

Abstract
Current multimodality therapy consisting of surgery, chemotherapy and radiation will fail in approximately 40% of patients with pediatric sarcomas and results in substantial long-term morbidity in those who are cured. Immunotherapeutic regimens for the treatment of solid tumors typically generate antigen-specific responses too weak to overcome considerable tumor burden and tumor suppressive mechanisms and are in need of adjuvant assistance. Previous work suggests that inhibitors of DASH (Dipeptidyl peptidase IV activity and/or structural homologues) enzymes can mediate tumor regression via immune-mediated mechanisms. Here we demonstrate that the DASH inhibitor, ARI-4175, can induce regression and eradication of well-established solid tumors, both as a single agent and as an adjuvant to a dendritic cell (DC) vaccine and adoptive cell therapy (ACT) in mice implanted with the M3-9-M rhabdomyosarcoma (RMS) cell line. Treatment with effective doses of ARI-4175 correlated with recruitment of myeloid (CD11b+) cells, particularly myeloid dendritic cells (DCs), to secondary lymphoid tissues and with reduced frequency of intratumoral monocytic (CD11b+Ly6-ChiLy6-Glo) myeloid-derived suppressor cells. In immunocompetent mice, combining ARI-4175 with a DC vaccine or ACT with tumor-primed T cells produced significant improvements in tumor responses against well-established M3-9-M tumors. In M3-9-M-bearing immunodeficient (Rag1-/-) mice, ACT combined with ARI-4175 produced greater tumor responses and significantly improved survival compared to either treatment alone. These studies warrant the clinical investigation of ARI-4175 for treatment of sarcomas and other malignancies particularly as an adjuvant to tumor vaccines and ACT.
Introduction
Pediatric sarcomas are heterogeneous solid tumors that arise from bone or soft tissues and include osteosarcoma, Ewing's sarcoma, and rhabdomyosarcoma (RMS). RMS accounts for over half of the soft tissue tumors occurring in individuals under the age of 201. Dramatic improvements in surgery, multi-agent chemotherapy and radiotherapy have resulted in improvements in overall survival for children and young adults with pediatric RMS from 25% in the 1970s to about 55-60% for patients treated on current cooperative group studies2. However, approximately 40% of cases recur and will ultimately be fatal in almost all of these patients including the majority presenting with metastatic disease. For these individuals there has been little improvement in outcome over recent decades. Moreover, patients who are cured experience substantial life-long morbidity including a markedly increased risk for secondary malignancies and other health problems3,4. Thus, more effective therapies for RMS will be required for further improvements in overall survival and quality of life after treatment.
The feasibility of cancer immunotherapy has recently gained considerable support from the success of proof-of-concept clinical trials 5. One approach to using the immune system to eradicate tumors is adoptive cell therapy (ACT), in which T cells are isolated from the patient, expanded and activated ex vivo, and infused back into the patient 6. Indeed, adoptive T-cell transfer in the M3-9-M model of embryonal RMS has been shown to reduce primary tumor burden and to have significant impact on growth of metastases but was unable to produce a cure7. There is also considerable interest in the use of vaccines that can strengthen tumor immunity sufficiently to achieve clinically meaningful responses 8. Dendritic cells (DCs) pulsed with lysates of RMS tumors have been shown to stimulate CD4+ T cell responses against multiple tumor antigens in murine RMS 9. Furthermore, favorable outcomes have been observed in patients receiving peptide-based vaccines combined with autologous T cells10. However, improvements in the effectiveness of immunotherapy for pediatric sarcomas are needed.
The immunosuppressive nature of the tumor microenvironment presents a major challenge to the efficacy of immunotherapy with ACT and therapeutic vaccines. In order to eradicate the tumor, T cells must home to the tumor, where they may encounter multiple immunosuppressive mechanisms that defend the tumor against immune attack. One mechanism involves the accumulation of subsets of suppressive immune cells in the tumor microenvironment including regulatory T cells (TReg), regulatory B cells and myeloid derived suppressor cell (MDSCs) 11,12. These suppressive cells have been shown to inhibit the function of adaptive immune cells targeting tumors in multiple models 13. In addition, increased numbers of regulatory T cells has been shown to be associated with worse prognosis in multiple epithelial cancers 13,14. More recently, infiltration of sarcomas with myeloid cells has also been associated with worse outcome 15. Thus, targeting suppressive immune cells has the potential to improve the efficacy of cancer immunotherapy. Indeed, immune checkpoint blockade has been shown to be effective in a number of recent clinical trials 16-18.
The first-generation compound, L-valinyl-L-boroproline (PT-100, talabostat), is known to inhibit the proteolytic activity of DASH (Dipeptidyl peptidase IV activity and/or structural homologues) enzymes including dipeptidyl peptidase (DPP)-IV-like subfamily of serine proteases comprising DPP4/CD26, DPP2, DPP8, DPP9, prolyl endopeptidase (PREP), and fibroblast activation protein (FAP) 19. The DPPs and FAP selectively cleave active forms of certain peptides after proline separated by one residue from the N-terminus, and FAP and PREP can also cleave after internal prolines 20. We and others have shown that oral administration of PT-100 mediates complete regression in multiple tumor models 21,22. In addition, PT-100 achieved partial responses in patients with stage IV melanoma, and both partial and complete responses in non-small cell lung carcinoma 23,24. Previous studies with PT-100 demonstrated that it appears to stimulate tumor immunity via the upregulation of cytokine and chemokine expression in tumor and lymphoid tissues 21. More recently, we have shown that PT-100 produces increased DC trafficking and accelerates CD4+ and CD8+ T-cell priming against a tumor-associated antigen in mice22.
Our primary objective in the present study was to determine whether ARI-4175, which is pan-inhibitor of DASH enzymes, could produce tumor regression in a preclinical model of RMS. Using the well-established MB49 immunogenic tumor model 25 in tandem with the M3-9-M RMS model7, we have demonstrated the efficacy of ARI-4175 in treatment of both early and late stage tumors. To help elucidate some of the mechanism behind the anti-tumor effects of ARI-4175, we analyzed the responses of various immune cell subsets to ARI-4175 in vivo. Our results suggest that the modulation of myeloid (m)DC and myeloid-derived suppressor cells (MDSC) by ARI-4175 in tumor-bearing mice might contribute to improved tumor immunity. Importantly for the possible future clinical development of ARI-4175, we have demonstrated the ability of ARI-4175 to enhance the activity of ACT and DC vaccination, resulting in regression of established RMS, which is resistant to treatment with ACT or DC vaccine when either is used alone.
Materials & Methods
Cell lines
MB49 26, M3-9-M 7, and 76-927 cells were cultured in 10% complete mouse media (CMM: RPMI-1640 with 10% heat inactivated FCS, 1% HEPES buffer, 1% penicillin/streptomycin/L-glutamine, 1% Non-essential amino acids and 2-mercaptoethanol 50umol/L) at 37°C in 5% CO2 (Invitrogen, Carlsbad CA). MB49 was administered to mice subcutaneously on the flank at a dose of 106 unless stated otherwise. M3-9-M and 76-9 were orthotopically administered by intramuscular injection into the gastrocnemius at 106 or 5×105 as indicated. All tumor injections were given in a total volume of 0.2mL RPMI.
