

Abstract
Background
The acute response to cardiac resynchronization therapy (CRT) has been shown to be due to three mechanisms: resynchronization of ventricular contraction, efficient preloading of the ventricles by a properly timed atrial contraction, and mitral regurgitation reduction. However, the contribution of each of the three mechanisms to the acute response of CRT, specifically stroke work improvement, has not been quantified.
Objective
The goal of this study was to use an MRI-based anatomically accurate 3D model of failing canine ventricular electromechanics to quantify the contribution of each of the three mechanisms to stroke work improvement and identify the predominant mechanisms.
Methods
An MRI-based electromechanical model of the failing canine ventricles assembled previously by our group was further developed and modified. Three different protocols were used to dissect the contribution of each of the three mechanisms to stroke work improvement.
Results
Resynchronization of ventricular contraction did not lead to significant stroke work improvement. Efficient preloading of the ventricles by a properly timed atrial contraction was the predominant mechanism underlying stroke work improvement. Stroke work improvement peaked at an intermediate AV delay, as it allowed ventricular filling by atrial contraction to occur at a low diastolic LV pressure but also provided adequate time for ventricular filling before ventricular contraction. Diminution of mitral regurgitation by CRT led to stroke work worsening instead of improvement.
Conclusion
Efficient preloading of the ventricles by a properly timed atrial contraction is responsible for significant stroke work improvement in the acute CRT response.
Keywords: Heart failure, Cardiac resynchronization therapy, Dyssynchronous heart failure, Left bundle branch block, Stroke work
Introduction
Heart failure is a major cause of morbidity and mortality,1 contributing significantly to global health expenditure. A large number of heart failure patients exhibit a left bundle branch block (LBBB) type of electrical activation. In such patients, the left ventricle (LV) is activated through the septum from the right ventricle (RV), resulting in a delayed onset of LV contraction relative to that of the RV.2 Cardiac resynchronization therapy (CRT), which employs bi-ventricular pacing to re-coordinate the contraction of the heart, has been demonstrated to be a valuable therapeutic option for such patients.3 Even though CRT has been shown to improve heart failure symptoms and reduce hospitalization for most of the patients, one third of them fail to benefit from the therapy,3 reflecting an incomplete understanding of the mechanisms underlying the response to CRT. Gaining a better understanding of these mechanisms will help optimize the delivery of CRT so that the benefit of the therapy is extended to a larger patient population.
CRT typically involves implantation of three leads, one into right atrium (RA), and two in the RV and LV, respectively. The two ventricular electrodes provide stimuli simultaneously to elicit a synchronous contraction. Optimization of the pacing time interval between activating the RA and the subsequent activation of both ventricles (atrioventricular, AV, delay) has been shown to additionally improve the acute hemodynamic response of the heart.4 Heart failure patients often exhibit mitral regurgitation as a result of ventricular dilation and increased chamber sphericity.5 CRT has also been shown to acutely reduce mitral regurgitation severity in the dyssynchronous failing (dyssynchronous heart failure, DHF) ventricles5–7 by increasing the transmitral pressure gradient (the systolic LV-LA pressure difference)5 and diminishing the dyssynchrony of papillary muscle contraction.6, 7 Overall, the acute response of the DHF ventricles to CRT is thus believed to be due to 3 main mechanisms: resynchronization of ventricular contraction, efficient preloading of the ventricles by a properly timed atrial contraction, and reduced mitral regurgitation.8
The acute response to CRT is manifested by an augmentation in stroke work.9 However, the contribution of each of the three mechanisms to stroke work improvement during the acute response to CRT has not been quantified and therefore, the predominant mechanism underlying this improvement has not been identified. Currently, it is difficult to isolate and quantify the contribution of each of the three mechanisms to CRT-induced stroke work increase through experimental methods.10, 11 Therefore, a computational approach was undertaken in this study. A magnetic resonance image (MRI)-based electromechanical model of the DHF ventricles was developed to determine the contribution of each of the three proposed mechanisms to CRT-induced acute stroke work augmentation and to identify the predominant mechanism.
