of these sites identified Met476 as the most prominent modification, with 150 μM hypochlorite producing 73 % oxidation of the Met476 residue. Met476 is located within the αC region of the Aα chain and is thought to contribute to lateral aggregation of fibrin fibers [57]. Impairment of lateral aggregation by oxidation of Met476 may result in clots with thinner fibers and a higher fiber density, ultimately reducing fibrinolysis and weaker clot structure (Table 1).
Fibrinogen nitration
Protein 3-nitrotyrosine has been identified as both a marker and a functional post-translational modification of proteins during disease processes. Nitrated fibrinogen, (primarily tyrosine nitration within the β chain of the molecule) was initially identified in the plasma of subjects with acute respiratory distress syndrome [58]. Nitrated fibrinogen has been detected in the plasma of subjects with lung cancer [39] and with end stage renal disease [59]. Elevated levels of nitrated fibrinogen have been quantified in coronary artery disease subjects as compared with healthy controls [60]. Increased levels of plasma nitrated fibrinogen were also documented in subjects with venous thromboembolism (VTE) subjects compared with non-VTE subjects [61]. When compared with the lowest quartile, subjects in the highest quartile of nitrated fibrinogen had a significantly increased risk of VTE [61]. In previous studies aiming to identify biomarkers of VTE, MPO was significantly elevated in PE subjects, suggesting that nitrative and oxidative intermediates are produced during VTE [62]. Together these studies may indicate that nitrated fibrinogen is a potential diagnostic biomarker for VTE. Overall, increased circulating levels of nitrated fibrinogen have been associated with arterial [60] and venous thrombotic diseases [61], as well as those at risk for thrombosis, such as smokers [32]. Neutrophils and monocytes, which are capable of producing nitrating intermediates [63], are key mediators in the initial phases of venous thrombus formation [64], and the progression of the thrombus during atherosclerosis [65, 66]. Therefore appreciating the potential consequences of fibrinogen nitration will be of interest and could potentially provide mechanistic insights that link oxidative modifications and risk for thrombosis.
The functional effects of fibrinogen nitration have been explored both in vitro, [60] in smokers, [32] and in healthy subjects exposed to low levels of lipopolysaccharide [67]. When fibrinogen was nitrated in vitro with either 3-morpholinosydnonimine (SIN-1), which produces nitric oxide and superoxide that react to form peroxynitrite, or MPO, hydrogen peroxide, and nitrite, a decrease in lag time and an increase in both the rate of fibrin polymerization and final absorbance was observed during turbidity assays when compared with unmodified fibrinogen [60]. In contrast, fibrinogen treated with MPO and hydrogen peroxide alone (oxidizing conditions), impaired fibrin polymerization, consistent with the aforementioned exposure to HOCl [56]. The clot structure formed from nitrated fibrinogen was composed of thick, twisted fiber bundles and large pores, whereas fibrinogen treated with MPO and hydrogen peroxide generated a homogenous structure composed of thin fibers. Even though nitration and oxidation of fibrinogen produce disparate effects on clot structure, G′ was similar between the two groups and significantly lower than control, indicating a decrease in clot stiffness. Although no differences in lysis rate or lysis product formed were seen between control and nitrated fibrinogen in vitro, injection of microemboli composed of fibrin treated with SIN-1 into mice followed by bolus tPA injection showed enhanced lytic susceptibility compared to emboli of nascent fibrin. No differences were observed between nitrated and control fibrinogen for FpA or FpB release, FXIII crosslinking, or platelet aggregation. Expanding these findings, Parastatidis et al. [68] generated isolated fibrinogen from subjects with coronary artery disease that were depleted of nitrated fibrinogen molecules by the use of immunoaffinity removal procedures. As a control, identical plasma samples that retained the nitrated molecules were also generated by immunodepletion with a nonspecific immunoglobulin. Thrombin-induced fibrin clot formation showed a significant increase in the rate of fibrin polymerization and maximum absorbance in turbidity assays in fibrinogen that retained the nitrated fibrinogen molecules as compared to those where nitrated fibrinogen molecules were removed.
Smoking is a risk factor for thrombosis [69], and has been associated with increased production of oxidants [32, 70]. In smokers, elevated nitrated fibrinogen levels correlated with a dose-dependent increase in the rate of fibrin polymerization that was reversed when nitrated molecules were removed via immunoprecipitation with anti-nitrotyrosine antibodies [32]. Scanning electron micrographs showed fiber clustering that was also reversed by immunodepletion. Nitrated fibrinogen levels were positively correlated with G′, G″, tan δ, and inversely correlated with fibrinolysis rate. Tyrosine residues Tyr292 and Tyr422 were identified by mass spectrometry as the primary nitrated residues in a majority of the smoker samples analyzed. These residues are located within the carboxy terminus of the Bβ chain of fibrinogen near hole ‘B’, which may be involved in lateral aggregation (Figure 1; Table 1). Use of the ‘B’ knob mimetic peptide accelerated lateral aggregation in all samples, but was positively correlated with nitrated fibrinogen levels. The effects of nitration in vivo in smokers show some similarities to the in vitro modified fibrinogen, like increased rate of fibrin polymerization, as well as some disparities. These differences might be related to the tyrosines susceptible to nitration in each system. Additional studies to identify nitrated tyrosines within fibrinogen in vitro would be necessary to explore this hypothesis.