Mice
C57BL/6 mice were purchased from the Animal Production Facility at NCI-Frederick and housed until 6 weeks of age. B6.129SvRag1tn1Mom/J mice were originally purchased from Jackson Laboratories (Bar Harbor, ME). Animals were kept in a specific pathogen-free facility at the NIH (Bethesda, MD) and were handled according to protocols approved by the NCI Animal Care and Use Committee.
ARI-4175
ARI-4175 was produced by the Tufts University School of Medicine Biochemistry Department (Boston, MA). Concentrated aliquots of ARI-4175 (100mM) were made by dissolving ARI-4175 in 0.1N HCl. Aliquots were diluted immediately prior to administration with saline for a final concentration of 1mg/mL. ARI-4175 was typically administered to mice daily by oral gavage (0.2cc) at a dose of 200 μg on a schedule of five days on followed by two days off unless otherwise stated. Normal saline was given orally as a control (0.2cc).
Dendritic cell vaccination
Donor C57BL/6 female mice were sacrificed and bone marrow was harvested, pooled, processed into a single cell suspension, and ACK lysed to remove red blood cells. Lineage negative cells were isolated with the AutoMACs by using a Lineage cell depletion kit (Miltenyi Biotech, Auburn, CA). Cells were maintained for 8 days in 10% CMM supplemented with GM-CSF and IL-4 (PeproTech, Inc., Rocky Hill, NJ) at 37 °C and 5% CO2. On day 6 of culture, cells were pulsed with irradiated M3-9-M cells at a 1:1 ratio to DCs. DCs were activated with 20 μg/ml of αCD40 (clone I-C10, R&D Systems, Minneapolis, MN) on day 7 and administered intraperitoneally to recipient mice on day 8 (106/0.2cc).
Adoptive Transfer of T cells
Donor C57BL/6 female mice were primed and subsequently boosted 7 days later with irradiated M3-9-M (5×106/0.2cc) or RPMI (0.2cc) by intraperitoneal injection. One week following the boost, spleens and axillary, brachial, and inguinal LNs from donor mice were harvested, pooled, processed into a single cell suspension, and ACK lysed to remove red blood cells. From this suspension, T cells were purified with the AutoMACs using a Pan T cell isolation kit II (Miltenyi Biotech, Auburn, CA). In C57BL/6 recipients, purified T cells (5.7×106) were adoptively transferred on day 10 after tumor challenge. In Rag1-/- recipients, purified T cells (8×106) were adoptively transferred one day after tumor challenge.
Flow Cytometry
Tissues were processed into single cell suspensions and ACK lysed to deplete red blood cells. Before staining for cell surface markers, FcγIII/II receptors were blocked (2.4G2). Cells were labeled with one of the following antibody panels: (1) Ly6-G-FITC, CD11c-PE-Cy7, CD11b-PerCP-Cy5.5 Ly6-C-APC; (2) Gr-1-FITC, CD11c-PE, CD11b-PerCP-Cy5.5, CD4-PE-Cy7, F4/80-APC, CD8a-PacificBlue; (3) Ly6-G-FITC, IL-4Rα-PE, Ly6-C-PerCP-Cy5.5, CD11c-APC, CD11b-PacificBlue; (4) CD4-FITC, CD8a-PE, B220-PerCP-Cy5.5, NK1.1-APC. Cells were incubated with antibodies for 30 minutes at 4°C and then washed and analyzed in FACS buffer (0.1% sodium azide, 0.5% fetal bovine serum, 2mM EDTA). Flow cytometry was performed using a seven channel LSRFortessa® and FACSDiva software (BD, Franklin Lakes, NJ). Further analysis was completed using FlowJo software, version 9.1 for Macintosh (Tree Star Inc., Ashland, OR).
MDSC in vitro Suppression Assay
C57BL/6 mice were given 1×106 M3-9-M tumor followed by either saline or ARI-4175 treatment from day 3-7. On day 10 spleens were harvested, Gr1+ cells were isolated using magnetic selection (Miltenyi Biotech) and cultured with purified CD4+/CD8+ T-cells labeled with CellTrace Violet (Invitrogen) at a ratio of 1:3 and stimulated with 5×104 CD3/CD28 beads. Cells were plated in 96 well round bottom plate in CMM containing physiologic levels of L-arginine (150uM). Cells were harvested on day 4 and analyzed by flow cytometry for dye dilution. Suppression was calculated by comparing the fraction of dividing cells in wells containing Gr1+ cells to the fraction dividing the absence of Gr1+ cells.
Real Time PCR
MDSC were isolated on day 8 using Gr1+ magnetic bead separation (Milteyni Biotech) from the spleens of mice given 1×106 M3-9-M cells and either saline or ARI-4175 on days 3-7. RNA was extracted using RNAqueous4PCR kit (Ambion) and cDNA was synthesized using RT2 SYBR Green ROX qPCR Mastermix (Qiagen). RT2 Profiler PCR Array for mouse chemokines and receptors was used to examine the differences in gene expression between untreated and 4175 treated MDSC samples. All samples were run on StepOnePlus real-time PCR system (Applied Biosystems) and analyzed using the ΔΔCT method.
Statistical Analysis
All statistical analysis was performed using GraphPad Prism software, version 4.0c (GraphPad Software Inc, La Jolla, CA). Two-tailed Mann-Whitney tests were used to examine the statistical differences between two treatment groups. One-way ANOVA (Kruskal-Wallis test and Dunn's Multiple Comparison test) was used to test significance of tumor volumes between 3 or more groups in tumor growth experiments. For tumor growth analysis with only two groups, two-tailed Mann-Whitney tests were performed on data points from groups on a specific day, as indicated. A score of p<0.05 for all tests was considered significant.
Results
Orally administered ARI-4175 induces tumor regression in multiple tumor models and results in immunologic memory that protects against rechallenge
We first tested ARI-4175 in mice transplanted with the highly immunogenic bladder epithelia carcinoma cell line, MB49, which expresses the male HY minor histocompatibility antigen. Starting on day 3 after inoculation of MB49 subcutaneously in the flank, ARI-4175 was administered daily at a dose of 200 μg per mouse by oral gavage following a schedule of 5 consecutive days of dosing interspersed by 2 days of no treatment (hereafter referred to as the early treatment schedule). As shown in Figure 1A, 2 weeks of ARI-4175 mediated complete regression in all mice. We next compared the antitumor activity of ARI-4175 when administered by i.p. injection versus gavage in order to determine the appropriate route of administration for use in subsequent studies. Comparison of tumor volumes between mice that received ARI-4175 orally and mice that received ARI-4175 i.p. revealed no difference in the potency of the antitumor effect (data not shown). We subsequently used gavage as the primary route of administration because of the desirability of an orally bioavailable agent for potential clinical application. Next, we tested the antitumor activity of ARI-4175 in two syngeneic models in which C57BL/6 mice were orthotopically implanted in the gastrocnemius muscle with M3-9-M and 76-9 RMS cell lines, 27. M3-9-M also expresses the HY antigen complex, whereas 76-9 does not (data not shown). Following tumor inoculation, 200 μg of ARI-4175 was administered by the early treatment schedule for 3 weeks. As shown in Figures 1B and 1C, ARI-4175 produced regression in 3/6 mice bearing 76-9 (p=0.002 compared to saline) and completely eradicated M3-9-M in 6/6 mice.
Figure 1. Early treatment with ARI-4175 induces regression in multiple tumor models and induces tumor-specific immunological memory.