Methods
MRI-based electromechanical model of the DHF canine ventricles
To achieve the goals of this study, we employed an MRI-based electromechanical model of the DHF canine ventricles (Fig. 1) developed previously by our group.12 The model and the advancements implemented in it for the present research are described briefly below. The ventricular geometry and fiber-sheet architecture of the model were constructed from high-resolution MR and diffusion tensor (DT) MR images of DHF canine ventricles. The model consisted of coupled electrical and mechanical components, and a lumped-parameter representation of the circulatory system. Mathematical description of the electrical component of the model involved the use of the monodomain representation of cardiac tissue; the ventricular mechanics component was based on the continuum mechanics equations, with the myocardium assumed to be orthotropic, hyperelastic nearly-incompressible material.13–15
Figure 1.
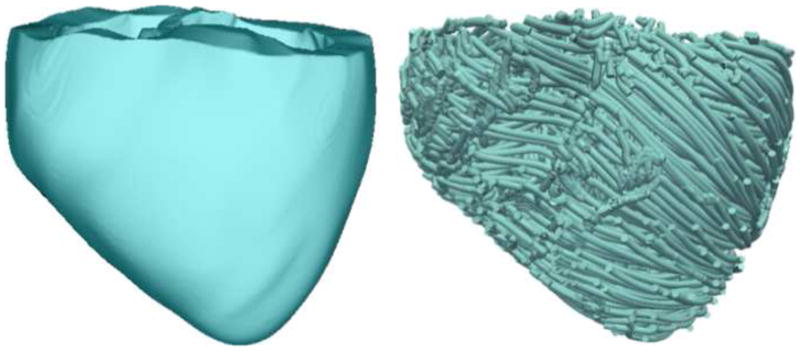
Geometry (left) and fiber orientation (right) of the DHF canine ventricular model.
For computational tractability, the electrical and mechanical components of the electromechanical model were weakly coupled. The local electrical activation times calculated from the electrical component of the model determined the instants when the Ca transient, which served as an input into the Rice et al. myofilament model16 in the mechanics component of the model, was initiated at the Gaussian points of each mechanical mesh element. The electrical component of the ventricular electromechanical model employed the Luo–Rudy dynamic model, LRd,17 to represent membrane kinetics; generic mammalian membrane kinetics models, such as LRd, are often used in whole-heart electromechanical models.18, 19 LRd is considered to be of medium-to-low complexity and is thus a reasonable trade-off in large-scale electromechanical simulations.
The formulation of the Ca transient in the myofilament model was modified to reflect abnormal Ca handling associated with DHF20 (detail in Supplementary Material, Appendix 1A). Furthermore, the values of the scaling factor for tension, of the crossbridge attachment rate constant to the first strongly-bound state, and of the scaling factor for all crossbridge cycling rates were adjusted to represent behavior in the canine ventricles; these were set to 205, 5000s−1 and 0.4, respectively, so that ejection fraction, LV peak pressure, and maximal rise in LV pressure matched values observed experimentally in failing canine ventricles.21 Additionally, the elastance of the atria in the lumped-parameter model of the circulatory system22 was increased fourfold so that atrial contraction accounted for 10% of ventricular filling, as reported for the failing canine ventricles.23 Mitral regurgitation was represented in the model of the circulatory system by allowing for backward flow from LV to the left atrium (LA) when LV pressure was higher than LA pressure (Supplementary Material, Appendix 1B).
To simulate LBBB activation of the canine ventricles, the RV endocardial surface of the ventricles was stimulated at a basic pacing cycle length of 500ms21 at discrete locations as if the electrical activation was emanating from the activation of the corresponding branch of the Purkinje network; the locations and timings were based on experimental findings.24 LBBB was simulated with an AV delay of 140ms, as recorded in DHF canine ventricles.21
CRT simulation
The model of the DHF ventricles described above was subjected to CRT, modeled as follows: RA pacing was represented by the onset of atrial contraction via initiating the activation function for atrial elastance in the model of the circulatory system; at a certain AV delay, the ventricles were paced simultaneously from the RV apex and the LV lateral wall.
