

Abstract
Increasing antibiotic resistance in Gram-negative bacteria, particularly in Pseudomonas aeruginosa, Acinetobacter baumannii and Klebsiella pneumoniae, presents a global medical challenge. No new antibiotics will be available for these ‘superbugs’ in the near future due to the dry antibiotic discovery pipeline. Colistin and polymyxin B are increasingly used as the last-line therapeutic options for treatment of infections caused by multidrug-resistant Gram-negative bacteria. This article surveys the significant progress over the last decade in understanding polymyxin chemistry, mechanisms of antibacterial activity and resistance, structure–activity relationships and pharmacokinetics/pharmacodynamics. In the ‘Bad Bugs, No Drugs’ era, we must pursue structure–activity relationship-based approaches to develop novel polymyxin-like lipopeptides targeting polymyxin-resistant Gram-negative ‘superbugs’. Before new antibiotics become available, we must optimize the clinical use of polymyxins through the application of pharmacokinetic/pharmacodynamic principles, thereby minimizing the development of resistance.
Keywords: colistin, lipid A, lipopolysaccharide, pharmacokinetic/pharmacodynamic, polymyxin, resistance, structure–activity relationship
The WHO has identified antibiotic resistance as one of the three greatest threats to human health [1]. Since the 1990s, there has been a marked decline in discovery of novel antibiotics and, unfortunately, a remarkable increase in bacterial resistance to current antibiotics. The world is now facing an enormous and growing threat from the emergence of bacteria that are resistant to almost all available antibiotics [1,2]. As highlighted by the Infectious Diseases Society of America in the ‘Bad Bugs, No Drugs’ paper, “as antibiotic discovery stagnates, a public health crisis brews” [1,2]. The situation is especially worrying with multidrug-resistant (MDR) Gram-negative bacteria (notably Pseudomonas aeruginosa, Acinetobacter baumannii and Klebsiella pneumoniae), against which no new antibiotics will be available for many years to come [3]. It is precisely this scenario, highlighted in the ‘Bad Bugs Need Drugs’ campaign, which led the Infectious Diseases Society of America to place these three pathogens on a ‘hit list’ of the top-priority, dangerous, MDR microorganisms [2].
Polymyxins: last-line therapy against Gram-negative ‘superbugs’
Increasingly, clinicians worldwide are confronted by the reality of infections with Gram-negative pathogens that are resistant to all antibiotics except polymyxins [4]. As a poignant example, since the first report of NDM-1-producing K. pneumoniae in the Indian subcontinent in December 2009 [5], a major international problem has arisen owing to the rapid spread of NDM-producing Enterobacteriaceae (mainly K. pneumoniae and Escherichia coli) to many countries [4,6]. Now NDM-producing MDR clinical isolates have been reported in more than 20 countries in all continents [4,6]. Many of these NDM-producing MDR isolates are only susceptible to polymyxins. The incidence of resistance to polymyxins is currently relatively low [7]; however, resistance in Gram-negative pathogens can emerge in vitro [8–10] and also in patients, owing to suboptimal use [2,11]. According to the results from a recent global antimicrobial surveillance program (SENTRY, 2006–2009), even though a trend towards greater resistance was observed in the Asia Pacific and Latin American regions, the polymyxins showed excellent in vitro activity against the vast majority of Gram-negative bacilli pathogens [7]. Without new antibiotics, polymyxins are increasingly used as the last-line therapy.
There are two polymyxins available for clinical use, colistin (i.e., polymyxin E) and polymyxin B (Table 1), and cross-resistance exists [12]. They were discovered in the 1940s and were never subjected to contemporary drug-development procedures. They have a narrow antibacterial spectrum, mainly against Gram-negatives [12,13]. Clinical use of colistin and polymyxin B waned in the 1970s due to the early experience of nephrotoxicity and neurotoxicity after intravenous administration; however, the rapid increase in resistance to all other antibiotics has necessitated their resurgence in the clinic [12]. Parenteral colistin is much more commonly used internationally, although injectable polymyxin B is available in a number of countries, such as Brazil, Singapore and the USA; in these three countries, both antibiotics are available [13].
Table 1.
Structures of known polymyxin B and polymyxin E.
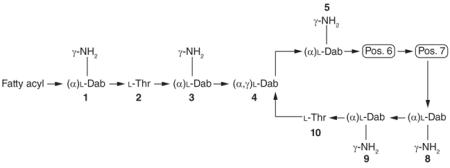
| Polymyxin | Fatty acyl group | Pos. 6 | Pos. 7 |
|---|---|---|---|
| B1 | (S)-6-methyloctanoyl | d-Phe | Leu |
| B1-Ile | (S)-6-methyloctanoyl | d-Phe | Ile |
| B2 | 6-methylheptanoyl | d-Phe | Leu |
| B3 | Octanoyl | d-Phe | Leu |
| B4 | Heptanoyl | d-Phe | Leu |
| B5 | Nonanoyl | d-Phe | Leu |
| B6 | 3-hydroxy-6-methyloctanoyl | d-Phe | Leu |
| E1 | (S)-6-methyloctanoyl | d-Leu | Leu |
| E2 | 6-methylheptanoyl | d-Leu | Leu |
| E3 | Octanoyl | d-Leu | Leu |
| E4 | Heptanoyl | d-Leu | Leu |
| E7 | 7-methyloctanoyl | d-Leu | Leu |
| E1-Ile | (S)-6-methyloctanoyl | d-Leu | Ile |
| E1-Val | (S)-6-methyloctanoyl | d-Leu | Val |
| E1-Nva | (S)-6-methyloctanoyl | d-Leu | Nva |
| E2-Ile | 6-methylheptanoyl | d-Leu | Ile |
| E2-Val | 6-methylheptanoyl | d-Leu | Val |
| E8-Ile | 7-methylnonanoyl | d-Leu | Ile |
Pos.: Amino acid position.
Chemical structure of the polymyxins
To understand the mechanisms of antibacterial activity of the polymyxins and their resistance, it is crucial to have knowledge of their chemical structures. Polymyxins are nonribosomal cyclic lipopeptides that can be characterized by the general structure illustrated in Table 1. Their decapeptide sequence contains an intramolecular cyclic heptapeptide loop between the amino group of the side chain of the diaminobutyric acid (Dab) residue at position 4 and the carboxyl group of the C-terminal threonine residue at position 10. Polymyxins also have several other distinguishing structural features, including: five nonproteogenic Dab residues, which makes them polycationic at pH 7.4; hydrophobic residues at positions 6 and 7; and an N-terminal fatty acyl group. Like many other antimicrobial peptides, this mixture of lipophilic and hydrophilic groups makes polymyxins amphipathic, a chemophysical property that is essential for their antibacterial activity, as discussed below.
To date, several distinct groups of polymyxins have been isolated and structurally identified from Paenibacillus polymyxa, with each group being structurally defined by the unique amino acid residues present in their sequences. Each of these distinct groups of polymyxins have been labeled with a letter, and each group can contain several individual lipopeptide components that differ from one another in the chemical structure of the fatty acyl group they present at their N-terminus. The individual lipopeptide components of each polymyxin group are labeled with a number. This nomenclature is illustrated in Table 1 with the polymyxin B and E groups. Of the different ‘polymyxin’ groups identified to date, only the components of the polymyxin B and E (i.e., colistin) groups have undergone extensive structural analysis, very likely due to the fact that both polymyxins are used in the clinic. The commercial products of polymyxin B and E (used as an inactive prodrug colistin methanesulfonate) for parenteral use in patients usually contain several different components, with polymyxin B1 and B2, and polymyxin E1 and E2 (Table 1) as the two major components (usually accounting for >80%) [12,14–18]. Therefore, the remaining discussion will focus only on the chemical structures of the lipopeptide components of these two groups of polymyxin.