(A) Female C57BL/6 mice were challenged subcutaneously with MB49 (1×106 cells) on day 0 and treated with saline or 200 μg ARI-4175 by oral gavage on days 3-7 and 10-14. Two weeks of ARI-4175 treatment produced complete regression in all mice (n=7, p<0.001). Representative of 4 independent experiments. (B and C) Female C57BL/6 mice were challenged intramuscularly with the murine RMS lines, M3-9-M (1×106 cells, n=10/group, representative of 3 independent experiments) or 76-9 (5×105 cells, n=6/group, representative of 2 independent experiments) on day 0 and treated with saline or 200 μg ARI-4175 by oral gavage on days 3-7, 10-14, and 17-21. ARI-4175 induced regression in all mice challenged with M3-9-M (B) and in 3/6 mice challenged with 76-9 (C). (D) Female C57BL/6 mice were challenged with M3-9-M as in (B) and orally dosed with ARI-4175 on days 3-7. Treatment resumed on day 14 and continued on a schedule of 5 days treatment with 2 days rest until day 42 when the mice exhibited complete tumor regression. On day 56, mice were rechallenged with M3-9-M (5×105 cells) in the opposite leg. With no further treatment following rechallenge, ARI-4175-treated survivors exhibited initial tumor growth followed by rejection, indicative of immunological memory, whereas M3-9-M grew rapidly in naïve controls. * p<0.05, ***p<0.01 Representative of 2 independent experiments. (E) Rag1-/- mice were challenged with M3-9-M (1×105 cells, n=5/group) and treated with saline or 200 μg ARI-4175 by oral gavage on days 3-7 and 10-14 as in figure (B).
In order for the first generation DPP4-like serine protease inhibitor, PT-100, to induce tumor regression and rejection in vivo, CD4+ and CD8+ T cells and CD11c+ DCs are required22. Hypothesizing that the antitumor activity of ARI-4175 might also involve adaptive immunity, we investigated mice that had rejected primary M3-9-M tumors in response to early treatment with ARI-4175 for resistance to rechallenge with M3-9-M on day 56 following initial tumor challenge. After a short period of initial tumor growth, the rechallenged mice rapidly rejected the secondary tumors without any further ARI-4175 treatment (Figure 1D, n = 7, p < 0.05) but were unable to reject 76-9 (data not shown), which is consistent with tumor-specific immunological memory response to M3-9-M tumor antigens. Finally, we challenged Rag1-/- mice with M-3-9M and treated with ARI-4175. As shown in Figure 1E, the absence of adaptive immune cells completely abrogated the anti-tumor activity of ARI-4175 confirming that tumor eradication is immunologically mediated and that direct antitumor activity of ARI-4175 is not sufficient.
The anti-tumor effect of ARI-4715 correlates with a dose-dependent increase in recruitment of myeloid cells to secondary lymphoid tissues
In order to further investigate how ARI-4175 mediates tumor regression we analyzed blood cells and lymphoid tissues for quantitative changes in leukocytes and immune cell subsets. Female C57BL/6 mice were inoculated s.c. with MB49 (1×106) and administered saline or ARI-4175 at daily doses of 5 and 200 μg by the early treatment schedule. After 9 days, blood was collected for complete blood cell counts (CBC), and tumor-draining lymph nodes (TDLNs) and spleen were harvested for FACS analysis. The total and differential white blood cell counts and erythroid cell numbers in the blood were unaltered by ARI-4175 treatment (data not shown). However, there was an increase in the platelet count from approximately 2 × 105/μL in saline-treated mice to 7 × 105/μL in both 5 μg and 200 μg ARI-4175 treated mice (n = 5 per group, p < 0.01 for both doses compared to saline, Figure 2A). In both TDLNs and spleens of mice treated with 200 μg ARI-4175, the absolute numbers and frequencies of CD11b+CD11c+ myeloid (m)DCs (Figure 2B and 2C) and CD11b+CD11c- myeloid cells (Figure 2D) increased significantly but were not affected at the 5 μg dose level. Plasmacytoid (p)DCs (CD11b-CD11c+) increased in frequency and absolute number in TDLNs of mice treated with 200 μg ARI-4175 and increased in frequency, but not in absolute number, in the spleen (Figure 2E). To further analyze the myeloid cell populations affected by ARI-4175, we investigated the expression of Ly6 C and Ly6 G on gated CD11b+CD11c- cells. These markers define monocytic (CD11b+Ly6ChiLy6Glo) and granulocytic subsets (CD11b+Ly6 CloLy6 Ghi) of myeloid derived suppressor cells (MDSCs). In the mice that received 200 μg ARI-4175, the most significant change occurred in the TDLN where there was a substantial shift toward the granulocytic MDSCs (Figure 2F). Changes in splenic MDSCs were more modest but there was a significant decrease in the granulocytic fraction.
Figure 2. ARI-4175 modulates the myeloid cell populations in secondary lymphoid tissues.


Mice were injected with MB49 and treated with ARI-4175 as described in Figure 1. (A) On day 9 complete blood counts showed a significant increase in platelet count at both the 5 μg and 200 μg ARI-4175 doses. (B) Splenocytes from tumor-challenged mice analyzed on day 11 of ARI-4715 treatment for expression of CD11c and CD11b showed an increase in CD11b+ cells at the 200 μg dose. (C-E) Changes in the frequency and absolute number of CD11b+CD11c+ mDCs (C), CD11b+CD11c- myeloid cells (D), and CD11b-CD11c+ pDCs (E) on day 11 in spleen and TDLN of ARI-4175 treated MB49-challenged mice. (F) Further characterization of CD11b+CD11c- myeloid cells in the spleen and TDLN demonstrated an increase in proportion of Ly6CloLy6Ghi granulocytic myeloid cells and a decrease in proportion of Ly6ChiLy6Glo monocytic cells in TDLN with ARI-4175 treatment. A modest decrease in proportion of Ly6CloLy6Ghi granulocytic myeloid occurred in the spleen. * p<0.05, **p<0.01. n=6 mice per group. Data representative of 3 independent experiments.
With the goal of identifying a potential biomarker for the ARI-4175 anti-tumor effect and establishing the active dose level, we investigated the effects of early treatment with 5, 10, 50, 100 and 200 μg doses of ARI-4175 in the MB49 tumor model. A cohort of mice was sacrificed on day 11 for FACS analysis of TDLNs and spleen, and the remaining mice continued to receive treatment until day 14 and were monitored for tumor growth. ARI-4175 had no effect on MB49 tumor growth at the 5 and 10 μg dose levels; however, evidence of anti-tumor activity became apparent at 50 μg, and tumor regression was observed at 100 and 200 μg (Figure 3A). ARI-4175 was well tolerated up to at least 200 μg daily, at which dose complete tumor regression was consistently achieved. There were no changes in total numbers of CD4+ or CD8+ T cells (Figure 3B) or in the frequency of naïve (CD44-CD62L+), central memory (CD44+CD62L+) or effector memory (CD44+CD62L-) CD4+ (Figure 3C) or CD8+ subsets (data not shown) in spleen or TDLN of ARI-4175 treated mice at any dose. There were also no observed changes in B cells or NK cells related to ARI-4175 dose or tumor volume (data not shown). Treatment with ARI-4175 increased the absolute number of CD11b+ CD11c+ mDCs and CD11b+ CD11c- myeloid cells in TDLNs and spleen in a dose-dependent manner, and the increases became significant at dose levels that were sufficient to produce a tumor response (Figure 3D and E ). When the CD11b+ CD11c- cells were further analyzed for expression of the F4/80 macrophage marker and the Gr-1 granulocyte marker the frequency of the F4/80+ Gr-1+ double positive subset was found to increase significantly in response to ARI-4175 in a dose-dependent manner in TDLNs and with a non-significant trend towards increased frequency in spleen (Figure 3F).