Stroke work was calculated by integrating the area within the pressure-volume loop. Stroke work improvement (or worsening) following CRT was defined as the percentage increase (or decrease) of stroke work as a result of CRT relative to that in the DHF ventricles. Previous research has demonstrated that a physiologically meaningful CRT response is associated with a minimum of 15% stroke work improvement.9
Simulation protocol (I): Quantifying stroke work improvement as a result of resynchronization of ventricular contraction alone
Experimental evidence has demonstrated that both ventricular filling following atrial contraction and mitral regurgitation reduction affect ventricular hemodynamics.25, 26 Therefore, to quantify how much stroke work improvement resulted from CRT-induced resynchronization of ventricular contraction alone, DHF ventricular activation and CRT were simulated without accounting for atrial contraction and in the absence of mitral regurgitation (the DHF model in this case is only that of LBBB activation). This was achieved by setting the activation function for atrial elastance and the scaling factor for mitral valve resistance to 0 and infinity, respectively, in the lumped-parameter model of the circulatory system. The stroke work following CRT was compared to that under LBBB activation to determine whether there is a stroke work improvement due to resynchronization of ventricular contraction alone.
Simulation protocol (II): Quantifying stroke work improvement as a result of efficient preloading of the ventricles by a properly timed atrial contraction
Clinical and experimental studies have found that varying the pacing time interval between RA activation and ventricular activation (AV delay) in CRT affects the preloading of the ventricles and thus ventricular hemodynamics performance.4, 25, 27, 28 To understand how the duration of the AV delay imposed by the device affects the preloading of the ventricles and thus stroke work improvement following CRT, the DHF canine ventricular model with mitral regurgitation eliminated from it was employed in this protocol; device-imposed AV delays were varied between 0 and 140ms. This allowed us to determine the additional CRT stroke work improvement contributed by the efficient preloading of the ventricles due to a properly timed atrial contraction, over the baseline stroke work improvement resulting from resynchronization of ventricular contraction alone.
Simulation protocol (III): Quantifying stroke work improvement as a result of mitral regurgitation reduction by CRT
CRT has been shown to reduce mitral regurgitation to a different extent,6 which would then result in different changes in stroke work.29 Accordingly, we employed the original DHF canine ventricular model to simulate different degrees of CRT-induced mitral regurgitation reduction in this study: the scaling factor for mitral valve resistance was chosen (values are presented in Table 2) so that mitral regurgitant fraction either remained at 31% or was reduced to 15% or 0% as a result of CRT. The AV delay that gave rise to the maximal stroke work improvement as determined from simulation protocol II was used in the CRT simulations.
Table 2.
Stroke work improvement in simulation protocol III
| Stroke work for LBBB activation (kPa*mL) | CRT reduces mitral regurgitant ratio to | Scaling factor for mitral valve resistance | Stroke work following CRT (kPa*mL) | Stroke work improvement |
|---|---|---|---|---|
| 163.2 | 31% | 710 | 231.6 | 41.9% |
| 15% | 1670 | 209.5 | 28.4% | |
| 0% | infinity | 190.6 | 16.8% |
Results
Figure 2 portrays the 3D distribution maps of fiber strain at end-diastole, mid-systole and late-systole in the DHF canine ventricular model. Dyssynchronous activation is evident by the fact that when the septal wall contracted, the LV lateral wall was pre-stretched at mid-systole and the lateral wall shortened at late-systole. These 3D deformation patterns are consistent with those found experimentally in DHF ventricles.30
Figure 2.
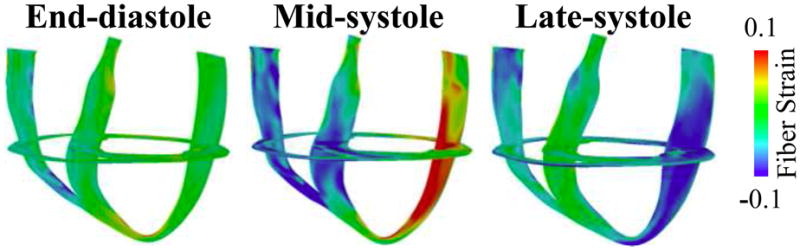
Distribution of fiber strain at three different time instants in the failing canine ventricular model with simulated LBBB electrical activation.