The relationship between inflammation and fibrinogen nitration was explored by administering low levels of lipopolysaccharide to healthy human subjects, which resulted in increased production of several protein mediators of inflammation such as TGF-β, C-reactive protein, and myeloperoxidase (MPO) [67]. Nitrated fibrinogen was also increased in this challenge model, peaking at 72-hrs post-injection, much later than the other inflammatory mediators. In addition, nitration of fibrinogen post-lipopolysaccharide injection resulted in increased rate of clot formation. This study implies that nitration results as a direct consequence of inflammatory challenge, nitrating intermediates are produced for prolonged periods of time following insult, and tyrosine nitration affects fibrinogen function, providing evidence for the role of inflammation and protein nitration in thrombotic disorders.
Although the aforementioned studies demonstrated that nitrated fibrinogen accelerated clot formation kinetics, some studies have reported opposing effects [71, 72]. Peroxynitrite treatment of fibrinogen dose-dependently decreased the rate of fibrin polymerization, increased lag time, and decreased maximum absorbance during clot polymerization assays [71, 72]. Additionally, platelet aggregation and adhesion were dose-dependently impaired by peroxynitrite treatment of fibrinogen compared to nascent fibrinogen [73]. However, plasmin degradation of fibrin clots was slower in treated fibrinogen, which is consistent with previous nitrated fibrinogen studies [32]. Peroxynitrite is an oxidant and a nitrating agent capable of oxidizing cysteine and tryptophan residues and producing dityrosine crosslinks and carbonyl-modified residues [74, 75]. Previous studies have reported that oxidized fibrinogen that was subsequently nitrated resulted in a reversal of the impaired fibrin polymerization, producing polymerization rates similar to nitration alone [60]. Thus, the effects of peroxynitrite on clotting kinetics may be due to the effects of other oxidative modifications, rather than solely nitration [47, 60]. These modifications may occur in fibrin monomer association sites [48], or within the fibrinogen integrin αIIbβ3 binding sites located within the γ chain [34]. Again, these studies indicate that in vitro modeling of the functional effects of fibrinogen oxidative modifications need to carefully consider the sites, magnitude and type of modification accounting for the presence of other off-target modifications.
Cysteine modifications of fibrinogen
All 58 cysteine residues in fibrinogen are disulfide bonded [76]. However, several disulfide bonds, including γ Cys23-Aα Cys45, Aα Cys442-Aα Cys472, γ Cys326-γ Cys339 and the symmetrical bonds Aα Cys28, γ Cys8, and γ Cys9 were capable of being reduced in vitro by thioredoxin and dithiothreitol [77, 78]. Reduction by thioredoxin or dithiothreitol resulted in prolonged clotting time in the absence of effects of FpA release, indicating alterations in fibrin polymer formation [77, 78]. In addition, plasminogen was more activated in the presence of partially reduced fibrinogen compared with non-reduced [79].
The low molecular weight thiol, glutathione (GSH) and its S-nitrosated analogue S-nitrosoglutathione (GSNO) are capable of undergoing disulphide exchange reactions with disulphide-bonded cysteine residues, generating S-glutathionylated cysteine. In addition, GSNO can modify reduced cysteine residues to S-nitrosocysteine. In vitro treatment of purified fibrinogen with GSNO, GSH, or oxidized glutathione (GSSG) dose dependently delayed clotting time, reduced the rate of fibrin polymerization, and reduced final turbidity [80]. The authors proposed that the effects of the GSH derivatives were due to S-glutathionylation reactions within the αC region of fibrinogen, although the S-glutathionylated sites were not identified by sequencing or mass spectrometry [80, 81]. Akhter et al. also reported impaired fibrin polymerization upon exposure of fibrinogen to GSNO. However, CD spectroscopy and tryptophan quenching experiments showed that GSNO did not participate in S-nitrosation or S-glutathionylation reactions, but instead, reversibly interacted with the αC region of fibrinogen, causing allosteric conformational changes in the protein that affected polymerization [81]. Additional studies have investigated the role of GSNO treatment on fibrin clot structure. Fiber density decreased and fiber diameter increased following GSNO treatment of PPP [82].
However, at high concentrations of GSNO (3.75 mM), fibrin did not polymerize, and instead formed aggregate clusters. Although the authors did not perform turbidity assays, previous studies have found that the formation of clusters and increased fiber diameter both resulted in increased turbidity [32], which would be in contrast to the previously described reports [80, 81]. These conflicting results may be due to GSNO interactions or reactions with other proteins within the plasma, specifically those that play a role in fibrin clot formation, such as thrombin [19], FXIII [19], and FXII [83].