Figure 3. ARI-4175 induces dose-dependent anti-tumor activity and myeloid cell expansion in lymph nodes.
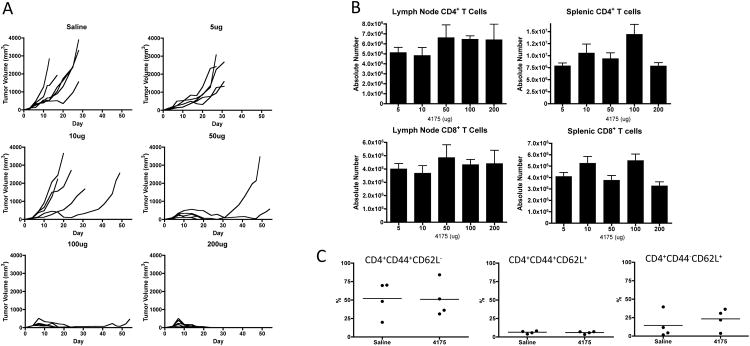

Following subcutaneous MB49 challenge (106 cells), C57BL/6 mice were gavaged with saline (n=6) or ARI-4175 at doses: 5 μg, 10 μg, 50 μg, 100 μg, or 200 μg (n=5-6/group). (A) Individual tumor curves demonstrate anti-tumor activity of ARI-4175 at doses of ≥ 50 μg without noticeable toxicity. (B-E) C57BL/6 mice were orally gavaged as described in (A), sacrificed on day 11, and tissues were harvested, processed, and stained for FACS analysis. Absolute number of CD4+ or CD8+ T cells (B) or phenotype of CD4 + T cell subsets (C) were not altered by ARI-4175 treatment. (D and E) Myeloid cells (CD11b+CD11c-) and mDCs (CD11b+CD11c+) showed dose-dependent increases in lymphoid tissues of ARI-4175 treated mice. (F) F4/80+Gr-1+ myeloid cells increased in frequency in a dose-dependent manner in lymph nodes, but not in spleens, of ARI-4175-treated mice. * p<0.05, **p<0.01. Data representative of 2 (Figure 3B-F) or 4 (Figure 3A) experiments.
ARI-4175 treatment decreases the frequency of monocytic MDSCs and the suppressive function of immature myeloid cells within the tumor
Based on the changes in myeloid cell subsets in secondary lymphoid tissues we hypothesized that ARI-4175 might affect the tumor-infiltrating myeloid cell population, subsets of which can negatively impact adaptive immune responses against tumors. Mice were challenged with MB49, administered ARI-4175 or saline by the early treatment schedule, and sacrificed for analysis on day 9 when tumor sizes were similar in both groups (as shown in Figure 1). Tumors were harvested and single-cell suspensions were analyzed by flow cytometry. CD11b+ CD11c- myeloid cells were subgated into Ly6ChiLy6Glo (monocytic) and Ly6CloLy6Ghi (granulocytic) subsets. As seen in the TDLNs (Figure 2G), there was a marked decrease in the proportion of tumor infiltrating monocytic MDSCs in mice treated with ARI-4175 (Figure 4A and B) accompanied by a proportional increase in the frequency of the granulocytic fraction (Figure 4A and C). In the spleen, there was an increase in monocytic MDSCs with a modest, but non-significant, decrease in granulocytic MDSCs. The changes appeared to be dose dependent and generally achieved significance at the 100- and 200-μg dose levels of ARI-4175 that are required to produce tumor regression.
Figure 4. Monocytic, but not granulocytic, MDSCs are reduced within tumors of ARI-4175-treated mice.

(A-C) Female C57BL/6 mice were challenged with MB49 and treated as described in Figure 3A. On day 11, spleens and tumors were harvested and stained for FACS analysis. (A) Monocytic MDSCs were identified by gating on CD11b+CD11c- cells followed by analysis of Ly6-C versus Ly6-G levels. Cells in the upper left-hand quadrant (Ly6-ChiLy6-Glo) are referred to as monocytic MDSCs, and cells in the lower right-hand quadrant (Ly6-CloLy6-Ghi), as granulocytic MDSCs. (B) Monocytic MDSCs within the tumor decreased significantly at the 100 μg and 200 μg doses of ARI-4175 as compared to lower doses. The opposite trend was observed in the spleen, where a significant increase in monocytic MDSCs occurred at the 200 μg ARI-4175 dose. (C) Granulocytic MDSCs exhibited a significant increase within the tumor at the two highest doses of ARI-4175, and they seemed to decrease with dose within the spleen, although not significantly. * p<0.05, **p<0.01 n=6 mice/group. Data representative of 2 independent experiments. (D) Female C57BL/6 mice were challenged with M3-9-M (1×106 cells, n=6/group) and treated with saline or 200-μg ARI-4175 by oral gavage on days 3-7 and days 10-14. On Day 14, Gr1+ cells were isolated from the spleen (n=6 mice/group) and analyzed by qPCR. Tables show genes with expression differences in the treated mice of 5 fold or more compared to saline treated. (E) Female C57BL/6 mice were challenged with M3-9-M (1×106 cells, n=6/group) and treated with saline or 200-μg ARI-4175 by oral gavage on days 3-7 and days 10-14. On Day 14, Gr1+ cells were isolated from the spleen (n=6 mice/group), pooled and added CFSE-labeled T cells in triplicate at a ratio of 1:3. Suppression was determined as described in methods. * p<0.05
We hypothesized that changes in MDSC subsets were partially due to alterations in myeloid cell trafficking. To test this, Gr1+ cells from the spleens of M3-9-M tumor-bearing mice were analyzed for chemokine and chemokine receptor gene expression. As shown in figure 4D, ARI-4175 induced changes in the expression of a number of chemokine receptors and ligands further suggesting that ARI-4175 may alter the chemokine-mediated trafficking pattern of suppressive myeloid cells. Finally, the effect of 4175 on the overall suppressive capacity of tumor-infiltrating myeloid cells in mice bearing M3-9-M tumors was assessed. Gr1+ myeloid cells were isolated from spleens ARI-4175 or saline treated mice and tested for the ability to suppress T cell proliferation in vitro. As shown in Figure 4E, Gr1+ myeloid cells from tumor-bearing mice suppress T cell proliferation but this suppression is statistically reduced with ARI-4175 treatment. Taken together, these results suggest that at least some of the anti-tumor activity of ARI-4175 results from trafficking and functionality of suppressive myeloid cells.