Structurally, the lipopeptides of the polymyxin B group are generally defined by the presence of a d-phenylalanine residue at position 6 and a leucine residue at position 7. All amino acid residues are of the l-configuration, except for the d-phenylalanine at position 6. To date, seven individual polymyxin B components have been identified (Table 1). Of these seven lipopeptides, six contain structurally different branched and non-branched N-terminal fatty acyl groups varying in length from 7 to 9 carbons, which have been labeled polymyxin B1–B6. The 6-methyloctanoyl fatty acyl group of polymyxin B1 and B1-Ile (i.e., polymyxin B7) has a stereocenter at C6, which has been identified as being the (S)-configuration. Polymyxin B6 is unique in that its fatty acyl chain contains a 3-hydroxy group, which is not present in the fatty acyl chains of the other polymyxin B peptides. This unique fatty acyl group also has two stereocenters at C3 and C6; however, the absolute stereochemistry of these two stereocenters is yet to be reported. Interestingly, polymyxin B1-Ile contains the same N-terminal 6-methyloctanoyl fatty acyl group as polymyxin B1 but has an isoleucine residue at position 7.
The polymyxin E group of lipopeptides is generally defined by the presence of a d-leucine residue at position 6 and a leucine residue at position 7 (Table 1). Similarly to the polymyxin B structural series, all of the amino acid residues in the polymyxin E series are of the l-configuration except for the d-leucine at position 6. Like the polymyxin B lipopeptides, the individual components of the polymyxin E group (polymyxin E1, E2, E3, E4, E7 and E8-Ile) contain structurally distinct branched and nonbranched N-terminal fatty acyl groups, varying in length from seven to nine carbons. The 6-methyloctanoyl fatty acyl group of polymyxin E1, E1-Val, E1-Ile and E1-Nva contains a stereocenter at C6, which has been identified as being the (S)-configuration. Several polymyxin E lipopeptides (polymyxin E1-Val, E1-Ile, E1-Nva, E2-Val, E2-Ile and E8-Ile) have the same N-terminal 6-methyloctanoyl fatty acyl group, but contain structurally different branched-chain amino acid residues (valine, norvaline and isoleucine) at position 7 (Table 1). To date, polymyxin E5 and E6 have not been reported in the literature.
Mechanism of antibacterial activity of polymyxins
Understanding the mechanism of polymyxin antibacterial activity also requires knowledge of the outer membrane architecture of Gram-negative bacteria. The outer membrane constitutes a permeability barrier to various noxious substances, including numerous antimicrobials [19]. Polymyxins exert their antimicrobial action via direct interaction with the lipid A component of the lipopolysaccharide (LPS) (Figure 1).
Figure 1. Molecular models of the complex between polymyxin B1 and the lipid A structure from Klebsiella pneumoniae.

(A) The lipid A molecule is shown in space-filling representation and polymyxin B1 is shown in stick representation. (B) The chemical structure of the lipid A molecule. (C) The key electrostatic interactions between positively charged Dab residues on polymyxin and the negatively charged lipid A phosphoresters.
Dab: Diaminobutyric acid.
The complex asymmetrical structure of the outer membrane comprises an inner phospholipid leaflet, as well as an outer leaflet that predominantly contains LPS, proteins and phospholipids. Structurally, LPS is composed of three domains: the variable O-antigen chain (encompassing repeated saccharide units); a coreoligosaccharide region; and the conserved lipid A moiety (Figure 1). Lipid A is intercalated within the outer leaflet, functioning as a hydrophobic anchor [20]. The consensus structure of lipid A is represented by a β-1′-6-linked d-glucosamine disaccharide that is phosphorylated at the 1′- and 4′-positions [19]. The saturated lipid A hydrocarbon chains are tightly packed together within the membrane through van der Waals forces, while divalent cations (Mg2+ and Ca2+) associated with lipid A phosphoresters function to bridge adjacent LPS molecules [20,21]. The barrier function of the outer membrane is further accentuated by a highly repulsive anionic charge conveyed by lipid A phosphorester moieties, as well as phosphate and carboxylate functionalities within the core and O-antigen sugars [20,21].
The precise mode of action of polymyxins remains contentious and, based upon biophysical studies, a number of models have been proposed [22]. The consensus view is that polymyxins are membrane surface active and that lipid A is an important polymyxin-binding target in the outer membrane of Gram-negative species [22]. A long-accepted model, termed the ‘self-promoted uptake’ pathway, purports that the amphipathic nature of polymyxins is crucial to enable uptake of the polymyxin molecule across the outer membrane barrier [23]. In this model, protonation of free Ψ-amines present on the Dab residues of polymyxins (Table 1) at physiological pH provides a means of electrostatic attraction to anionic lipid A phosphates. The resultant displacement of divalent cations (Ca2+ and Mg2+) that stabilize the LPS leaflet allows the hydrophobic N-terminal fatty acyl tail, and d-Phe-l-Leu or d-Leu-l-Leu motifs (of polymyxin B and colistin, respectively; Table 1) to be inserted into the outer membrane (Figure 1). Insertion of the fatty acyl chain and the position 6–7 hydrophobic motif acts to weaken the packing of adjacent lipid A fatty acyl chains, causing expansion of the outer membrane. Subsequent events are not completely understood; however, polymyxin-mediated fusion of the inner leaflets of the outer membrane and the outer leaflet of the cytoplasmic membrane surrounding the periplasmic space is believed to induce phospholipid exchange, resulting in an osmotic imbalance that culminates in cell death [23]. The elucidation of the 3D nuclear magnetic resonance solution state structure of polymyxin B in complex with LPS revealed that the polymyxin B molecule is folded such that the polar and hydrophobic domains form two distinct faces, thereby conferring structural amphipathicity (Figure 1) [24]. This amphipathicity, and possibly their ability to form pore-like aggregates, may be responsible for their outer membrane-permeabilizing action. In vitro evidence suggests that the two processes of permeabilizing the outer membrane and bacterial killing activity may be entirely uncoupled [25].
In addition, polymyxins have been shown to inhibit alternative nicotinamide adenine dinucleotide dehydrogenase and malate:quinone oxidoreductase in Mycobacterium smegmatis [26]; no such enzymatic study has been reported in Gram-negatives. A recent preliminary biochemical study reported that rapid killing of A. baumannii by polymyxins is mediated by a hydroxyl radical death pathway [27].
Mechanisms of polymyxin resistance in Gram-negative bacteria
It is becoming increasingly apparent that polymyxin resistance in Gram-negative bacteria involves the multitier upregulation of a number of regulatory systems (Figure 2) [28,29]. LPS remodeling is an important survival strategy for Gram-negative bacteria [20]. Accordingly, the most common polymyxin resistance mechanism in P. aeruginosa, Salmonella enterica serovar Typhimurium, E. coli, A. baumannii and K. pneumoniae is due to modifications of lipid A phosphates with positively charged groups, such as 4-amino-4-deoxy-l-arabinose and/or phosphoethanolamine [30,31]. The first step in the action of polymyxins on the Gram-negative bacterial outer membrane involves an electrostatic interaction between the positive charge of the five Dab residues of the polymyxin molecule and the negatively charged phosphate groups on lipid A [32]. Therefore, by reducing the net negative charge of the outer membrane via the aforementioned lipid A modifications, the bacterial cell is able to avoid the initial electrostatic attraction of the polymyxin molecule to its surface (Figure 1) [33–36]. In many Gram-negative bacterial species, resistance to cationic antimicrobial peptides is mediated by two-component regulatory systems, such as PhoP–PhoQ [35,36]. Such a system is also employed by the bacterial cell in survival situations under growth conditions of low Mg2+, which can potentially destabilize the outer membrane due to the decrease in the bridging action of divalent cations between LPS molecules. Normally, under optimal growth conditions, the PhoP–PhoQ remains repressed in high (mM) Mg2+ environments and can be activated under conditions of low (μM) Mg2+ [34]. In S. enterica, PhoP–PhoQ acts as a master regulator of virulence and evasion of killing by many cationic antimicrobial peptides [35,37]. In response to low Mg2+ or sublethal concentrations of cationic antimicrobial peptides, PhoQ, an inner membrane sensor kinase, phosphorylates the cognate response regulator PhoP, leading to activation of PmrA–PmrB. Subsequently, PmrA–PmrB activates the expression of genes that encode the enzymes that are required for the above-mentioned modifications of lipid A [35,37]. Of note, it has been reported that cationic antimicrobial peptides can directly activate the PmrA–PmrB system [38]. In P. aeruginosa, the regulation of these LPS modifications in response to polymyxins involves at least two additional two-component systems. This would suggest that homologous, as yet undiscovered, systems may also exist in other Gram-negative species [39]. Another lipid A modification that has been associated with increased polymyxin resistance of Gram-negative bacteria is the decoration of the lipid A with additional fatty acyl chains [40]. This mechanism appears to render the outer membrane less penetrable for the N-terminal fatty acyl chains and position 6–7 hydro phobic motifs of the polymyxin molecule (Table 1). In terms of higher-order structures on the LPS molecule, the core and O-antigen polysaccharide appear to contribute to polymyxin resistance, as rough mutants that express truncated versions of these structures display an increased susceptibility to polymyxins [28,41]. Given the important role of LPS for polymyxin activity, it is not surprising that a novel polymyxin resistance mechanism reported in A. baumannii involves complete loss of LPS production [42]. In order to compensate for the decreased outer membrane integrity due to the LPS loss, polymyxin-resistant A. baumannii strains upregulate the expression of genes of biosynthetic systems responsible for phospholipid, lipoprotein and poly-β-1,6-N-acetylglucosamine, thereby consolidating the cell envelope structure [43].