To date, neither S-glutathionylated nor S-nitrosated fibrinogen has been detected in vivo. The in vitro effects of glutathione analogs on fibrinogen function may suggest that fibrinogen becomes modified or interacts with excess glutathione produced in response to production of oxidants by neutrophils and macrophages during thrombosis or other inflammatory conditions [84]. Future studies focusing on the role of glutathione and fibrinogen in may help clarify the roles of the oxidant/antioxidant systems in thrombosis and inflammation.
Conclusion
Oxidative modifications of fibrinogen have the potential to influence the kinetics of fibrin formation as well as the structure and biomechanical properties of fibrin, ultimately producing dysfunctional hemostatic clots. Some of these modifications have been identified in vivo, providing further evidence for a relationship between the production of oxidants and thrombosis. However, the precise mapping of sites of in vivo modifications has not been fully achieved, creating considerable reservations about the significance of these modifications in physiological or pathological settings. Moreover, the in vitro exposures do not always reproduce the in vivo modifications. As such caution should be exercised in interpreting the functional effects of these modifications on fibrinogen function. An additional question concerning the effects of oxidative modifications on the interactions of fibrinogen with other proteins, such as the platelet integrin receptor αIIbβ3 remains. Future studies should aim to identify the sites of oxidative modifications in vivo and, if possible, employ fibrinogen extracted from humans or animal models to determine the effects on fibrinogen function and protein interactions. These studies can then lead towards a greater understanding of the role of fibrinogen modifications in health and disease.
Abbreviations
- FpA/FpB
Fibrinopeptide A/B
- PPP
Platelet poor plasma
- PTM
Post-translational modification
- tPA
Tissue plasminogen activator
- VTE
Venous thromboembolism
- MPO
Myeloperoxidase
- GSH
Glutathione
- GSSG
Oxidized glutathione
- GSNO
S-nitrosoglutathione
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Whipple GH, Hurwitz SH. Fibrinogen of the blood as influenced by the liver necrosis of chloroform poisoning. J Exp Med. 1911;13:136–161. doi: 10.1084/jem.13.1.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stein TP, Leskiw MJ, Wallace HW. Measurement of half-life human plasma fibrinogen. Am J Physiol. 1978;234:D504–10. doi: 10.1152/ajpendo.1978.234.5.E504. [DOI] [PubMed] [Google Scholar]
- 3.Hall CE, Slayter HS. The fibrinogen molecule: Its size, shape, and mode of polymerization. J Biophys Biochem Cytol. 1959;5:11–16. doi: 10.1083/jcb.5.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kollman JM, Pandi L, Sawaya MR, Riley M, Doolittle RF. Crystal structure of human fibrinogen. Biochemistry. 2009;48:3877–3886. doi: 10.1021/bi802205g. [DOI] [PubMed] [Google Scholar]
- 5.Yang Z, Kollman JM, Pandi L, Doolittle RF. Crystal structure of native chicken fibrinogen at 2.7 A resolution. Biochemistry. 2001;40:12515–12523. doi: 10.1021/bi011394p. [DOI] [PubMed] [Google Scholar]
- 6.Brown JH, Volkmann N, Jun G, Henschen-Edman AH, Cohen C. The crystal structure of modified bovine fibrinogen. Proc Natl Acad Sci U S A. 2000;97:85–90. doi: 10.1073/pnas.97.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lorand L. ‘Fibrino-peptide’; new aspects of the fibrinogen-fibrin transformation. Nature. 1951;167:992–993. doi: 10.1038/167992a0. [DOI] [PubMed] [Google Scholar]
- 8.Blomback B, Hessel B, Hogg D, Therkildsen L. A two-step fibrinogen--fibrin transition in blood coagulation. Nature. 1978;275:501–505. doi: 10.1038/275501a0. [DOI] [PubMed] [Google Scholar]
- 9.Blomback B, Hessel B, Iwanaga S, Reuterby J, Blomback M. Primary structure of human fibrinogen fibrinIclevage of fibrinogen with cyanogen bromide isolation and characterization of NH 2 -terminal fragments of the (“A”) chain. J Biol Chem. 1972;247:1496–1512. [PubMed] [Google Scholar]