ARI-4175 produces significant tumor responses in a model of established RMS, both as a single agent and as an adjuvant to a DC vaccine or adoptively transferred tumor antigen-primed T cells
In order to investigate whether ARI-4175 could produce regression of established tumors, treatment of C57BL/6 mice orthotopically inoculated with M3-9-M cells (106) was delayed until day 10 and continued for 5 weeks. As in the early treatment schedule, 200-μg of ARI-4175 was administered by daily gavage for periods of 5 consecutive days interspersed by 2 days of no treatment. Although M3-9-M tumor volumes approached 2000 mm3 at the start of ARI-4175 treatment, 5/7 mice completely rejected their tumors and survived (p < 0.01; Figure 5A).
Figure 5. Inhibition of established M3-9-M tumors by ARI-4175 in combination with DC vaccination or ACT.

(A) Individual tumor-growth curves in female C57BL/6 mice that were challenged intramuscularly with M3-9-M (1×106 cells) on day 0. Treatment with 200 μg ARI-4175 started on day 10 when tumors were well established, and ended on day 42. Although two ARI-4175-treated mice had to be euthanized around day 20 due to large tumor burden, tumors in the remaining mice regressed completely and did not recur after termination of treatment. Data representative of 3 independent experiments. (B and C) M3-9-M challenge and ARI-4175 treatment follow the schedule described in (A), except that ARI-4175 treatment was discontinued one week earlier on day 35. An RPMI sham- or a DC-vaccine (106) was administered intraperitoneally on day 14. (B) Individual tumor curves demonstrating eradiation of tumors in 2/7 mice with ARI-4175 alone and 5/7 mice with a combination of ARI-4175 and DC vaccination. There was no effect with DC vaccination alone. (C) Survival plot showing significantly improved survival with ARI-4175 alone or ARI-4175 plus vaccine (both <0.001 compared to saline). The survival between the ARI-4175+DC Vaccination was significantly improved compared to ARI-4175 plus sham vaccination (p<0.05). Data representative of 2 independent experiments. (D and E) M3-9-M challenge and ARI-4175 treatment follow the same schedule described in (B and C). T cells (5.7×106) from either primed or naïve donors were administered IV by tail vein injection on day 10. (D) Individual tumor curves show complete regressions in 5/10 mice treated with ARI-4175 and naïve T cells and in 8/10 mice treated with ARI-4175 and primed T cells. (E) When compared with saline controls, both ARI-4175 treated groups had significantly improved survival compared to saline (p<0.05 for ARI-4175 plus naïve T cells vs saline and p<0.001 for ARI-4175 plus primed T cells vs saline), however the difference between these groups was not significant. Data representative of 2 independent experiments.
The first generation DASH inhibitor, PT-100, has been shown to produce tumor regression and rejection by activating endogenous tumor immunity and by enhancing responses to a tumor vaccine 21,28. The establishment of tumor-specific immunological memory in mice that had rejected M3-9-M tumors as a result of ARI-4175 treatment suggests that tumor immunity also contributes to the antitumor activity of this agent. Therefore, we hypothesized, that, ARI-4175 might improve the response to therapeutic vaccination against M3-9-M. Because ARI-4175 was effective as a single agent, even when treatment was delayed until the tumors were established, we reduced the period of administration to 4 weeks in order to investigate the interaction with a DC-based M3-9-M tumor vaccine (M3-9-M DC vaccine). The vaccine was produced from C57BL/6 bone marrow cells cultured with GM-CSF and IL-4 for one week, loaded with irradiated M3-9-M tumor cells, and incubated with anti-CD40 antibody to activate the differentiated DCs. The M3-9-M DC vaccine (106 cells per mouse) and sham vaccine (unloaded, activated DCs) were administered by IP injection on day 14 after tumor inoculation either alone or with 4-week course of ARI-4175 started on day 10 after tumor inoculation. A 4-week course of ARI-4175 alone was sufficient to mediate regression in only 2/7 mice (Figure 5B and C). Although the M3-9-M DC vaccine is sufficient to protect against tumor challenge 7, it is insufficient as a therapeutic vaccine. However, the addition of DC vaccine to ARI-4175 improved the therapeutic efficacy resulting in eradication of tumors in 5/7 mice.
The apparent adjuvant effect of ARI-4175 in combination with the M3-9-M DC vaccine suggested enhancement of T cell-mediated anti-tumor immunity by ARI-4175. Thus, we next investigated whether ARI-4175 could enhance tumor responses produced by the adoptive transfer of T cells primed against tumor antigens. Donor mice were primed with irradiated M3-9-M tumor cells and boosted one week later, and their splenic T cells (5.7 × 106) were isolated and transferred intravenously to tumor-bearing recipients that had been inoculated with M3-9-M tumor cells 14 days earlier. ARI-4175 was given for 4 weeks, starting on day 10 after tumor inoculation. ARI-4175 combined with T cells from unprimed donors eradicated tumors in 3/7 mice, and when combined with tumor-primed T cells, 5/7 mice were cured (Figure 5D and E).
Adoptive cell therapy combined with ARI-4175 significantly improves tumor responses in lymphopenic recipients
The effectiveness of adoptive immunotherapy is increased in the setting of lymphopenia 29. In order to investigate whether the efficacy of ACT accompanied by treatment with ARI-4175 might be improved in a lymphopenic environment, we utilized Rag1-/- mice as tumor-bearing recipients of tumor-primed T cells. T cells (8 ×106) were isolated from the spleens of naïve or M3-9-M tumor-primed female C57BL/6 mice and transferred to Rag1-/- female mice that had been inoculated with M3-9-M tumor cells (1×105) one day earlier. The mice were then treated with 100-μg ARI-4175 or saline for five weeks (Figure 6A). In the absence of host-derived T cells, early initiation of ARI-4175 treatment by itself had no effect on tumor growth, further confirming the role lymphocytes play in the therapeutic effect of this compound (Figure 6B-D). The transfer of naïve T cells did not improve the efficacy of ARI-4175 at the dose used. Primed T cells alone appeared to modestly, but not significantly, delay tumor growth in some mice (Figure 6C) but all succumbed to tumor. However, the addition of ARI-4175 to the transfer of primed T cells produced a significant delay in tumor development and an apparent cure in some mice.
Figure 6. As an adjuvant to ACT in immunodeficient hosts, ARI-4175 inhibits M3-9-M tumors and increases survival.

(A) Female Rag1-/- mice were challenged intramuscularly with M3-9-M (1×105) on day 0. Purified splenic T cells (8×106) from M3-9-M primed or naïve C57BL/6 mice were adoptively transferred to Rag1-/- mice by i.v. tail injections one day after tumor challenge. Mice were treated with 100 μg ARI-4175 or saline for 5 weeks starting on day 3. (B) Individual tumor curves demonstrating regression of tumors only with ARI-4175 plus primed T cells. (C) Mice given primed T cells seemed to exhibit slower tumor growth compared to controls, however only mice that received both primed T cells and ARI-4175 exhibited a clear decrease in tumor volume. (D) Mice given ARI-4175 and primed T cells had significantly improved survival compared to all other treatment groups. **p<0.01, ***p<0.001. Data representative of 2 independent experiments.