The results from simulation protocol I show that resynchronization of ventricular contraction, which resulted in a reduction of total activation time from 151ms in the DHF ventricles to 109ms following CRT, led to only 8.7% stroke work improvement, from 137.2 to 149.1 kPa*mL. Since stroke work improvement only above 15% is considered meaningful,9 our simulations concluded that resynchronization of ventricular contraction (i.e. reduction of total activation time) alone does not give rise to meaningful stroke work improvement.
The simulation results from protocol II demonstrated that stroke work improvement increased when AV delay decreased from 140ms to 40ms; when AV delay further shortened reaching 0ms, stroke work improvement decreased (Table 1). The maximum stroke work improvement was 40.5% achieved at an optimal AV delay of 40ms (Table 1). These results demonstrate that optimizing AV delay is critical to achieving a significant stroke work improvement.
Table 1.
| AV delay (ms) | 140 | 110 | 70 | 40 | 0 |
|---|---|---|---|---|---|
| Stroke work for CRT where only the RA was paced (kPa* mL) | 135.7* | 152.9 | 170.9 | 176.2* | 167.2* |
| Stroke work improvement for CRT where only the RA was paced | 0.0% | 12.7% | 25.9% | 29.8% | 23.2% |
| Stroke work for CRT simulations in protocol II (kPa* mL) | 156.6 | 174.1 | 189.5 | 190.6 | 173.4 |
| Stroke work improvement for CRT simulations in protocol II | 15.4% | 28.3% | 39.6% | 40.5% | 27.8% |
indicates the cases to which traces presented in Figure 3 correspond.
Since resynchronization of ventricular contraction did not result in marked stroke work improvement, while optimizing AV delay led to a significant stroke work improvement, it is possible that marked stroke work could be obtained through optimizing AV delay alone, without resynchronization of ventricular contraction (i.e. without reduction in total activation time). To test this hypothesis, we employed the DHF model from simulation protocol II; instead of pacing from RA and both ventricles for CRT, we only paced from RA and varied the time interval between RA pacing and LBBB electrical activation in the ventricles from 0 to 140ms. Stroke work improvement peaked at 29.8% when AV delay was 40ms (Table 1). This demonstrates that optimizing AV delay alone leads to significant stroke work improvement. Adding the 29.8% stroke work improvement by optimizing AV delay alone to the stroke work improvement resulting solely from resynchronization of ventricular contraction (8.7%) amounts to a total stroke work improvement of 38.5%, which is similar to that obtained under simulation protocol II with an AV delay of 40ms (40.5%). This shows that resynchronization of ventricular contraction (reduction in total activation time) contributed to the increased stroke work improvement, but not significantly.
To understand how optimizing AV delay alone, without resynchronization of ventricular contraction, increases stroke work improvement significantly, we examined the pressure and volume changes obtained in simulations in which we only paced the RA and changed the time interval between RA pacing and LBBB electrical activation in the ventricles (Fig. 3). Comparing LV pressures from simulations with AV delays of 40ms and 140ms, it can be seen that when AV delay was short, atrial contraction occurred when LV pressure was lower. As a result, the pressure against which LV filling by atrial contraction occurred was lower with an AV delay of 40ms. This led to an increase in LV end-diastolic volume and a larger stroke work improvement when AV delay was shortened from 140ms to 40ms (Fig. 3B, Table 1). However, when AV delay was shortened further to 0ms, even though the diastolic LV pressure was lower than that for AV delay of 40ms, LV filling time decreased by 20ms (Fig. 3A). As a result, the LV end-diastolic volume and thus the stroke work improvement were smaller than when AV delay was 40ms (Fig. 3B, Table 1). Therefore, by optimizing AV delay, preloading of the ventricles became more efficient and stroke work improvement increased. The mechanism underlying significant stroke work improvement following CRT was thus the efficient preloading of the ventricles by a properly timed atrial contraction.
Figure 3.