Figure 2. Key mechanisms of polymyxin resistance in Gram-negative bacteria.

The pink shading indicates molecular determinants of polymyxin resistance.
LPS: Lipopolysaccharide; NAG: N-acetylglucosamine; NAM: N-acetylmuramic acid; OMP: Outer membrane protein.
It is noteworthy that a number of unique and often species-specific polymyxin resistance mechanisms do not involve the LPS-binding pathway. The capsular polysaccharide levels on K. pneumoniae have been shown to coincide with polymyxin resistance [44]. Polymyxin resistance in a number of Gram-negative bacterial species has been associated with alterations in the expression of outer membrane proteins, including efflux pumps. In P. aeruginosa biofilms, colistin resistance in a metabolically active subpopulation was found to coincide with the overexpression of the mexAB–oprM efflux pump system [45]. Polymyxin resistance in P. aeruginosa has also been associated with changes in the expression of the outer membrane protein OprH, which is purported to perform a membrane stabilization role under conditions of Mg2+ starvation [36]. In K. pneumoniae, a deficiency in the outer membrane protein OmpA, which mediates adhesion to eukaryotic cells, has been associated with an increased susceptibility to polymyxin B [46]. Moreover, the AcrAB–TolC energy-driven efflux pump has been linked to polymyxin resistance and efflux from K. pneumoniae and E. coli cells [47,48]. In Burkholderia vietnamien, a multidrug efflux pump NorM has been shown to contribute to polymyxin resistance [49].
In addition, hopanoids are sterol-like compounds that are believed to perform a barrier function in the outer membrane of certain bacterial species [50]. In a recent study, it was shown that hopanoid deficiency in Burkholderia multivorans was coincident with increased polymyxin susceptibility, suggesting hopanoids contribute to the intrinsic resistance of Burkholderia bacteria to polymyxins [51]. Increased polymyxin susceptibility in two intrinsically resistant species, Burkholderia cepacia and Proteus mirabilis, has been associated with defects in UDP-glucose dehydrogenase and UDP-glucose phosphorylase, enzymes involved in the biosynthesis of the LPS precursor, UDP-glucose [52,53]. In addition, the expression of periplasmic proteases has been shown to be a component of the intrinsic polymyxin phenotype of B. cenocepacia [29,54].
Structure–activity relationships of polymyxins
From the foregoing appreciation of the mechanisms of polymyxin activity and resistance, it is evident that discussions of polymyxin structure–activity relationship (SAR) require structural knowledge of the polymyxin–lipid A complex. Such knowledge is also critical for efforts to develop novel polymyxin analogs with activity against polymyxin-resistant isolates. Structural information for the interaction between polymyxins and lipid A at the molecular level has been well characterized by nuclear magnetic resonance techniques [24,55]. The nuclear magnetic resonance model of the polymyxin B–lipid A complex shows that, in general, the complex is stabilized by a combination of electrostatic and hydrophobic interactions (Figure 1). The positively charged side chains of Dab1 and Dab5 bond with the negative charge on the 4′-phosphate head group of lipid A, while those of Dab8 and Dab9 bond with the 1-phosphate head. The buckled configuration of the cyclic peptide portion forces the lipid A-binding surface of the polymyxin molecule to one face of the molecule. The polymyxin B–lipid A model suggests that the loss of a secondary 3′-myristate fatty acyl chain leads to a reduced hydrophobic surface area for interaction with the d-Phe6-l-Leu7, and the N-terminal fatty acyl chain of polymyxin B (Figure 3). More specifically, the 3′-secondary myristate fatty acyl chain forms hydrophobic contacts with the octanoic acyl chain of polymyxins. The model of the polymyxin B–lipid A complex implies the modifications on the lipid A phosphates (e.g., addition of 4-amino-4-deoxy-l-arabinose) commonly observed in polymyxin-resistant strains block the electrostatic interaction between the lipid A phosphates and the positively charged Dab residues, thereby destabilizing the complex (Figure 3). The polymyxin molecule can essentially be divided into hydrophobic and polar domains, namely the N-terminal fatty acyl chain and position 6–7 motif (hydrophobic), and the Dab and Thr residue segments (polar). The cyclic heptapeptide and linear tripeptide provide an integral scaffolding function that involves maintaining the optimal distances between each domain, thereby giving the structure its amphipathicity, a property that is indispensible for polymyxin antibacterial activity [24,55].
Figure 3. A selection of the reported medicinal chemistry modifications that have been made to the polymyxin core structural domains.

The hydrophobic N-terminal fatty acyl chain (red), the linear tripeptide segment (green), the hydrophobic motif at positions 6 and 7 (blue) and the heptapeptide backbone (pink) are shown. Modifications to the diaminobutyric acid residue positions are not depicted.
The intent of this section is to provide an overview that highlights the key aspects of the polymyxin SAR; for a comprehensive treatise on the current state of development of polymyxin analogs, please refer to [22]. The current understanding of the polymyxin SAR is that both electrostatic and hydrophobic interactions with the lipid A are critical for antimicrobial activity. The polymyxin molecule consists of five key domains: the hydrophobic N-terminal fatty acyl chain; the positive charge of the Dab side chains; the linear tripeptide segment; the hydrophobic motif at positions 6 and 7 in the cyclic heptapeptide ring; and the heptapeptide backbone (Table 1). Most medicinal chemistry strategies for improving the antibacterial activity and toxicity of polymyxins have included modifications to the above-mentioned domains.
The hydrophobic N-terminal fatty acyl chain
The availability of large quantities of polymyxin B/colistin and ease of semisynthesis has meant that most medicinal chemistry programs have focused on generating N-terminal analogs of the polymyxin molecule [56–62]. The available SAR data indicate that a hydrophobic substituent at the N-terminus of the polymyxin molecule is indispensable for antimicrobial activity. A comparison across all N-terminal analogs documented to date reveals that the degree of antibacterial activity observed for these analogs appears to coincide with the length and bulkiness of the N-terminal substituent. As per the naturally occurring polymyxins, the ideal fatty acyl chain length correlating with superior antimicrobial activity appears to be C7–C9. The introduction of longer- or shorter-chain N-terminal fatty acyl chains has been shown to negatively impact on the antimicrobial activity. However, the LPS-binding affinity of the polymyxin molecule appears to correlate with the hydrophobicity of the N-terminal substituent [58,63]. Interestingly, the Cubist Pharmaceuticals polymyxin clinical candidate CB-182,804 contained a shorter N-terminal 2-chlorophenyl carbamate group, yet had comparable in vitro and in vivo antibacterial activity with polymyxin B and colistin [62]. Furthermore, there has been a recent report describing des-fatty acyl polymyxin analogs that display selective antimicrobial activity against P. aeruginosa [64].