Discussion
We have demonstrated that oral administration of ARI-4175 can produce tumor regression and eradication in mice transplanted with the MB49 bladder tumor cell line and the M3-9-M or 76-9 RMS cell lines. It was previously reported that a functional adaptive immune system was required for the earlier compound, PT-100, to produce tumor rejection in syngeneic tumor transplant models 21. We found that the antitumor effect of ARI-4175 was completely absent in Rag1-/- mice transplanted with the M3-9-M cell line, indicating a similar dependence on adaptive immunity. Protective immunological memory was also established in mice that had rejected primary M3-9-M tumors as a result of an initial course of treatment with ARI-4175. The memory response was able to prevent the development of tumors after secondary challenge with M3-9-M tumor cells without any requirement for additional treatment with ARI-4175. The M3-9-M tumor cell line was derived from a RMS that arose in a hepatocyte growth factor transgenic (HGFTg+) p53+/- mouse that was full backcrossed to the C57BL/6 mouse strain 7,30. When C57BL/6 mice are transplanted with M3-9-M cells, aggressive tumors develop that have been reported to resemble embryonal RMS in human patients, both morphologically and in the transcriptional signature of gene expression 7. Immunotherapeutic approaches that demonstrate high efficacy in preclinical tumor models very often fail to produce clinically meaningful responses in human cancer 31. This has been attributed in established cancer to the development of an immunosuppressive microenvironment that involves DC-mediated T-cell tolerance, MDSCs, immunosuppressive tumor-associated fibroblasts, and the predominance of TReg cells over TEff cells that are competent to perform the functions required for tumor killing 12,32-34. Therefore, it is noteworthy that ARI-4175 could produce tumor regression when used to treat mice bearing established M3-9-M tumors that approached 2000 mm3 in volume.
Changes in the content of myeloid cells and DCs in lymphoid and tumor tissues appeared to accompany the tumor response produced by ARI-4175 in the M3-9-M RMS model. These changes are provocative in the light of the ability of ARI-4175 to stimulate tumor immunity, because DC act as professional antigen-presenting cells required for the cross-priming of T-cell responses against tumor antigens 35, and myeloid cells appear to include subsets belonging to the population of MDSCs that are thought to dampen the activation of T-cell responses against tumor antigens in the tumor microenvironment 12. DCs migrate in response to a cognate interaction between CCR7 of the DCs and chemotactic gradients of CCL19 and CCL21 cytokines. The ability of tumor cells to prevent the maturation and the function of DCs can contribute to the immune escape of tumors. Tumors appear to inhibit the migration of DCs by the release of prostaglandin E2 (PGE2) that signals IL-10-dependent downregulation of CCL19 36,37, and by the release of liver X receptor (LXR) ligands that inhibit expression of CCR7 on maturing DCs 38. ARI-4175 produced significant increases in the numbers of mDCs and pDCs resident in TDLNs, and of mDCs in spleen, suggesting that the agent might restore the migration of DCs that is otherwise defective in the tumor-bearing mouse. MDSCs comprise myeloid progenitors and immature myeloid cells identified by co-expression of cell surface CD11b and Gr-1 and the absence of cell-surface markers specific for mature cell types, such as the CD11c marker of mDCs 39,40. ARI-4175 appeared to have differential effects on the absolute numbers and frequencies of the granulocytic and monocytic MDSCs identified by the Ly-6 markers in tumor and lymphoid tissues. In MB49 tumor-bearing mice, ARI-4175 produced overall increases in the absolute number and frequency of CD11b+CD11c- myeloid cells in spleen and TDLNs; but the frequencies of CD11b+Ly-6ChiLy-6Glo monocytic MDSCs and CD11b+Ly-6CloLy-6Ghi granulocytic MDSCs in TDLNs, tumor and splenic tissues were altered differently: the frequency of monocytic MDSCs decreased significantly in TDLNs and tumor tissues but increased in the spleen, whereas the frequency of granulocytic MDSCs increased significantly in TDLNs and tumor tissue but decreased in the spleen. It has been reported in ex vivo assays that monocytic MDSCs are more potent suppressors of antigen-stimulated T cell responses than granulocytic MDSCs, and, moreover, the monocytic MDSCs appear to undergo differentiation into macrophage-like cells with highly potent antiproliferative effects against activated T cells 41. In addition, the reduced suppressive capacity of splenic myeloid cells in the spleens of ARI-4175 treated mice suggests function changes induced by DASH inhibition. Consequently, it is highly plausible that part of the immunostimulatory activity of ARI-4175 is due to relief of immune suppression by modulation of trafficking and/or suppressive capacity of myeloid cell populations, a novel feature of DASH inhibition as an adjuvant.
The results obtained with ARI-4715 in combination with a DC vaccine and adoptively transferred tumor antigen-primed T cells in the M3-9-M RMS model suggest that ARI-4175 might be used clinically for the enhancement of immunotherapy with cancer vaccines and ACT. The US Food and Drug Administration approved the sipuleucel-T (Provenge) DC-based vaccine, for treatment for of advanced prostate cancer in April 2010 42. Despite this success, the development of DC-based cancer vaccines continues to be impeded by lack of knowledge of the optimal tumor antigens for DC loading ex vivo and the lack of optimal adjuvants 5. In the M3-9-M DC vaccine used in our studies, we have surmounted the issue of optimal antigen selection by loading the DCs with apoptotic tumor bodies, thereby enabling presentation of multiple antigenic peptides in association with MHC class I and II proteins. The apparent ability of ARI-4175 to enhance tumor responses in combination with M3-9-M DC vaccine suggests that ARI-4175 might provide a new type of adjuvant that can be administered orally and tested in combination with a variety of cancer vaccines in the clinic. Rosenberg et al have discovered that adoptive transfer of tumor-infiltrating lymphocytes isolated from the patient in combination with IL-2 treatment can produce durable and complete tumor regression in up to 40% of patients with metastatic melanoma when used after nonmyeloablative lymphodepletion with chemotherapy 43. A similar ACT approach with adoptively transferred autologous T cells transduced with a HLA class I-restricted T-cell receptor recognizing a NY-ESO-1 cancer/testis peptide epitope produced objective clinical responses in 4 out of 6 patients with synovial cell sarcoma and 5 out of 11 patients with melanoma, including 2 complete regressions of greater than 1 year in duration 44. Although these results are impressive, additional immunotherapy might be required to further improve the durable response rate. The significant improvement in survival that we obtained by adding ARI-4175 to ACT with M3-9-M tumor-primed T cells in lymphopenic Rag1-/- mice provides a rationale for clinical investigation of the activity of ARI-4175 combined with ACT in the setting of lymphopenia produced by chemotherapy.
Historically, clinical development of anticancer agents has depended on Response Criteria in Solid Tumors (RECIST) for the demonstration of the efficacy of investigational anticancer agents in human cancer patients 45,46. However, because measurable tumor responses to immunotherapy may be delayed for months while the human patient develops immunity, prolonged survival may be a more appropriate measure of clinical benefit. Indeed, this appeared to be the case in trials of sipuleucel-T 47. In addition, tumor biopsies have revealed that tumor infiltration by immune cells can be accompanied by edema followed by fibrosis that might interfere with the use of RECIST to evaluate clinical responses to immunotherapy 48. Consequently, for any future clinical development of ARI-475 it will be important to have biological markers available that are predictive of clinical benefit. We have demonstrated that significant increases in the content of mDCs and myeloid cells in TDLNs appear to correlate with tumor regression in response to ARI-4175. In phase 1 safety trials of ARI-4175, analysis of these cell types in biopsies of TDLNs might give an early indication of whether immune modulation can be achieved at dose levels that are well tolerated.