- 10.Bettelheim FR. The clotting of fibrinogen II. fractionation of peptide material liberated. Biochim Biophys Acta. 1956;19:121–130. doi: 10.1016/0006-3002(56)90393-6. [DOI] [PubMed] [Google Scholar]
- 11.Budzynski AZ, Olexa SA, Pandya BV. Fibrin polymerization sites in fibrinogen and fibrin fragments. Ann N Y Acad Sci. 1983;408:301–314. doi: 10.1111/j.1749-6632.1983.tb23253.x. [DOI] [PubMed] [Google Scholar]
- 12.Pratt KP, Cote HC, Chung DW, Stenkamp RE, Davie EW. The primary fibrin polymerization pocket: Three-dimensional structure of a 30-kDa C-terminal gamma chain fragment complexed with the peptide gly-pro-arg-pro. Proc Natl Acad Sci U S A. 1997;94:7176–7181. doi: 10.1073/pnas.94.14.7176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spraggon G, Everse SJ, Doolittle RF. Crystal structures of fragment D from human fibrinogen and its crosslinked counterpart from fibrin. Nature. 1997;389:455–462. doi: 10.1038/38947. [DOI] [PubMed] [Google Scholar]
- 14.Krakow W, Endres GF, Siegel BM, Scheraga HA. An electron microscopic investigation of the polymerization of bovine fibrin monomer. J Mol Biol. 1972;71:95–103. doi: 10.1016/0022-2836(72)90403-2. [DOI] [PubMed] [Google Scholar]
- 15.Fowler WE, Hantgan RR, Hermans J, Erickson HP. Structure of the fibrin protofibril. Proc Natl Acad Sci U S A. 1981;78:4872–4876. doi: 10.1073/pnas.78.8.4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hewat EA, Tranqui L, Wade RH. Electron microscope structural study of modified fibrin and a related modified fibrinogen aggregate. J Mol Biol. 1983;170:203–222. doi: 10.1016/s0022-2836(83)80233-2. [DOI] [PubMed] [Google Scholar]
- 17.Hantgan RR, Hermans J. Assembly of fibrin A light scattering study. J Biol Chem. 1979;254:11272–11281. [PubMed] [Google Scholar]
- 18.Ng AS, Lewis SD, Shafer JA. Quantifying thrombin-catalyzed release of fibrinopeptides from fibrinogen using high-performance liquid chromatography. Methods Enzymol. 1993;222:341–358. doi: 10.1016/0076-6879(93)22023-9. [DOI] [PubMed] [Google Scholar]
- 19.Ryan EA, Mockros LF, Weisel JW, Lorand L. Structural origins of fibrin clot rheology. Biophys J. 1999;77:2813–2826. doi: 10.1016/S0006-3495(99)77113-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Collet JP, Park D, Lesty C, Soria J, Soria C, Montalescot G, Weisel JW. Influence of fibrin network conformation and fibrin fiber diameter on fibrinolysis speed: Dynamic and structural approaches by confocal microscopy. Arterioscler Thromb Vasc Biol. 2000;20:1354–1361. doi: 10.1161/01.atv.20.5.1354. [DOI] [PubMed] [Google Scholar]
- 21.Gerth C, Roberts WW, Ferry JD. Rheology of fibrin clotsII linear viscoelastic behavior in shear creep. Biophys Chem. 1974;2:208–217. doi: 10.1016/0301-4622(74)80046-3. [DOI] [PubMed] [Google Scholar]
- 22.Brown AE, Litvinov RI, Discher DE, Purohit PK, Weisel JW. Multiscale mechanics of fibrin polymer: Gel stretching with protein unfolding and loss of water. Science. 2009;325:741–744. doi: 10.1126/science.1172484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lorand L, Jacobsen A. Accelerated lysis of blood clots. Nature. 1962;195:911–912. doi: 10.1038/195911b0. [DOI] [PubMed] [Google Scholar]
- 24.Nakamura S, Iwanaga S, Suzuki T. On the activation of bovine plasma factor XIII amino acid sequence of the peptide released by thrombin and the terminal residues of the subunit polypeptides. J Biochem. 1975;78:1247–1266. doi: 10.1093/oxfordjournals.jbchem.a131023. [DOI] [PubMed] [Google Scholar]
- 25.Sobel JH, Gawinowicz MA. Identification of the alpha chain lysine donor sites involved in factor XIIIa fibrin cross-linking. J Biol Chem. 1996;271:19288–19297. doi: 10.1074/jbc.271.32.19288. [DOI] [PubMed] [Google Scholar]
- 26.Cottrell BA, Strong DD, Watt KW, Doolittle RF. Amino acid sequence studies on the alpha chain of human fibrinogen exact location of cross-linking acceptor sites. Biochemistry. 1979;18:5405–5410. doi: 10.1021/bi00591a023. [DOI] [PubMed] [Google Scholar]
- 27.Robbins KC, Summaria L, Hsieh B, Shah RJ. The peptide chains of human plasmin mechanism of activation of human plasminogen to plasmin. J Biol Chem. 1967;242:2333–2342. [PubMed] [Google Scholar]