The positive charge of the Dab side chains
The importance of the cationic character of the five Dab residues (at physiological pH) for conferring the antimicrobial activity of polymyxins has been well documented [65]. Based upon the SAR literature, it can be discerned that key features of the Dab residues that are important for LPS-binding affinity and antibacterial activity include: the cationic charge and two-carbon length of the Dab side chain; and the specific sequence of the Dabs, which presents the correct spatial distribution and orientation of the positive charges for electrostatic interactions with the phosphates of lipid A. Attempts to derivatize or substitute the Dab residues and/or reduce the overall positive charge of the polymyxin molecule have been met with variable success [66]. In general, apart from Dab1, the other Dab positions – particularly those within the cyclic heptapeptide – are essential for polymyxin antibacterial activity.
The linear tripeptide segment
The linear tripeptide segment (Dab1-Thr2-Dab3) serves to bridge the heptapeptide cyclic core of the polymyxin molecule to the N-terminal fatty acyl chain (Table 1). In terms of its functional role, the linear tripeptide segment appears to contribute two positive charges towards the binding interaction with lipid A. The molecular model of the polymyxin B–LPS complex indicates that hydrogen bonding between the side chain of Thr2 and the side chain of Dab3, and the amide nitrogen of Thr2 and the main chain carbonyl of Dab4, reorientates the tripeptide segment towards the heptapeptide (Figure 1). Notably, a number of medicinal chemistry strategies have involved exploring the SAR of the linear tripeptide segment by examining the impact of amino acid substitutions or deletions [58,66,67]. From the SAR data in the literature, two main conclusions can be drawn: the tripeptide segment can only be truncated by deletion of Dab1 with a negligible loss of antibacterial activity; and only conservative amino acid substitutions are tolerated.
The hydrophobic motif at positions 6 & 7 in the cyclic heptapeptide ring
The d-Phe6-l-Leu7 segment in the polymyxin heptapeptide ring forms a hydrophobic domain and β-turn-forming element that is highly conserved across the naturally occurring polymyxins (Table 1), and appears to be important for antibacterial activity and plasma protein binding. The introduction of fatty acyl amino acid derivatives at these positions appears to improve antimicrobial activity and LPS binding; albeit, this is accompanied by a concomitant increase in plasma protein binding. By comparison, the introduction of hydrophilic groups or β-turn mimetics appears to negatively impact antimicrobial activity [56,68].
The heptapeptide backbone
The Nγ-amino side chain of Dab4 is deacylated by the C-terminal Thr10 to form a 23-membered lactam ring. The molecular model of the polymyxin B–LPS complex shows how the precise 23-atom size of the heptapeptide ring acts as a scaffold for electrostatic and hydrophobic LPS contact points. The available SAR data demonstrate that the 23-atom size of the native polymyxin ring provides the most ideal structural configuration for potent antimicrobial activity, and that deletions or expansion of the ring size impact negatively on antimicrobial activity [69].
Pharmacokinetics/pharmacodynamics of polymyxins
Probably because colistin is used much more widely than polymyxin B, most modern pharmacology information on polymyxins is for colistin. Over the last decade, the pharmacokinetics/pharmacodynamics (PK/PD) of colistin has been examined using in vitro and mouse infection models, and such pharmacological information is employed for optimizing its clinical use [13,70–73]. Colistin exhibits rapid, concentration-dependent bacterial killing with negligible post-antibiotic effects [10,74]. Colistin heteroresistance, the presence of resistant subpopulations within a strain that is considered susceptible based upon its MIC, has been reported in clinical isolates of A. baumannii [75,76], K. pneumoniae [10] and P. aeruginosa [77]. The potential for resistant subpopulations to rapidly amplify upon exposure to polymyxins has been demonstrated in an in vitro PK/PD model that mimics clinical dosing regimens in humans [8,9]. Our recent preclinical studies conducted in both in vitro PK/PD and animal thigh and lung infection models have, for the first time, elucidated that the area under free plasma concentration–time curve to MIC ratio is the PK/PD index that best correlates with colistin antibacterial activity against P. aeruginosa and A. baumannii [78,79]. This has allowed the identification of area under free plasma concentration–time curve to MIC ratio targets to achieve various magnitudes of bacterial killing.
In the NIH-funded, multicenter, multinational project the current authors are undertaking on the PK/PD/toxicodynamics of colistin in critically ill patients, it is evident that the currently recommended intravenous dosage regimens of the prodrug colistin methanesulfonate (sodium) are suboptimal in many patients [71]. The average steady-state plasma concentrations of formed colistin (the antibacterial entity) with intravenous colistin methanesulphonate dosage regimens (75–410 mg colistin base activity per day) specified in the product information were in the range of 0.48–9.38 mg/l (median: 2.36 mg/l) (Figure 4); other groups reported similar findings in a much smaller number of patients [72,80]. For information on the suggested loading dose and daily maintenance doses of colistin methanesulfonate in patients with various renal functions, please see [71]. It should be noted that conversion of the prodrug colistin methanesulfonate (CMS) to the antibacterial entity colistin is very slow, therefore plasma concentrations of formed colistin are well below the MIC breakpoint (2 mg/l) after the first dose [72]. This results in an unintended delay in achieving adequate exposure for treatment. Based upon the recent PK data on colistin in critically ill patients [71,72,80–82] and PK/PD relationships in animal models [78,79], it is evident that colistin monotherapy is not likely to be reliably efficacious with currently recommended daily doses of CMS, especially for patients with moderate-to-good renal function and/or for causative pathogens with MICs of ≥1.0 mg/l [71]. In marked contrast to colistin, there is very limited information on PK of polymyxin B in patients [15,83–85]. Intravenous polymyxin B is mainly used in the USA, Brazil, Singapore and Malaysia [15]. Even though polymyxin B and colistin look alike, it is extremely important to note that they differ in the method of administration to patients. Unlike colistin, which is available for clinical use as an inactive prodrug CMS [86], polymyxin B is directly administered as its active form (i.e., sulfate). This almost certainly affects their clinical effectiveness, as polymyxin B is able to provide more rapid and higher concentration–time exposure after initiating an intravenous regimen [84]. It seems clear that large clinical PK/PD/toxicodynamic investigations on polymyxin B and its comparison with colistin/CMS are urgently required.
Figure 4. Steady-state plasma concentration–time profiles of formed colistin in 105 critically ill patients (89 not on renal replacement, 12 on intermittent hemodialysis and four on continuous renal replacement therapy).
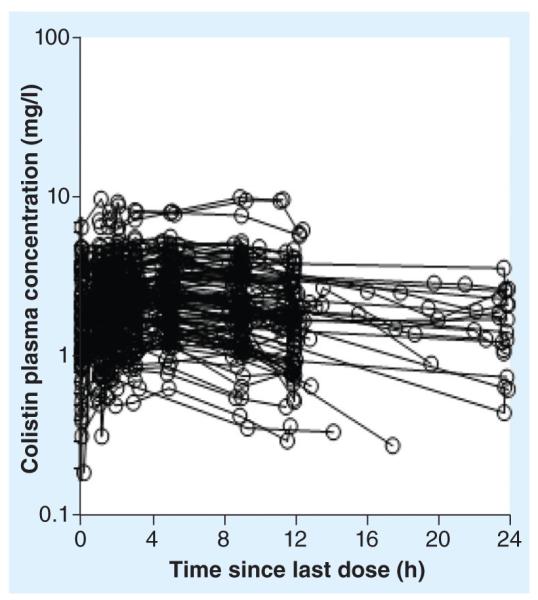
Physician-selected colistin methanesulfonate dosage intervals ranged from 8 to 24 h and hence the interdosing blood sampling interval spanned the same range.