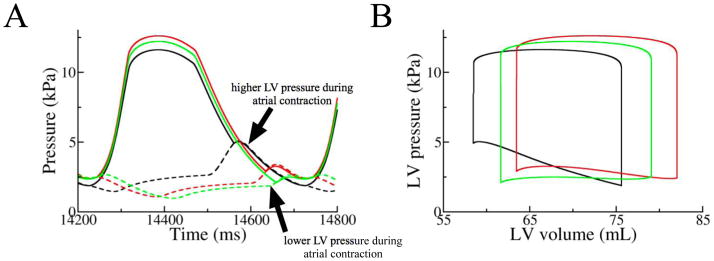
A) LA (broken lines) and LV (solid lines) pressures and B) pressure-volume loops from the simulations in which we only paced the RA and varied the time interval, 140 (black), 40 (red) and 0 ms (green), between RA pacing and LBBB electrical activation in the ventricles.
Our simulations found that diminution of mitral regurgitation associated with CRT did not lead to additional stroke work improvement; it actually caused a worsening in stroke work. Indeed, the results from simulation protocol III showed that if CRT did not reduce mitral regurgitation, stroke work improvement was 41.9%. However, if CRT reduced mitral regurgitation, stroke work improvement became less than that; decreasing mitral regurgitant ratio from 31% to 15% and 0% by CRT gave rise to only 28.4% and 16.8% stroke work improvement, respectively (Table 2).
In the DHF ventricular model from protocol III, 7.6mL and 16.5mL of blood were ejected by the LV into the LA and aorta, respectively. Stroke work for the DHF ventricles was 163.2kPa*mL: the work done by the LV to eject blood into the LA and the aorta were −17.3kPa*mL (the reason the work done by the LV to eject blood into the LA was negative was due to the fact that the mitral inflow volume was more than the regurgitant volume) and 180.5kPa*mL, respectively. If CRT reduced mitral regurgitant ratio to 0%, 19mL of blood was ejected by the LV into the aorta. Stroke work for the ventricles following CRT was 190.6kPa*mL: the work done by the LV to eject blood into the LA and aorta were −49.7kPa*mL and 240.2kPa*mL, respectively. Therefore, the 27.4kPa*mL (190.6kPa*mL-163.2kPa*mL) CRT-induced stroke work increase was due to 59.7kPa*mL (240.2kPa*mL-180.5kPa*mL) more work done by the LV to eject blood into the aorta but 32.4kPa*mL (−49.7kPa*mL-(−17.2kPa*mL)) less work done to eject blood into the LA.
Since efficient preloading of the ventricles by a properly timed atrial contraction results in significant stroke work changes in the acute response of CRT, atrial contraction was eliminated from the DHF model to dissect the mechanism by which mitral regurgitation decrease following CRT led to stroke work worsening. The scaling factor for mitral valve resistance was set to infinity so that mitral regurgitant ratio was reduced to 0% in CRT. LV (solid lines) and LA (broken lines) pressures from both LBBB activation (black) and CRT (red) simulations are plotted in Figure 4A. Mitral regurgitation in the DHF ventricles gave rise to elevation of atrial pressure (Fig. 4A) due to blood leaking back into the LA during ventricular systole. The elevated atrial pressure was then transmitted to the LV during filling so that LV end-diastolic volume was increased by 3mL (Fig. 4B). The net result was an increase in stroke volume and stroke work with mitral regurgitation (Fig. 4B). Thus diminution of mitral regurgitation by CRT led to stroke work worsening.
Figure 4.
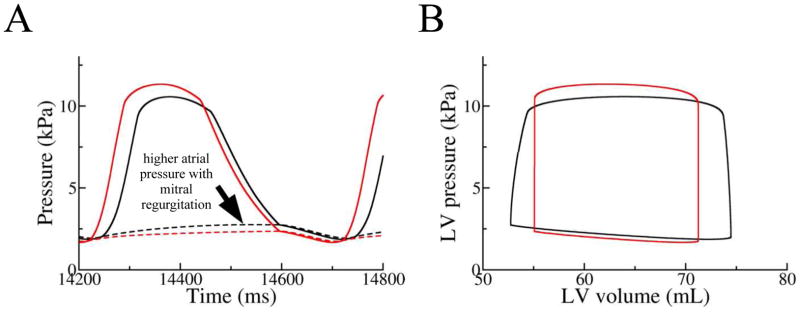
A) LA (broken line) and LV (solid line) pressures and B) pressure-volume loops from the LBBB and CRT simulations using the model from protocol III with atrial contraction eliminated.