The stimulation of endogenous tumor immunity by PT-100, was accompanied by upregulated expression of cytokines and chemokines mRNA in tumors and TDLNs 21. At the time it was hypothesized that PT-100 might promote T-cell priming and activation by increasing the levels of chemokines responsible for the trafficking of T cells and antigen-presenting cells between the tumor and the TDLNs; however, no direct evidence was provided for this. Similar to PT-100, ARI-4175 is a DASH inhibitor. In the studies reported here, we have found that the trafficking of mDCs, pDCs and MDSCs appears to be modified by ARI-4175. Indeed, treatment of tumor bearing hosts with ARI-4175 induced changes in the chemokine and chemokine receptor gene expression profile of Gr1+ myeloid cells. Sedo and Malik have suggested that conserved prolines might act as regulatory elements in biologically active peptides for which post-prolyl peptidases provide checkpoints 49. Interestingly, the trafficking of MDSCs is guided by interaction of their CCR2 with gradients of CCL2 and CCL7 chemokines 50, both of which contain prolines at the second position from the amino terminus of the mature peptide 51. CCL2 and CCL7 are, therefore, potentially cleaved in vivo by DPP4, and it has been reported that deletion of the first two residues results in the conversion of CCL2 from an agonist into an antagonist of CCR2 51. It seems feasible that ARI-4175 might preserve the chemotactic activity of CCL2 and CCL7 by inhibiting DPP4 proteolytic activity, and thereby alter the trafficking of MDSCs. Another member of the DPP4-like protease subfamily, FAP, is selectively expressed in the stroma of many solid tumors 52; therefore, ARI-4175 might also modulate chemokine activity in the tumor microenvironment by inhibition of FAP 53. In a preliminary study, Jesson et al reported that PT-100 could stimulate the transcriptional upregulation of cytokines and chemokines in cultures of macrophages, and that this response could also be initiated by selective inhibitors of the intracellular DPP4-like proteases, DPP8 and DPP9 54. It is possible that ARI-4175 and PT-100 might promote tumor immunity by a combination of transcriptional and post-translational effects on chemokines mediated via the inhibition of different members of the DPP4-like subfamily. We have synthesized a panel of selective pharmacological inhibitors of individual DPP4-like proteases, and we are using these to probe the roles DPP4, DPP8/9 and FAP in the regulation of tumor immunity.
Acknowledgments
Support: This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Disclosures: The content of this publication does not necessarily reflect the views and policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Financial Disclosure: BJ and WWB receive consulting fees and grant support from Arasaph Pharmaceuticals. All other authors have no financial disclosures to declare.
References
- 1.Parham DM, Ellison DA. Rhabdomyosarcomas in adults and children: an update. Arch Pathol Lab Med. 2006;130:1454–65. doi: 10.5858/2006-130-1454-RIAACA. [DOI] [PubMed] [Google Scholar]
- 2.Malempati S, Hawkins DS. Rhabdomyosarcoma: review of the Children's Oncology Group (COG) Soft-Tissue Sarcoma Committee experience and rationale for current COG studies. Pediatr Blood Cancer. 2012;59:5–10. doi: 10.1002/pbc.24118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oeffinger KC, Mertens AC, Sklar CA, et al. Chronic health conditions in adult survivors of childhood cancer. N Engl J Med. 2006;355:1572–82. doi: 10.1056/NEJMsa060185. [DOI] [PubMed] [Google Scholar]
- 4.Punyko JA, Mertens AC, Gurney JG, et al. Long-term medical effects of childhood and adolescent rhabdomyosarcoma: a report from the childhood cancer survivor study. Pediatr Blood Cancer. 2005;44:643–53. doi: 10.1002/pbc.20310. [DOI] [PubMed] [Google Scholar]
- 5.Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480:480–9. doi: 10.1038/nature10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosenberg SA. Cell transfer immunotherapy for metastatic solid cancer--what clinicians need to know. Nature reviews Clinical oncology. 2011;8:577–85. doi: 10.1038/nrclinonc.2011.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meadors JL, Cui Y, Chen QR, et al. Murine rhabdomyosarcoma is immunogenic and responsive to T-cell-based immunotherapy. Pediatr Blood Cancer. 2011;57:921–9. doi: 10.1002/pbc.23048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nature reviews Cancer. 2012;12:265–77. doi: 10.1038/nrc3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rodeberg DA, Erskine C, Celis E. In vitro induction of immune responses to shared tumor-associated antigens in rhabdomyosarcoma. Journal of pediatric surgery. 2007;42:1396–402. doi: 10.1016/j.jpedsurg.2007.03.041. [DOI] [PubMed] [Google Scholar]
- 10.Mackall CL, Rhee EH, Read EJ, et al. A pilot study of consolidative immunotherapy in patients with high-risk pediatric sarcomas. Clin Cancer Res. 2008;14:4850–8. doi: 10.1158/1078-0432.CCR-07-4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kerkar SP, Restifo NP. Cellular Constituents of Immune Escape within the Tumor Microenvironment. Cancer research. 2012;72:3125–30. doi: 10.1158/0008-5472.CAN-11-4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nature reviews Immunology. 2009;9:162–74. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nature reviews Immunology. 2006;6:295–307. doi: 10.1038/nri1806. [DOI] [PubMed] [Google Scholar]
- 14.Curiel TJ, Coukos G, Zou L, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nature medicine. 2004;10:942–9. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 15.Finkelstein SE, Iclozan C, Bui MM, et al. Combination of external beam radiotherapy (EBRT) with intratumoral injection of dendritic cells as neo-adjuvant treatment of high-risk soft tissue sarcoma patients. International journal of radiation oncology, biology, physics. 2012;82:924–32. doi: 10.1016/j.ijrobp.2010.12.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hodi FS, O'Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–65. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–54. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Connolly BA, Sanford DG, Chiluwal AK, et al. Dipeptide boronic acid inhibitors of dipeptidyl peptidase IV: determinants of potency and in vivo efficacy and safety. Journal of medicinal chemistry. 2008;51:6005–13. doi: 10.1021/jm800390n. [DOI] [PubMed] [Google Scholar]
- 20.Rosenblum JS, Kozarich JW. Prolyl peptidases: a serine protease subfamily with high potential for drug discovery. Curr Opin Chem Biol. 2003;7:496–504. doi: 10.1016/s1367-5931(03)00084-x. [DOI] [PubMed] [Google Scholar]
- 21.Adams S, Miller GT, Jesson MI, Watanabe T, Jones B, Wallner BP. PT-100, a small molecule dipeptidyl peptidase inhibitor, has potent antitumor effects and augments antibody-mediated cytotoxicity via a novel immune mechanism. Cancer research. 2004;64:5471–80. doi: 10.1158/0008-5472.CAN-04-0447. [DOI] [PubMed] [Google Scholar]