Reproduced with permission from [71].
Unfortunately, adequate exposure to formed colistin in patients cannot be achieved by simply increasing the daily dose of colistin methanesulfonate. Even with the currently recommended dosage regimens, approximately 50% of critically ill patients in our NIH project developed nephrotoxicity while receiving colistin methanesulfonate; similar nephrotoxicity rates have been reported recently by others for CMS/colistin [72,80] and polymyxin B (~60%) [87]. The fact that approximately one in two patients experience this major dose-limiting adverse effect highlights the urgency to discover novel polymyxin-like lipopeptides with significantly less nephrotoxicity. Research on the relationship between polymyxin structure and apoptotic effect on kidney tubular cells is being conducted in our group.
Future perspective
Significant progress in understanding the mechanisms of antibacterial activity and resistance of polymyxins and their PK/PD has been recently made and provided important pharmacological information to clinicians for optimizing their clinical use. Considering that no new antibiotics will be available against MDR Gram-negative ‘superbugs’ for many years to come, polymyxin B and colistin will continue to be used as a last-line therapeutic option. However, reports of resistance to polymyxins are becoming more commonplace in the clinical setting. Inevitably, resistance to the current polymyxins will present a significant global health challenge, as resistance to polymyxins means that virtually no antibiotics will be available for treatment of life-threatening infections caused by polymyxin-resistant ‘superbugs’. Therefore, development of a new generation of polymyxins is urgently required, along with a better understanding of the mechanisms of polymyxin antibacterial activity and resistance. In contrast to the majority of current empirical drug discovery programs for new polymyxins, designing novel polymyxin-like lipopeptides using SAR models to target polymyxin resistance holds the key to success. Modern peptide chemistry has made the synthesis of virtually any polymyxin analog possible, and integration of molecular microbiology and antimicrobial PK/PD within drug discovery and development procedures will significantly facilitate such drug-development programs. It is expected that, similarly to other major classes of antibiotics, new-generation polymyxins will be available for the treatment of infections caused by Gram-negative ‘superbugs’, which are resistant to all current antibiotics.
Executive summary.
Chemical structure of the polymyxins
▪ Polymyxins are nonribosomal polycationic cyclic lipopeptides of which several different groups have been identified based on their unique amino acid sequences.
▪ The chemical structures of the polymyxin B and polymyxin E groups of lipopeptides and their commercial preparations for clinical use have been the most extensively studied polymyxins to date.
Mechanisms of polymyxin activity against Gram-negative bacteria & resistance
▪ Even though the detailed mechanisms of polymyxin antibacterial activity are unknown, their initial interaction with the lipid A of lipopolysaccharide is essential.
▪ Polymyxin resistance in Gram-negative bacteria involves the multitier upregulation of a number of two-component regulatory systems.
▪ Lipopolysaccharide remodeling represents by far the most common mechanism of polymyxin resistance.
Structure–activity relationships of polymyxins
▪ Empirical medicinal chemistry strategies for improving the antibacterial activity and toxicity profile of polymyxins have focused on the five key domains of the polymyxin molecule, and a number of such programs have been met with variable success.
▪ Recent structure–activity relationship knowledge holds the promise of the development of a new generation of polymyxins with superior activity against strains already resistant to current polymyxins.
Pharmacokinetics/pharmacodynamics of polymyxins
▪ Area under free plasma concentration–time curve to MIC ratio is the most predictive pharmacokinetics/pharmacodynamics index for the in vivo efficacy of polymyxins, and the currently recommended dosage regimens of colistin are suboptimal.
Acknowledgments
The authors are supported by Award Numbers R01AI098771 (all authors), R01AI079330 and R01AI070896 (RL Nation and J Li) from the National Institute of Allergy and Infectious Diseases (NIH). J Li is an Australian National Health and Medical Research Council Senior Research Fellow and T Velkov is an Australian National Health and Medical Research Council Industry Career Development Award Fellow.
No writing assistance was utilized in the production of this manuscript.
Footnotes
Disclaimer The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the NIH.
Financial & competing interests disclosure The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
Papers of special note have been highlighted as:
▪ of interest
▪▪ of considerable interest
- 1.Infectious Diseases Society of America The 10 × ‘20 Initiative: pursuing a global commitment to develop 10 new antibacterial drugs by 2020. Clin. Infect. Dis. 2010;50(8):1081–1083. doi: 10.1086/652237. [DOI] [PubMed] [Google Scholar]
- 2.Talbot GH, Bradley J, Edwards JE, Jr, Gilbert D, Scheld M, Bartlett JG. Bad bugs need drugs: an update on the development pipeline from the Antimicrobial Availability Task Force of the Infectious Diseases Society of America. Clin. Infect. Dis. 2006;42(5):657–668. doi: 10.1086/499819. [DOI] [PubMed] [Google Scholar]
- 3.Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 2007;6(1):29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- 4.Kumarasamy KK, Toleman MA, Walsh TR, et al. Emergence of a new antibiotic resistance mechanism in India, Pakistan, and the UK: a molecular, biological, and epidemiological study. Lancet Infect. Dis. 2010;10(9):597–602. doi: 10.1016/S1473-3099(10)70143-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yong D, Toleman MA, Giske CG, et al. Characterization of a new metallo-beta-lactamase gene, bla(NDM-1), and a novel erythromycin esterase gene carried on a unique genetic structure in Klebsiella pneumoniae sequence type 14 from India. Antimicrob. Agents Chemother. 2009;53(12):5046–5054. doi: 10.1128/AAC.00774-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cornaglia G, Giamarellou H, Rossolini GM. Metallo-beta-lactamases: a last frontier for beta-lactams? Lancet Infect. Dis. 2011;11(5):381–393. doi: 10.1016/S1473-3099(11)70056-1. [DOI] [PubMed] [Google Scholar]
- 7.Gales AC, Jones RN, Sader HS. Contemporary activity of colistin and polymyxin B against a worldwide collection of Gram-negative pathogens: results from the SENTRY Antimicrobial Surveillance Program (2006–09) J. Antimicrob. Chemother. 2011;66(9):2070–2074. doi: 10.1093/jac/dkr239. [DOI] [PubMed] [Google Scholar]
- 8.Bergen PJ, Li J, Nation RL, Turnidge JD, Coulthard K, Milne RW. Comparison of once-, twice- and thrice-daily dosing of colistin on antibacterial effect and emergence of resistance: studies with Pseudomonas aeruginosa in an in vitro pharmacodynamic model. J. Antimicrob. Chemother. 2008;61(3):636–642. doi: 10.1093/jac/dkm511. [DOI] [PubMed] [Google Scholar]
- 9.Tan CH, Li J, Nation RL. Activity of colistin against heteroresistant Acinetobacter baumannii and emergence of resistance in an in vitro pharmacokinetic/pharmacodynamic model. Antimicrob. Agents Chemother. 2007;51(9):3413–3415. doi: 10.1128/AAC.01571-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Poudyal A, Howden BP, Bell JM, et al. In vitro pharmacodynamics of colistin against multidrug-resistant Klebsiella pneumoniae. J. Antimicrob. Chemother. 2008;62(6):1311–1318. doi: 10.1093/jac/dkn425. [DOI] [PubMed] [Google Scholar]
- 11.Matthaiou DK, Michalopoulos A, Rafailidis PI, et al. Risk factors associated with the isolation of colistin-resistant Gram-negative bacteria: a matched case–control study. Crit. Care Med. 2008;36(3):807–811. doi: 10.1097/CCM.0B013E3181652FAE. [DOI] [PubMed] [Google Scholar]
- 12.Li J, Nation RL, Turnidge JD, et al. Colistin: the re-emerging antibiotic for multidrug-resistant Gram-negative bacterial infections. Lancet Infect. Dis. 2006;6(9):589–601. doi: 10.1016/S1473-3099(06)70580-1. [DOI] [PubMed] [Google Scholar]
- 13.Bergen PJ, Landersdorfer CB, Zhang J, et al. Pharmacokinetics and pharmacodynamics of ‘old’ polymyxins: what is new? Diagn. Microbiol. Infect. Dis. 2012;74(3):213–223. doi: 10.1016/j.diagmicrobio.2012.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li J, Nation RL, Milne RW, Turnidge JD, Coulthard K. Evaluation of colistin as an agent against multiresistant Gram-negative bacteria. Int. J. Antimicrob. Agents. 2005;25(1):11–25. doi: 10.1016/j.ijantimicag.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 15.Zavascki AP, Goldani LZ, Li J, Nation RL. Polymyxin B for the treatment of multidrug-resistant pathogens: a critical review. J. Antimicrob. Chemother. 2007;60:1206–1215. doi: 10.1093/jac/dkm357. [DOI] [PubMed] [Google Scholar]
- 16.Orwa JA, Govaerts C, Busson R, Roets E, Van Schepdael A, Hoogmartens J. Isolation and structural characterization of colistin components. J. Antibiot. 2001;54(7):595–599. doi: 10.7164/antibiotics.54.595. [DOI] [PubMed] [Google Scholar]
- 17.Orwa JA, Govaerts C, Busson R, Roets E, Van Schepdael A, Hoogmartens J. Isolation and structural characterization of polymyxin B components. J. Chromatogr. A. 2001;912(2):369–373. doi: 10.1016/s0021-9673(01)00585-4. ▪ Demonstrates the use of advanced analytical techniques for a thorough structural analysis of polymyxin components present in polymyxin preparations.