Discussion
This study identified the predominant mechanism underlying stroke work augmentation in the acute response of CRT by employing an MRI-based anatomically accurate 3D model of the DHF canine ventricular electromechanics. For the first time, the contribution of each of the three mechanisms, namely resynchronization of ventricular contraction, reduction of mitral regurgitation, and efficient preloading of the ventricles by a properly timed atrial contraction, to stroke work improvement was quantified and the mechanisms identified. Significant findings of this study include:
Resynchronization of ventricular contraction alone did not result in significant stroke work improvement.
Efficient preloading of the ventricles by a properly timed atrial contraction was the predominant mechanism underlying stroke work improvement.
Stroke work improvement peaked at an intermediate AV delay, as it allowed ventricular filling by atrial contraction to occur at a low diastolic LV pressure but also provided adequate time for ventricular filling before ventricular contraction.
Diminution of mitral regurgitation by CRT led to stroke work worsening instead of improvement.
The acute response to CRT
In this paper we demonstrated that the predominant mechanism responsible for the large stroke work increase following CRT was not resynchronization of ventricular contraction, but rather it was the result of efficient preloading of the ventricles. In fact, optimizing AV delay alone without resynchronization of ventricular contraction was enough to increase stroke work significantly. Previous modeling research has shown that another hemodynamic metric - maximal rise in LV pressure increase in the acute response of CRT could be attributed to resynchronization of ventricular contraction; however, representation of atrial contraction was not incorporated into the model and the contribution of ventricular preloading to hemodynamics was not investigated in that study.31
Our simulation results demonstrated that when the AV delay was in the intermediate range, stroke work improvement was larger than when the AV delay was short or long. If mitral regurgitation reduction by CRT was small, stroke work augmentation was more than that if mitral regurgitation reduction by CRT was large. Therefore, we surmise that the reason why some patients do not respond to CRT, specifically do not register a significant stroke work improvement following CRT might be due to the fact that they had a non-optimal device-imposed AV delay or/and large degree of mitral regurgitation reduction as a result of CRT.
Optimal AV delay
We demonstrated that the AV delay needed to achieve maximum stroke work improvement in the acute response of CRT had an intermediate value (40ms in the tested range of 0–140ms). Clinical and experimental studies have found that the optimal AV delay in terms of hemodynamic metrics other than stroke work, such as maximal cardiac output,25 maximal rise in LV increase,4, 27, 28 aortic systolic pressure increase27 and pulse pressure improvement27 also have an intermediate value. The reason why these hemodynamic metrics were reduced at short and long AV delays was that with a short AV delay, atrial contraction occurred simultaneously with ventricular contraction, resulting in inefficient ventricular filling,25 consistent with our analysis; when the AV delay was too long, preloading of the LV was impaired due to diastolic mitral regurgitation.25 We showed that with a long AV delay, especially at relatively fast pacing rates (2Hz), preloading of the LV was impaired due to the high diastolic pressure against which atrial contraction occurred, which made ventricular filling by atrial contraction inefficient. It is possible that with a longer pacing cycle length, there would be more time for ventricular relaxation. Therefore, we expect that the LV pressure against which LV filling by atrial contraction occurs would not be significantly higher with a longer AV delay (140ms) compared to that at a shorter AV delay (40ms), as found here. As a result, preloading of the ventricles would not be as significantly impaired as in this study, and therefore, stroke work improvement would be larger at a longer AV delay. The contribution of efficient preloading of the ventricles by a properly timed atrial contraction to stroke work improvement could thus be less with a larger pacing cycle length.