- 22.Walsh MP, Duncan B, Larabee S, et al. Val-BoroPro Accelerates T Cell Priming via Modulation of Dendritic Cell Trafficking Resulting in Complete Regression of Established Murine Tumors. PLoS One. 2013;8:e58860. doi: 10.1371/journal.pone.0058860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eager RM, Cunningham CC, Senzer N, et al. Phase II trial of talabostat and docetaxel in advanced non-small cell lung cancer. Clin Oncol (R Coll Radiol) 2009;21:464–72. doi: 10.1016/j.clon.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 24.Eager RM, Cunningham CC, Senzer NN, et al. Phase II assessment of talabostat and cisplatin in second-line stage IV melanoma. BMC Cancer. 2009;9:263. doi: 10.1186/1471-2407-9-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang X, Shi X, Li J, et al. A novel therapeutic vaccine of mouse GM-CSF surface modified MB49 cells against metastatic bladder cancer. The Journal of urology. 2012;187:1071–9. doi: 10.1016/j.juro.2011.10.126. [DOI] [PubMed] [Google Scholar]
- 26.Melchionda F, McKirdy MK, Medeiros F, Fry TJ, Mackall CL. Escape from immune surveillance does not result in tolerance to tumor-associated antigens. J Immunother. 2004;27:329–38. doi: 10.1097/00002371-200409000-00001. [DOI] [PubMed] [Google Scholar]
- 27.Weigel BJ, Rodeberg DA, Krieg AM, Blazar BR. CpG oligodeoxynucleotides potentiate the antitumor effects of chemotherapy or tumor resection in an orthotopic murine model of rhabdomyosarcoma. Clin Cancer Res. 2003;9:3105–14. [PubMed] [Google Scholar]
- 28.Blood (52nd ASH Annual Meeting Abstracts) Orlando, FL. Washington (DC): ASH; 2008. Dec, Dipeptidyl peptidase inhibition accelerates dendritic cell cross priming of tumor-reactive T cells resulting in repression of established tumors [abstract] pp. 4–7. 2008. Abstract nr 2579. (Accessed at http://ash.confex.com/ash/2008/webprogram/paper14667.html) [Google Scholar]
- 29.Cui Y, Zhang H, Meadors J, Poon R, Guimond M, Mackall CL. Harnessing the physiology of lymphopenia to support adoptive immunotherapy in lymphoreplete hosts. Blood. 2009;114:3831–40. doi: 10.1182/blood-2009-03-212134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sharp R, Recio JA, Jhappan C, et al. Synergism between INK4a/ARF inactivation and aberrant HGF/SF signaling in rhabdomyosarcomagenesis. Nature medicine. 2002;8:1276–80. doi: 10.1038/nm787. [DOI] [PubMed] [Google Scholar]
- 31.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nature medicine. 2004;10:909–15. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hurwitz AA, Watkins SK. Immune suppression in the tumor microenvironment: a role for dendritic cell-mediated tolerization of T cells. Cancer immunology, immunotherapy: CII. 2012;61:289–93. doi: 10.1007/s00262-011-1181-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kraman M, Bambrough PJ, Arnold JN, et al. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science. 2010;330:827–30. doi: 10.1126/science.1195300. [DOI] [PubMed] [Google Scholar]
- 34.Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limon P. The polarization of immune cells in the tumour environment by TGFbeta. Nature reviews Immunology. 2010;10:554–67. doi: 10.1038/nri2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Flinsenberg TW, Compeer EB, Boelens JJ, Boes M. Antigen cross-presentation: extending recent laboratory findings to therapeutic intervention. Clinical and experimental immunology. 2011;165:8–18. doi: 10.1111/j.1365-2249.2011.04411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ahmadi M, Emery DC, Morgan DJ. Prevention of both direct and cross-priming of antitumor CD8+ T-cell responses following overproduction of prostaglandin E2 by tumor cells in vivo. Cancer research. 2008;68:7520–9. doi: 10.1158/0008-5472.CAN-08-1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rossi DL, Vicari AP, Franz-Bacon K, McClanahan TK, Zlotnik A. Identification through bioinformatics of two new macrophage proinflammatory human chemokines: MIP-3alpha and MIP-3beta. J Immunol. 1997;158:1033–6. [PubMed] [Google Scholar]
- 38.Villablanca EJ, Raccosta L, Zhou D, et al. Tumor-mediated liver X receptor-alpha activation inhibits CC chemokine receptor-7 expression on dendritic cells and dampens antitumor responses. Nature medicine. 2010;16:98–105. doi: 10.1038/nm.2074. [DOI] [PubMed] [Google Scholar]
- 39.Gabrilovich DI, Bronte V, Chen SH, et al. The terminology issue for myeloid-derived suppressor cells. Cancer research. 2007;67:425. doi: 10.1158/0008-5472.CAN-06-3037. author reply 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008;181:5791–802. doi: 10.4049/jimmunol.181.8.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Movahedi K, Guilliams M, Van den Bossche J, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood. 2008;111:4233–44. doi: 10.1182/blood-2007-07-099226. [DOI] [PubMed] [Google Scholar]
- 42.Uren A, Merchant MS, Sun CJ, et al. Beta-platelet-derived growth factor receptor mediates motility and growth of Ewing's sarcoma cells. Oncogene. 2003;22:2334–42. doi: 10.1038/sj.onc.1206330. [DOI] [PubMed] [Google Scholar]
- 43.Rosenberg SA, Yang JC, Sherry RM, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clinical cancer research: an official journal of the American Association for Cancer Research. 2011;17:4550–7. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robbins PF, Morgan RA, Feldman SA, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29:917–24. doi: 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. Journal of the National Cancer Institute. 2000;92:205–16. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 46.Therasse P, Eisenhauer EA, Verweij J. RECIST revisited: a review of validation studies on tumour assessment. Eur J Cancer. 2006;42:1031–9. doi: 10.1016/j.ejca.2006.01.026. [DOI] [PubMed] [Google Scholar]
- 47.Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–22. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 48.Hodi FS, Butler M, Oble DA, et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:3005–10. doi: 10.1073/pnas.0712237105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sedo A, Malik R. Dipeptidyl peptidase IV-like molecules: homologous proteins or homologous activities? Biochim Biophys Acta. 2001;1550:107–16. doi: 10.1016/s0167-4838(01)00278-3. [DOI] [PubMed] [Google Scholar]
- 50.Sawanobori Y, Ueha S, Kurachi M, et al. Chemokine-mediated rapid turnover of myeloid-derived suppressor cells in tumor-bearing mice. Blood. 2008;111:5457–66. doi: 10.1182/blood-2008-01-136895. [DOI] [PubMed] [Google Scholar]
- 51.Fernandez EJ, Lolis E. Structure, function, and inhibition of chemokines. Annual review of pharmacology and toxicology. 2002;42:469–99. doi: 10.1146/annurev.pharmtox.42.091901.115838. [DOI] [PubMed] [Google Scholar]
- 52.Dolznig H, Schweifer N, Puri C, et al. Characterization of cancer stroma markers: in silico analysis of an mRNA expression database for fibroblast activation protein and endosialin. Cancer Immun. 2005;5:10. [PubMed] [Google Scholar]
- 53.Keane FM, Nadvi NA, Yao TW, Gorrell MD. Neuropeptide Y, B-type natriuretic peptide, substance P and peptide YY are novel substrates of fibroblast activation protein-alpha. The FEBS journal. 2011;278:1316–32. doi: 10.1111/j.1742-4658.2011.08051.x. [DOI] [PubMed] [Google Scholar]
- 54.Jesson MI, McLean PA, Miller GT, Adams S, Aubin J, Jones B. Immune mechanism of action of talabostat: a dipeptidyl peptidase targeting antitumor agent [abstract]. Proceedings of the 98th Annual Meeting of the American Association for Cancer Research; Los Angeles, CA. Philadelphia (PA). AACR; 2007. Apr, pp. 14–18. 2007, Abstract nr 1894. [Google Scholar]