- 18.Decolin D, Leroy P, Nicolas A, Archimbault P. Hyphenated liquid chromatographic method for the determination of colistin residues in bovine tissues. J. Chromatogr. Sci. 1997;35(12):557–564. doi: 10.1093/chromsci/35.12.557. [DOI] [PubMed] [Google Scholar]
- 19.Nikaido H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 2003;67(4):593–656. doi: 10.1128/MMBR.67.4.593-656.2003. ▪▪ Exceptional review on the permeability of the Gram-negative bacterial outer membrane.
- 20.Raetz CR, Whitfield C. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hancock RE. The bacterial outer membrane as a drug barrier. Trends Microbiol. 1997;5(1):37–42. doi: 10.1016/S0966-842X(97)81773-8. [DOI] [PubMed] [Google Scholar]
- 22.Velkov T, Thompson PE, Nation RL, Li J. Structure–activity relationships of polymyxin antibiotics. J. Med. Chem. 2010;53(5):1898–1916. doi: 10.1021/jm900999h. ▪▪ Landmark paper that provides a comprehensive treatise on the structure–activity relationship of polymyxins.
- 23.Hancock RE, Chapple DS. Peptide antibiotics. Antimicrob. Agents Chemother. 1999;43(6):1317–1323. doi: 10.1128/aac.43.6.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pristovsek P, Kidric J. Solution structure of polymyxins B and E and effect of binding to lipopolysaccharide: an NMR and molecular modeling study. J. Med. Chem. 1999;42(22):4604–4613. doi: 10.1021/jm991031b. ▪ Provides a detailed structural insight into the mechanism of polymyxin binding to lipid A.
- 25.Soon RL, Velkov T, Chiu F, et al. Design, synthesis and evaluation of a new fluorescent probe for measuring polymyxin–lipopolysaccharide binding interactions. Anal. Biochem. 2011;409(2):273–283. doi: 10.1016/j.ab.2010.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mogi T, Murase Y, Mori M, et al. Polymyxin B identified as an inhibitor of alternative NADH dehydrogenase and malate: quinone oxidoreductase from the Gram-positive bacterium Mycobacterium smegmatis. J. Biochem. 2009;146(4):491–499. doi: 10.1093/jb/mvp096. [DOI] [PubMed] [Google Scholar]
- 27.Sampson TR, Liu X, Schroeder MR, Kraft CS, Burd EM, Weiss DS. Rapid killing of Acinetobacter baumannii by polymyxins is mediated by a hydroxyl radical death pathway. Antimicrob. Agents Chemother. 2012;56(11):5642–5649. doi: 10.1128/AAC.00756-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fernandez L, Alvarez-Ortega C, Wiegand I, et al. Characterization of the polymyxin B resistome of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2013;57(1):110–119. doi: 10.1128/AAC.01583-12. ▪ Provides a global perspective on the bacterial cellular changes required to evolve polymyxin resistance.
- 29.Loutet SA, Mussen LE, Flannagan RS, Valvano MA. A two-tier model of polymyxin B resistance in Burkholderia cenocepacia. Environ. Microbiol. Rep. 2011;3(2):278–285. doi: 10.1111/j.1758-2229.2010.00222.x. [DOI] [PubMed] [Google Scholar]
- 30.Raetz CR, Reynolds CM, Trent MS, Bishop RE. Lipid A modification systems in Gram-negative bacteria. Annu. Rev. Biochem. 2007;76:295–329. doi: 10.1146/annurev.biochem.76.010307.145803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fernandez L, Jenssen H, Bains M, Wiegand I, Gooderham WJ, Hancock RE. The two-component system CprRS senses cationic peptides and triggers adaptive resistance in Pseudomonas aeruginosa independently of ParRS. Antimicrob. Agents Chemother. 2012;56(12):6212–6222. doi: 10.1128/AAC.01530-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clausell A, Garcia-Subirats M, Pujol M, Busquets MA, Rabanal F, Cajal Y. Gram-negative outer and inner membrane models: insertion of cyclic cationic lipopeptides. J. Phys. Chem. B. 2007;111(3):551–563. doi: 10.1021/jp064757+. [DOI] [PubMed] [Google Scholar]
- 33.Arroyo LA, Herrera CM, Fernandez L, Hankins JV, Trent MS, Hancock RE. The pmrCAB operon mediates polymyxin resistance in Acinetobacter baumannii ATCC 17978 and clinical isolates through phosphoethanolamine modification of lipid A. Antimicrob. Agents Chemother. 2011;55(8):3743–3751. doi: 10.1128/AAC.00256-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Breazeale SD, Ribeiro AA, McClerren AL, Raetz CR. A formyltransferase required for polymyxin resistance in Escherichia coli and the modification of lipid A with 4-amino-4-deoxy-l-arabinose. Identification and function of UDP-4-deoxy-4-formamido-l-arabinose. J. Biol. Chem. 2005;280(14):14154–14167. doi: 10.1074/jbc.M414265200. [DOI] [PubMed] [Google Scholar]
- 35.Gunn JS, Lim KB, Krueger J, et al. PmrA–PmrB-regulated genes necessary for 4-aminoarabinose lipid A modification and polymyxin resistance. Mol. Microbiol. 1998;27(6):1171–1182. doi: 10.1046/j.1365-2958.1998.00757.x. [DOI] [PubMed] [Google Scholar]
- 36.Macfarlane EL, Kwasnicka A, Ochs MM, Hancock RE. PhoP–PhoQ homologues in Pseudomonas aeruginosa regulate expression of the outer-membrane protein OprH and polymyxin B resistance. Mol. Microbiol. 1999;34(2):305–316. doi: 10.1046/j.1365-2958.1999.01600.x. [DOI] [PubMed] [Google Scholar]
- 37.Gunn JS. The Salmonella PmrAB regulon: lipopolysaccharide modifications, antimicrobial peptide resistance and more. Trends Microbiol. 2008;16(6):284–290. doi: 10.1016/j.tim.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 38.McPhee J, Lewenza S, Hancock R. Cationic antimicrobial peptides activate a two-component regulatory system, PmrA–PmrB, that regulates resistance to polymyxin B and cationic antimicrobial peptides in Pseudomonas aeruginosa. Mol. Microbiol. 2003;50(1):205–217. doi: 10.1046/j.1365-2958.2003.03673.x. [DOI] [PubMed] [Google Scholar]
- 39.Fernandez L, Gooderham WJ, Bains M, McPhee JB, Wiegand I, Hancock RE. Adaptive resistance to the ‘last hope’ antibiotics polymyxin B and colistin in Pseudomonas aeruginosa is mediated by the novel two-component regulatory system ParR–ParS. Antimicrob. Agents Chemother. 2010;54(8):3372–3382. doi: 10.1128/AAC.00242-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Llobet E, Campos MA, Gimenez P, Moranta D, Bengoechea JA. Analysis of the networks controlling the antimicrobial-peptide-dependent induction of Klebsiella pneumoniae virulence factors. Infect. Immun. 2011;79(9):3718–3732. doi: 10.1128/IAI.05226-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Loutet SA, Flannagan RS, Kooi C, Sokol PA, Valvano MA. A complete lipopolysaccharide inner core oligosaccharide is required for resistance of Burkholderia cenocepacia to antimicrobial peptides and bacterial survival in vivo. J. Bacteriol. 2006;188(6):2073–2080. doi: 10.1128/JB.188.6.2073-2080.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moffatt JH, Harper M, Harrison P, et al. Colistin resistance in Acinetobacter baumannii is mediated by complete loss of lipopolysaccharide production. Antimicrob. Agents Chemother. 2010;54(12):4971–4977. doi: 10.1128/AAC.00834-10. ▪ Using whole-genome sequencing, this study discovered a novel mechanism of polymyxin resistance that involves complete loss of lipopolysaccharide from the outer membrane.