Mitral regurgitation reduction due to CRT
CRT has been shown to acutely reduce mitral regurgitation severity in DHF patients.5–7 Two mechanisms have been proposed to explain how mitral regurgitation was reduced by CRT: CRT resulted in an increase in transmitral pressure gradient (the systolic LV-LA pressure difference), which effectively counteracted the increased tethering forces in heart failure that impaired mitral valve competence;5 and CRT diminished dyssynchrony of papillary muscle contraction in DHF, which led to reduced mitral regurgitation.6, 7 Our results demonstrate that mitral regurgitation gave rise to higher LA pressure, end-diastolic volume, stroke volume, and stroke work, in agreement with clinical and experimental results.26, 29, 32, 33 Inducing mitral regurgitation in dogs has been shown to result in increased LA pressure, end-diastolic volume, stroke volume, and stroke work.26, 29 End-diastolic volume, stroke volume and stroke work were higher in patients with mitral regurgitation than in the control subjects32 or in patients after surgical treatment of mitral valve disease.33
Clinical significance
Our findings indicate that in order to maximize, clinically, the acute CRT hemodynamic response in terms of improvement in stroke work, the best strategy would be to focus on optimizing device-imposed AV delay. Our simulations indicated that an intermediate AV delay (between 0ms but and the AV delay in LBBB activation) would be optimal.
Limitations
The model for mitral regurgitation in this study is relatively simple compared to existing models.34 However, there is no reason to expect that incorporating a more complex model for mitral regurgitation would alter the main conclusions of this simulation study regarding the predominant mechanism underlying stroke work improvement in the acute response of CRT.
Conclusion
In the present study, we quantified the contribution of each of the three mechanisms hypothesized to underlie stroke work improvement in the acute CRT response, namely resynchronization of ventricular contraction, improved preloading of the ventricles by a properly timed atrial contraction, and mitral regurgitation reduction. The predominant mechanism underlying significant stroke work improvement was found to be improved preloading of the ventricles by a properly timed atrial contraction.
Supplementary Material
Acknowledgments
This work was funded by National Institutes of Health (NIH) grant R01-HL103428, National Science Foundation Grants CBET-0933029 and IOS-1124804 to Natalia Trayanova, and by NIH fellowship F31-HL103090 to Jason Constantino. Natalia Trayanova is a cofounder of CardioSolv, LLC. CardioSolv was not involved in this research. Viatcheslav Gurev is employed by the IBM T.J. Watson Research Center. There are no patents, products in development or marketed products to declare.
Abbreviations
- AV delay
atrioventricular delay
- CRT
cardiac resynchronization therapy
- DHF
dyssynchronous heart failure
- DTMRI
diffusion tensor magnetic resonance image
- LA
left atrium
- LBBB
left bundle branch block
- LV
left ventricle
- MRI
magnetic resonance image
- RA
right atrium
- RV
right ventricle
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lloyd-Jones D, Adams R, Carnethon M, et al. Heart disease and stroke statistics--2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2009;119:e21–181. doi: 10.1161/CIRCULATIONAHA.108.191261. [DOI] [PubMed] [Google Scholar]
- 2.Wyndham CR, Smith T, Meeran MK, Mammana R, Levitsky S, Rosen KM. Epicardial activation in patients with left bundle branch block. Circulation. 1980;61:696–703. doi: 10.1161/01.cir.61.4.696. [DOI] [PubMed] [Google Scholar]
- 3.Abraham WT, Fisher WG, Smith AL, et al. Cardiac resynchronization in chronic heart failure. N Engl J Med. 2002;346:1845–1853. doi: 10.1056/NEJMoa013168. [DOI] [PubMed] [Google Scholar]
- 4.Kass DA, Chen CH, Curry C, et al. Improved left ventricular mechanics from acute VDD pacing in patients with dilated cardiomyopathy and ventricular conduction delay. Circulation. 1999;99:1567–1573. doi: 10.1161/01.cir.99.12.1567. [DOI] [PubMed] [Google Scholar]
- 5.Breithardt OA, Sinha AM, Schwammenthal E, et al. Acute effects of cardiac resynchronization therapy on functional mitral regurgitation in advanced systolic heart failure. J Am Coll Cardiol. 2003;41:765–770. doi: 10.1016/s0735-1097(02)02937-6. [DOI] [PubMed] [Google Scholar]