- 28.Hoylaerts M, Rijken DC, Lijnen HR, Collen D. Kinetics of the activation of plasminogen by human tissue plasminogen activator role of fibrin. J Biol Chem. 1982;257:2912–2919. [PubMed] [Google Scholar]
- 29.Bok RA, Mangel WF. Quantitative characterization of the binding of plasminogen to intact fibrin clots, lysine-sepharose, and fibrin cleaved by plasmin. Biochemistry. 1985;24:3279–3286. doi: 10.1021/bi00334a031. [DOI] [PubMed] [Google Scholar]
- 30.Tsurupa G, Medved L. Identification and characterization of novel tPA- and plasminogen-binding sites within fibrin(ogen) alpha C-domains. Biochemistry. 2001;40:801–808. doi: 10.1021/bi001789t. [DOI] [PubMed] [Google Scholar]
- 31.Collet JP, Allali Y, Lesty C, Tanguy ML, Silvain J, Ankri A, Blanchet B, Dumaine R, Gianetti J, Payot L, Weisel JW, Montalescot G. Altered fibrin architecture is associated with hypofibrinolysis and premature coronary atherothrombosis. Arterioscler Thromb Vasc Biol. 2006;26:2567–2573. doi: 10.1161/01.ATV.0000241589.52950.4c. [DOI] [PubMed] [Google Scholar]
- 32.Parastatidis I, Thomson L, Burke A, Chernysh I, Nagaswami C, Visser J, Stamer S, Liebler DC, Koliakos G, Heijnen HF, Fitzgerald GA, Weisel JW, Ischiropoulos H. Fibrinogen beta-chain tyrosine nitration is a prothrombotic risk factor. J Biol Chem. 2008;283:33846–33853. doi: 10.1074/jbc.M805522200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pieters M, Covic N, van der Westhuizen FH, Nagaswami C, Baras Y, Toit Loots D, Jerling JC, Elgar D, Edmondson KS, van Zyl DG, Rheeder P, Weisel JW. Glycaemic control improves fibrin network characteristics in type 2 diabetes - a purified fibrinogen model. Thromb Haemost. 2008;99:691–700. doi: 10.1160/TH07-11-0699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Farrell DH, Thiagarajan P. Binding of recombinant fibrinogen mutants to platelets. J Biol Chem. 1994;269:226–231. [PubMed] [Google Scholar]
- 35.Kannel WB, Wolf PA, Castelli WP, D’Agostino RB. Fibrinogen risk of cardiovascular disease the framingham study. JAMA. 1987;258:1183–1186. [PubMed] [Google Scholar]
- 36.Wilhelmsen L, Svardsudd K, Korsan-Bengtsen K, Larsson B, Welin L, Tibblin G. Fibrinogen as a risk factor for stroke and myocardial infarction. N Engl J Med. 1984;311:501–505. doi: 10.1056/NEJM198408233110804. [DOI] [PubMed] [Google Scholar]
- 37.Kaptoge S, Di Angelantonio E, Pennells L, Wood AM, White IR, Gao P, Walker M, Thompson A, Sarwar N, Caslake M, Butterworth AS, Amouyel P, Assmann G, Bakker SJ, Barr EL, Barrett-Connor E, Benjamin EJ, Bjorkelund C, Brenner H, Brunner E, Clarke R, Cooper JA, Cremer P, Cushman M, Dagenais GR, D’Agostino RBS, Dankner R, Davey-Smith G, Deeg D, Dekker JM, Engstrom G, Folsom AR, Fowkes FG, Gallacher J, Gaziano JM, Giampaoli S, Gillum RF, Hofman A, Howard BV, Ingelsson E, Iso H, Jorgensen T, Kiechl S, Kitamura A, Kiyohara Y, Koenig W, Kromhout D, Kuller LH, Lawlor DA, Meade TW, Nissinen A, Nordestgaard BG, Onat A, Panagiotakos DB, Psaty BM, Rodriguez B, Rosengren A, Salomaa V, Kauhanen J, Salonen JT, Shaffer JA, Shea S, Ford I, Stehouwer CD, Strandberg TE, Tipping RW, Tosetto A, Wassertheil-Smoller S, Wennberg P, Westendorp RG, Whincup PH, Wilhelmsen L, Woodward M, Lowe GD, Wareham NJ, Khaw KT, Sattar N, Packard CJ, Gudnason V, Ridker PM, Pepys MB, Thompson SG, Danesh J. C-reactive protein, fibrinogen, and cardiovascular disease prediction. N Engl J Med. 2012;367:1310–1320. doi: 10.1056/NEJMoa1107477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stec JJ, Silbershatz H, Tofler GH, Matheney TH, Sutherland P, Lipinska I, Massaro JM, Wilson PF, Muller JE, D’Agostino RBS. Association of fibrinogen with cardiovascular risk factors and cardiovascular disease in the framingham offspring population. Circulation. 2000;102:1634–1638. doi: 10.1161/01.cir.102.14.1634. [DOI] [PubMed] [Google Scholar]
- 39.Pignatelli B, Li CQ, Boffetta P, Chen Q, Ahrens W, Nyberg F, Mukeria A, Bruske-Hohlfeld I, Fortes C, Constantinescu V, Ischiropoulos H, Ohshima H. Nitrated and oxidized plasma proteins in smokers and lung cancer patients. Cancer Res. 2001;61:778–784. [PubMed] [Google Scholar]