- 43.Henry R, Vithanage N, Harrison P, et al. Colistin-resistant, lipopolysaccharide-deficient Acinetobacter baumannii responds to lipopolysaccharide loss through increased expression of genes involved in the synthesis and transport of lipoproteins, phospholipids and poly-β-1,6-N-acetylglucosamine. Antimicrob. Agents Chemother. 2012;56:59–69. doi: 10.1128/AAC.05191-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Llobet E, Tomas JM, Bengoechea JA. Capsule polysaccharide is a bacterial decoy for antimicrobial peptides. Microbiology. 2008;154(12):3877–3886. doi: 10.1099/mic.0.2008/022301-0. [DOI] [PubMed] [Google Scholar]
- 45.Pamp SJ, Gjermansen M, Johansen HK, Tolker-Nielsen T. Tolerance to the antimicrobial peptide colistin in Pseudomonas aeruginosa biofilms is linked to metabolically active cells, and depends on the pmr and mexAB-oprM genes. Mol. Microbiol. 2008;68(1):223–240. doi: 10.1111/j.1365-2958.2008.06152.x. [DOI] [PubMed] [Google Scholar]
- 46.Llobet E, March C, Gimenez P, Bengoechea JA. Klebsiella pneumoniae OmpA confers resistance to antimicrobial peptides. Antimicrob. Agents Chemother. 2009;53(1):298–302. doi: 10.1128/AAC.00657-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Padilla E, Llobet E, Domenech-Sanchez A, Martinez-Martinez L, Bengoechea JA, Alberti S. Klebsiella pneumoniae AcrAB efflux pump contributes to antimicrobial resistance and virulence. Antimicrob. Agents Chemother. 2010;54(1):177–183. doi: 10.1128/AAC.00715-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Warner DM, Levy SB. Different effects of transcriptional regulators MarA, SoxS and Rob on susceptibility of Escherichia coli to cationic antimicrobial peptides (CAMPs): Rob-dependent CAMP induction of the marRAB operon. Microbiology. 2010;156(Pt 2):570–578. doi: 10.1099/mic.0.033415-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fehlner-Gardiner CC, Valvano MA. Cloning and characterization of the Burkholderia vietnamiensis norM gene encoding a multidrug efflux protein. FEMS Microbiol. Lett. 2002;215(2):279–283. doi: 10.1111/j.1574-6968.2002.tb11403.x. [DOI] [PubMed] [Google Scholar]
- 50.Sahm H, Rohmer M, Bringer-Meyer S, Sprenger GA, Welle R. Biochemistry and physiology of hopanoids in bacteria. Adv. Microb. Physiol. 1993;35:247–273. doi: 10.1016/s0065-2911(08)60100-9. [DOI] [PubMed] [Google Scholar]
- 51.Malott RJ, Steen-Kinnaird BR, Lee TD, Speert DP. Identification of hopanoid biosynthesis genes involved in polymyxin resistance in Burkholderia multivorans. Antimicrob. Agents Chemother. 2012;56(1):464–471. doi: 10.1128/AAC.00602-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Loutet SA, Bartholdson SJ, Govan JR, Campopiano DJ, Valvano MA. Contributions of two UDP-glucose dehydrogenases to viability and polymyxin B resistance of Burkholderia cenocepacia. Microbiology. 2009;155(Pt 6):2029–2039. doi: 10.1099/mic.0.027607-0. [DOI] [PubMed] [Google Scholar]
- 53.Jiang SS, Lin TY, Wang WB, Liu MC, Hsueh PR, Liaw SJ. Characterization of UDP-glucose dehydrogenase and UDP-glucose pyrophosphorylase mutants of Proteus mirabilis: defectiveness in polymyxin B resistance, swarming, and virulence. Antimicrob. Agents Chemother. 2010;54(5):2000–2009. doi: 10.1128/AAC.01384-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Loutet SA, Valvano MA. Extreme antimicrobial peptide and polymyxin B resistance in the genus burkholderia. Front. Cell. Infect. Microbiol. 2011;1:6. doi: 10.3389/fcimb.2011.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mares J, Kumaran S, Gobbo M, Zerbe O. Interactions of lipopolysaccharide and polymyxin studied by NMR spectroscopy. J. Biol. Chem. 2009;284(17):11498–11506. doi: 10.1074/jbc.M806587200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.de Visser PC, Kriek NM, van Hooft PA, et al. Solid-phase synthesis of polymyxin B1 and analogues via a safety-catch approach. J. Pept. Res. 2003;61(6):298–306. doi: 10.1034/j.1399-3011.2003.00061.x. [DOI] [PubMed] [Google Scholar]
- 57.Okimura K, Ohki K, Sato Y, Ohnishi K, Sakura N. Semi-synthesis of polymyxin B (2–10) and colistin (2–10) analogs employing the trichloroethoxycarbonyl (Troc) group for side chain protection of alpha, gamma-diaminobutyric acid residues. Chem. Pharm. Bull. (Tokyo) 2007;55(12):1724–1730. doi: 10.1248/cpb.55.1724. [DOI] [PubMed] [Google Scholar]
- 58.Sakura N, Itoh T, Uchida Y, et al. The contribution of the N-terminal structure of polymyxin B peptides to antimicrobial and lipopolysaccharide binding activity. Bull. Chem. Soc. Jpn. 2004;77(10):1915–1924. [Google Scholar]
- 59.Chihara S, Ito A, Yahata M, Tobita T, Koyama Y. Chemical synthesis, isolation and characterization of α-N-fatty acyl colistin nonapeptide with special reference to the correlation between antimicrobial activity and carbon number of fatty acyl moiety. Agr. Biol. Chem. 1974;38(3):521–529. [Google Scholar]
- 60.Tsubery H, Ofek I, Cohen S, Fridkin M. N-terminal modifications of polymyxin B nonapeptide and their effect on antibacterial activity. Peptides. 2001;22(10):1675–1681. doi: 10.1016/s0196-9781(01)00503-4. [DOI] [PubMed] [Google Scholar]
- 61.Vaara M. The outer membrane permeability-increasing action of linear analogues of polymyxin B nonapeptide. Drugs Exp. Clin. Res. 1991;17(9):437–443. [PubMed] [Google Scholar]
- 62.Quale J, Shah N, Kelly P, et al. Activity of polymyxin B and the novel polymyxin analogue CB-182,804 against contemporary Gram-negative pathogens in New York City. Microb. Drug Resist. 2012;18(2):132–136. doi: 10.1089/mdr.2011.0163. [DOI] [PubMed] [Google Scholar]
- 63.Okimura K, Ohki K, Sato Y, Ohnishi K, Uchida Y, Sakura N. Chemical conversion of natural polymyxin B and colistin to their N-terminal derivatives. Bull. Chem. Soc. Jpn. 2007;80(3):543–552. [Google Scholar]
- 64.Katsuma N, Sato Y, Ohki K, Okimura K, Ohnishi K, Sakura N. Development of des-fatty acyl-polymyxin B decapeptide analogs with Pseudomonas aeruginosa-specific antimicrobial activity. Chem. Pharm. Bull. (Tokyo) 2009;57(4):332–336. doi: 10.1248/cpb.57.332. [DOI] [PubMed] [Google Scholar]
- 65.Barnett M, Bushby SR, Wilkinson S. Sodium sulphomethyl derivatives of polymyxins. Br. J. Pharmacol. Chemother. 1964;23:552–574. doi: 10.1111/j.1476-5381.1964.tb01610.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vaara M, Fox J, Loidl G, et al. Novel polymyxin derivatives carrying only three positive charges are effective antibacterial agents. Antimicrob. Agents Chemother. 2008;52(9):3229–3236. doi: 10.1128/AAC.00405-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kimura Y, Matsunaga H, Vaara M. Polymyxin B octapeptide and polymyxin B heptapeptide are potent outer membrane permeability-increasing agents. J. Antibiot. (Tokyo) 1992;45(5):742–749. doi: 10.7164/antibiotics.45.742. [DOI] [PubMed] [Google Scholar]
- 68.Kanazawa K, Sato Y, Ohki K, et al. Contribution of each amino acid residue in polymyxin B(3) to antimicrobial and lipopolysaccharide binding activity. Chem. Pharm. Bull. (Tokyo) 2009;57(3):240–244. doi: 10.1248/cpb.57.240. [DOI] [PubMed] [Google Scholar]
- 69.Tsubery H, Ofek I, Cohen S, Fridkin M. Structure activity relationship study of polymyxin B nonapeptide. Adv. Exp. Med. Biol. 2000;479:219–222. doi: 10.1007/0-306-46831-X_18. [DOI] [PubMed] [Google Scholar]
- 70.Nation RL, Li J. Colistin in the 21st century. Curr. Opin. Infect. Dis. 2009;22(6):535–543. doi: 10.1097/QCO.0b013e328332e672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Garonzik SM, Li J, Thamlikitkul V, et al. Population pharmacokinetics of colistin methanesulfonate and formed colistin in critically ill patients from a multicenter study provide dosing suggestions for various categories of patients. Antimicrob. Agents Chemother. 2011;55(7):3284–3294. doi: 10.1128/AAC.01733-10. ▪ Provides the first scientifically based dosage recommendations for colistin methanesulfonate for various categories of critically ill patients.
- 72.Plachouras D, Karvanen M, Friberg LE, et al. Population pharmacokinetic analysis of colistin methanesulphonate and colistin after intravenous administration in critically ill patients with Gram-negative bacterial infections. Antimicrob. Agents Chemother. 2009;53(8):3430–3436. doi: 10.1128/AAC.01361-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang H, Wu H, Ciofu O, Song Z, Hoiby N. In vivo pharmacokinetics/pharmacodynamics of colistin and imipenem in Pseudomonas aeruginosa biofilm infection. Antimicrob. Agents Chemother. 2012;56(5):2683–2690. doi: 10.1128/AAC.06486-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Owen RJ, Li J, Nation RL, Spelman D. In vitro pharmacodynamics of colistin against Acinetobacter baumannii clinical isolates. J. Antimicrob. Chemother. 2007;59:473–477. doi: 10.1093/jac/dkl512. [DOI] [PubMed] [Google Scholar]
- 75.Hawley JS, Murray CK, Jorgensen JH. Colistin heteroresistance in acinetobacter and its association with previous colistin therapy. Antimicrob. Agents Chemother. 2008;52(1):351–352. doi: 10.1128/AAC.00766-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yau W, Owen RJ, Poudyal A, et al. Colistin hetero-resistance in multidrug-resistant Acinetobacter baumannii clinical isolates from the western Pacific region in the SENTRY Antimicrobial Surveillance Programme. J. Infect. 2009;58(2):138–144. doi: 10.1016/j.jinf.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 77.Bergen PJ, Forrest A, Bulitta JB, et al. Clinically relevant plasma concentrations of colistin in combination with imipenem enhance pharmacodynamic activity against multidrug-resistant Pseudomonas aeruginosa at multiple inocula. Antimicrob. Agents Chemother. 2011;55(11):5134–5142. doi: 10.1128/AAC.05028-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dudhani RV, Turnidge JD, Coulthard K, et al. Elucidation of the pharmacokinetic/pharmacodynamic determinant of colistin activity against Pseudomonas aeruginosa in murine thigh and lung infection models. Antimicrob. Agents Chemother. 2010;54(3):1117–1124. doi: 10.1128/AAC.01114-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dudhani RV, Turnidge JD, Nation RL, Li J. fAUC/MIC is the most predictive pharmacokinetic/pharmacodynamic index of colistin against Acinetobacter baumannii in murine thigh and lung infection models. J. Antimicrob. Chemother. 2010;65(9):1984–1990. doi: 10.1093/jac/dkq226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Marchand S, Frat JP, Petitpas F, et al. Removal of colistin during intermittent haemodialysis in two critically ill patients. J. Antimicrob. Chemother. 2010;65(8):1836–1837. doi: 10.1093/jac/dkq185. [DOI] [PubMed] [Google Scholar]
- 81.Markou N, Fousteri M, Markantonis SL, et al. Colistin pharmacokinetics in intensive care unit patients on continuous venovenous haemodiafiltration: an observational study. J. Antimicrob. Chemother. 2012;67(10):2459–2462. doi: 10.1093/jac/dks257. [DOI] [PubMed] [Google Scholar]
- 82.Karvanen M, Plachouras D, Friberg LE, et al. Colistin methanesulfonate and colistin pharmacokinetics in critically ill patients receiving continuous venovenous hemodiafiltration. Antimicrob. Agents Chemother. 2013;57(1):668–671. doi: 10.1128/AAC.00985-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sandri AM, Landersdorfer CB, Jacob J, et al. Pharmacokinetics of polymyxin B in patients on continuous venovenous haemodialysis. J. Antimicrob. Chemother. 2013;68(3):674–677. doi: 10.1093/jac/dks437. [DOI] [PubMed] [Google Scholar]
- 84.Zavascki AP, Goldani LZ, Cao G, et al. Pharmacokinetics of intravenous polymyxin B in critically ill patients. Clin. Infect. Dis. 2008;47(10):1298–1304. doi: 10.1086/592577. [DOI] [PubMed] [Google Scholar]
- 85.Kwa AL, Lim TP, Low JG, et al. Pharmacokinetics of polymyxin B1 in patients with multidrug-resistant Gram-negative bacterial infections. Diagn. Microbiol. Infect. Dis. 2008;60(2):163–167. doi: 10.1016/j.diagmicrobio.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 86.Bergen PJ, Li J, Rayner CR, Nation RL. Colistin methanesulfonate is an inactive prodrug of colistin against Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2006;50(6):1953–1958. doi: 10.1128/AAC.00035-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kubin CJ, Ellman TM, Phadke V, Haynes LJ, Calfee DP, Yin MT. Incidence and predictors of acute kidney injury associated with intravenous polymyxin B therapy. J. Infect. 2012;65(1):80–87. doi: 10.1016/j.jinf.2012.01.015. [DOI] [PubMed] [Google Scholar]