- 40.Barua RS, Sy F, Srikanth S, Huang G, Javed U, Buhari C, Margosan D, Ambrose JA. Effects of cigarette smoke exposure on clot dynamics and fibrin structure: An ex vivo investigation. Arterioscler Thromb Vasc Biol. 2010;30:75–79. doi: 10.1161/ATVBAHA.109.195024. [DOI] [PubMed] [Google Scholar]
- 41.van Oijen M, Witteman JC, Hofman A, Koudstaal PJ, Breteler MM. Fibrinogen is associated with an increased risk of alzheimer disease and vascular dementia. Stroke. 2005;36:2637–2641. doi: 10.1161/01.STR.0000189721.31432.26. [DOI] [PubMed] [Google Scholar]
- 42.Thambisetty M, Simmons A, Hye A, Campbell J, Westman E, Zhang Y, Wahlund LO, Kinsey A, Causevic M, Killick R, Kloszewska I, Mecocci P, Soininen H, Tsolaki M, Vellas B, Spenger C, Lovestone S Emerging Risk Factors Collaboration, AddNeuroMed Consortium. Plasma biomarkers of brain atrophy in alzheimer’s disease. PLoS One. 2011;6:e28527. doi: 10.1371/journal.pone.0028527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu G, Zhang H, Zhang S, Fan X, Liu X. Plasma fibrinogen is associated with cognitive decline and risk for dementia in patients with mild cognitive impairment. Int J Clin Pract. 2008;62:1070–1075. doi: 10.1111/j.1742-1241.2007.01268.x. [DOI] [PubMed] [Google Scholar]
- 44.Hensley K, Hall N, Subramaniam R, Cole P, Harris M, Aksenov M, Aksenova M, Gabbita SP, Wu JF, Carney JM. Brain regional correspondence between alzheimer’s disease histopathology and biomarkers of protein oxidation. J Neurochem. 1995;65:2146–2156. doi: 10.1046/j.1471-4159.1995.65052146.x. [DOI] [PubMed] [Google Scholar]
- 45.Choi J, Malakowsky CA, Talent JM, Conrad CC, Gracy RW. Identification of oxidized plasma proteins in alzheimer’s disease. Biochem Biophys Res Commun. 2002;293:1566–1570. doi: 10.1016/S0006-291X(02)00420-5. [DOI] [PubMed] [Google Scholar]
- 46.Zieve PD, Solomon HM. Effect of hematoporphyrin and light on human fibrinogen. Am J Physiol. 1966;210:1391–1395. doi: 10.1152/ajplegacy.1966.210.6.1391. [DOI] [PubMed] [Google Scholar]
- 47.Inada Y, Hessel B, Blomback B. Photooxidation of fibrinogen in the presence of methylene blue and its effect on polymerization. Biochim Biophys Acta. 1978;532:161–170. doi: 10.1016/0005-2795(78)90459-2. [DOI] [PubMed] [Google Scholar]
- 48.Shimizu A, Saito Y, Inada Y. Distinctive role of histidine-16 of the B beta chain of fibrinogen in the end-to-end association of fibrin. Proc Natl Acad Sci U S A. 1986;83:591–593. doi: 10.1073/pnas.83.3.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Everse SJ, Spraggon G, Veerapandian L, Riley M, Doolittle RF. Crystal structure of fragment double-D from human fibrin with two different bound ligands. Biochemistry. 1998;37:8637–8642. doi: 10.1021/bi9804129. [DOI] [PubMed] [Google Scholar]
- 50.Stief TW. Oxidized fibrin stimulates the activation of pro-urokinase and is the preferential substrate of human plasmin. Blood Coagul Fibrinolysis. 1993;4:117–121. [PubMed] [Google Scholar]
- 51.Belisario MA, Di Domenico C, Pelagalli A, Della Morte R, Staiano N. Metal-ion catalyzed oxidation affects fibrinogen activity on platelet aggregation and adhesion. Biochimie. 1997;79:449–455. doi: 10.1016/s0300-9084(97)86155-x. [DOI] [PubMed] [Google Scholar]
- 52.Shacter E, Williams JA, Levine RL. Oxidative modification of fibrinogen inhibits thrombin-catalyzed clot formation. Free Radic Biol Med. 1995;18:815–821. doi: 10.1016/0891-5849(95)93872-4. [DOI] [PubMed] [Google Scholar]
- 53.Amici A, Levine RL, Tsai L, Stadtman ER. Conversion of amino acid residues in proteins and amino acid homopolymers to carbonyl derivatives by metal-catalyzed oxidation reactions. J Biol Chem. 1989;264:3341–3346. [PubMed] [Google Scholar]
- 54.Galanakis DK, Laurent P, Janoff A. Cigarette smoke contains anticoagulants against fibrin aggregation and factor XIIIa in plasma. Science. 1982;217:642–645. doi: 10.1126/science.6124042. [DOI] [PubMed] [Google Scholar]
- 55.Upchurch GR, Jr, Ramdev N, Walsh MT, Loscalzo J. Prothrombotic consequences of the oxidation of fibrinogen and their inhibition by aspirin. J Thromb Thrombolysis. 1998;5:9–14. doi: 10.1023/a:1008859729045. [DOI] [PubMed] [Google Scholar]
- 56.Weigandt KM, White N, Chung D, Ellingson E, Wang Y, Fu X, Pozzo DC. Fibrin clot structure and mechanics associated with specific oxidation of methionine residues in fibrinogen. Biophys J. 2012;103:2399–2407. doi: 10.1016/j.bpj.2012.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gorkun OV, Henschen-Edman AH, Ping LF, Lord ST. Analysis of A alpha 251 fibrinogen: The alpha C domain has a role in polymerization, albeit more subtle than anticipated from the analogous proteolytic fragment X. Biochemistry. 1998;37:15434–15441. doi: 10.1021/bi981551t. [DOI] [PubMed] [Google Scholar]
- 58.Gole MD, Souza JM, Choi I, Hertkorn C, Malcolm S, Foust RF, 3rd, Finkel B, Lanken PN, Ischiropoulos H. Plasma proteins modified by tyrosine nitration in acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol. 2000;278:L961–7. doi: 10.1152/ajplung.2000.278.5.L961. [DOI] [PubMed] [Google Scholar]
- 59.Piroddi M, Palmese A, Pilolli F, Amoresano A, Pucci P, Ronco C, Galli F. Plasma nitroproteome of kidney disease patients. Amino Acids. 2011;40:653–667. doi: 10.1007/s00726-010-0693-1. [DOI] [PubMed] [Google Scholar]
- 60.Vadseth C, Souza JM, Thomson L, Seagraves A, Nagaswami C, Scheiner T, Torbet J, Vilaire G, Bennett JS, Murciano JC, Muzykantov V, Penn MS, Hazen SL, Weisel JW, Ischiropoulos H. Pro-thrombotic state induced by post-translational modification of fibrinogen by reactive nitrogen species. J Biol Chem. 2004;279:8820–8826. doi: 10.1074/jbc.M306101200. [DOI] [PubMed] [Google Scholar]
- 61.Martinez M, Cuker A, Mills A, Lightfoot R, Fan Y, Tang WH, Hazen SL, Ischiropoulos H. Nitrated fibrinogen is a biomarker of oxidative stress in venous thromboembolism. Free Radic Biol Med. 2012;53:230–236. doi: 10.1016/j.freeradbiomed.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mitchell AM, Nordenholz KE, Kline JA. Tandem measurement of D-dimer and myeloperoxidase or C-reactive protein to effectively screen for pulmonary embolism in the emergency department. Acad Emerg Med. 2008;15:800–805. doi: 10.1111/j.1553-2712.2008.00204.x. [DOI] [PubMed] [Google Scholar]
- 63.Brennan ML, Wu W, Fu X, Shen Z, Song W, Frost H, Vadseth C, Narine L, Lenkiewicz E, Borchers MT, Lusis AJ, Lee JJ, Lee NA, Abu-Soud HM, Ischiropoulos H, Hazen SL. A tale of two controversies: Defining both the role of peroxidases in nitrotyrosine formation in vivo using eosinophil peroxidase and myeloperoxidase-deficient mice, and the nature of peroxidase-generated reactive nitrogen species. J Biol Chem. 2002;277:17415–17427. doi: 10.1074/jbc.M112400200. [DOI] [PubMed] [Google Scholar]
- 64.von Bruhl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, Khandoga A, Tirniceriu A, Coletti R, Kollnberger M, Byrne RA, Laitinen I, Walch A, Brill A, Pfeiler S, Manukyan D, Braun S, Lange P, Riegger J, Ware J, Eckart A, Haidari S, Rudelius M, Schulz C, Echtler K, Brinkmann V, Schwaiger M, Preissner KT, Wagner DD, Mackman N, Engelmann B, Massberg S. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209:819–835. doi: 10.1084/jem.20112322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, Pittet MJ. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Drechsler M, Megens RT, van Zandvoort M, Weber C, Soehnlein O. Hyperlipidemia-triggered neutrophilia promotes early atherosclerosis. Circulation. 2010;122:1837–1845. doi: 10.1161/CIRCULATIONAHA.110.961714. [DOI] [PubMed] [Google Scholar]
- 67.Heffron SP, Parastatidis I, Cuchel M, Wolfe ML, Tadesse MG, Mohler ER, 3rd, Ischiropoulos H, Rader DJ, Reilly MP. Inflammation induces fibrinogen nitration in experimental human endotoxemia. Free Radic Biol Med. 2009;47:1140–1146. doi: 10.1016/j.freeradbiomed.2009.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Parastatidis I, Thomson L, Fries DM, Moore RE, Tohyama J, Fu X, Hazen SL, Heijnen HF, Dennehy MK, Liebler DC, Rader DJ, Ischiropoulos H. Increased protein nitration burden in the atherosclerotic lesions and plasma of apolipoprotein A-I deficient mice. Circ Res. 2007;101:368–376. doi: 10.1161/CIRCRESAHA.107.157537. [DOI] [PubMed] [Google Scholar]
- 69.Anonymous. Oral contraceptives stroke in young women associated risk factors. JAMA. 1975;231:718–722. doi: 10.1001/jama.1975.03240190022010. [DOI] [PubMed] [Google Scholar]
- 70.Nakayama T, Church DF, Pryor WA. Quantitative analysis of the hydrogen peroxide formed in aqueous cigarette tar extracts. Free Radic Biol Med. 1989;7:9–15. doi: 10.1016/0891-5849(89)90094-4. [DOI] [PubMed] [Google Scholar]
- 71.Nowak P, Zbikowska HM, Ponczek M, Kolodziejczyk J, Wachowicz B. Different vulnerability of fibrinogen subunits to oxidative/nitrative modifications induced by peroxynitrite: Functional consequences. Thromb Res. 2007;121:163–174. doi: 10.1016/j.thromres.2007.03.017. [DOI] [PubMed] [Google Scholar]
- 72.Lupidi G, Angeletti M, Eleuteri AM, Tacconi L, Coletta M, Fioretti E. Peroxynitrite-mediated oxidation of fibrinogen inhibits clot formation. FEBS Lett. 1999;462:236–240. doi: 10.1016/s0014-5793(99)01500-8. [DOI] [PubMed] [Google Scholar]
- 73.Nowak P, Wachowicz B. Peroxynitrite-mediated modification of fibrinogen affects platelet aggregation and adhesion. Platelets. 2002;13:293–299. doi: 10.1080/0953770021000007230. [DOI] [PubMed] [Google Scholar]
- 74.Radi R, Beckman JS, Bush KM, Freeman BA. Peroxynitrite oxidation of sulfhydryls the cytotoxic potential of superoxide and nitric oxide. J Biol Chem. 1991;266:4244–4250. [PubMed] [Google Scholar]
- 75.Ischiropoulos H, al-Mehdi AB. Peroxynitrite-mediated oxidative protein modifications. FEBS Lett. 1995;364:279–282. doi: 10.1016/0014-5793(95)00307-u. [DOI] [PubMed] [Google Scholar]
- 76.HENSCHEN A. Number and reactivity of disulfide bonds in fibrinogen and fibrin. Arkiv Kemi. 1964;22:355–373. [Google Scholar]
- 77.Blomback B, Blomback M, Finkbeiner W, Holmgren A, Kowalska-Loth B, Olovson G. Enzymatic reduction of disulfide bonds in fibrin-ogen by the thioredoxin systemI identification of reduced bonds and studies on reoxidation process. Thromb Res. 1974;4:55–75. doi: 10.1016/0049-3848(74)90203-5. [DOI] [PubMed] [Google Scholar]
- 78.Procyk R, Blomback B. Disulfide bond reduction in fibrinogen: Calcium protection and effect on clottability. Biochemistry. 1990;29:1501–1507. doi: 10.1021/bi00458a022. [DOI] [PubMed] [Google Scholar]
- 79.Procyk R, Medved L, Engelke KJ, Kudryk B, Blomback B. Nonclottable fibrin obtained from partially reduced fibrinogen: Characterization and tissue plasminogen activator stimulation. Biochemistry. 1992;31:2273–2278. doi: 10.1021/bi00123a009. [DOI] [PubMed] [Google Scholar]
- 80.Geer CB, Stasko NA, Rus IA, Lord ST, Schoenfisch MH. Influence of glutathione and its derivatives on fibrin polymerization. Biomacromolecules. 2008;9:1876–1882. doi: 10.1021/bm800146j. [DOI] [PubMed] [Google Scholar]
- 81.Akhter S, Vignini A, Wen Z, English A, Wang PG, Mutus B. Evidence for S-nitrosothiol-dependent changes in fibrinogen that do not involve transnitrosation or thiolation. Proc Natl Acad Sci U S A. 2002;99:9172–9177. doi: 10.1073/pnas.142136499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bateman RM, Ellis CG, Suematsu M, Walley KR. S-nitrosoglutathione acts as a small molecule modulator of human fibrin clot architecture. PLoS One. 2012;7:e43660. doi: 10.1371/journal.pone.0043660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Konings J, Govers-Riemslag JW, Philippou H, Mutch NJ, Borissoff JI, Allan P, Mohan S, Tans G, Ten Cate H, Ariens RA. Factor XIIa regulates the structure of the fibrin clot independently of thrombin generation through direct interaction with fibrin. Blood. 2011;118:3942–3951. doi: 10.1182/blood-2011-03-339572. [DOI] [PubMed] [Google Scholar]
- 84.Bilzer M, Lauterburg BH. Glutathione metabolism in activated human neutrophils: Stimulation of glutathione synthesis and consumption of glutathione by reactive oxygen species. Eur J Clin Invest. 1991;21:316–322. doi: 10.1111/j.1365-2362.1991.tb01376.x. [DOI] [PubMed] [Google Scholar]