- 6.Kanzaki H, Bazaz R, Schwartzman D, Dohi K, Sade LE, Gorcsan J., 3rd A mechanism for immediate reduction in mitral regurgitation after cardiac resynchronization therapy: insights from mechanical activation strain mapping. J Am Coll Cardiol. 2004;44:1619–1625. doi: 10.1016/j.jacc.2004.07.036. [DOI] [PubMed] [Google Scholar]
- 7.Ypenburg C, Lancellotti P, Tops LF, et al. Acute effects of initiation and withdrawal of cardiac resynchronization therapy on papillary muscle dyssynchrony and mitral regurgitation. J Am Coll Cardiol. 2007;50:2071–2077. doi: 10.1016/j.jacc.2007.08.019. [DOI] [PubMed] [Google Scholar]
- 8.Abraham WT. Cardiac resynchronization therapy. Prog Cardiovasc Dis. 2006;48:232–238. doi: 10.1016/j.pcad.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 9.Helm RH, Byrne M, Helm PA, et al. Three-dimensional mapping of optimal left ventricular pacing site for cardiac resynchronization. Circulation. 2007;115:953–961. doi: 10.1161/CIRCULATIONAHA.106.643718. [DOI] [PubMed] [Google Scholar]
- 10.Amat-y-Leon F, Dhingra RC, Wu D, Denes P, Wyndham C, Rosen KM. Catheter mapping of retrograde atrial activation. Observations during ventricular pacing and AV nodal re-entrant paroxysmal tachycardia. Br Heart J. 1976;38:355–362. doi: 10.1136/hrt.38.4.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Messas E, Pouzet B, Touchot B, et al. Efficacy of chordal cutting to relieve chronic persistent ischemic mitral regurgitation. Circulation. 2003;108(Suppl 1):II111–115. doi: 10.1161/01.cir.0000087658.47544.7f. [DOI] [PubMed] [Google Scholar]
- 12.Constantino J, Hu Y, Trayanova NA. A computational approach to understanding the cardiac electromechanical activation sequence in the normal and failing heart, with translation to the clinical practice of CRT. Prog Biophys Mol Biol. 2012 doi: 10.1016/j.pbiomolbio.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu Y, Gurev V, Constantino J, Bayer JD, Trayanova NA. Effects of mechano-electric feedback on scroll wave stability in human ventricular fibrillation. PloS one. 2013;8:e60287. doi: 10.1371/journal.pone.0060287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gurev V, Lee T, Constantino J, Arevalo H, Trayanova NA. Models of cardiac electromechanics based on individual hearts imaging data: image-based electromechanical models of the heart. Biomech Model Mechanobiol. 2011;10:295–306. doi: 10.1007/s10237-010-0235-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gurev V, Constantino J, Rice JJ, Trayanova NA. Distribution of electromechanical delay in the heart: insights from a three-dimensional electromechanical model. Biophys J. 2010;99:745–754. doi: 10.1016/j.bpj.2010.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rice JJ, Wang F, Bers DM, de Tombe PP. Approximate model of cooperative activation and crossbridge cycling in cardiac muscle using ordinary differential equations. Biophys J. 2008;95:2368–2390. doi: 10.1529/biophysj.107.119487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Faber GM, Rudy Y. Action potential and contractility changes in [Na(+)](i) overloaded cardiac myocytes: a simulation study. Biophys J. 2000;78:2392–2404. doi: 10.1016/S0006-3495(00)76783-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Usyk TP, McCulloch AD. Relationship between regional shortening and asynchronous electrical activation in a three-dimensional model of ventricular electromechanics. J Cardiovasc Electrophysiol. 2003;14:S196–202. doi: 10.1046/j.1540.8167.90311.x. [DOI] [PubMed] [Google Scholar]
- 19.Jie X, Gurev V, Trayanova N. Mechanisms of mechanically induced spontaneous arrhythmias in acute regional ischemia. Circ Res. 2010;106:185–192. doi: 10.1161/CIRCRESAHA.109.210864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beuckelmann DJ, Nabauer M, Erdmann E. Intracellular calcium handling in isolated ventricular myocytes from patients with terminal heart failure. Circulation. 1992;85:1046–1055. doi: 10.1161/01.cir.85.3.1046. [DOI] [PubMed] [Google Scholar]
- 21.Leclercq C, Faris O, Tunin R, et al. Systolic improvement and mechanical resynchronization does not require electrical synchrony in the dilated failing heart with left bundle-branch block. Circulation. 2002;106:1760–1763. doi: 10.1161/01.cir.0000035037.11968.5c. [DOI] [PubMed] [Google Scholar]
- 22.Kerckhoffs RC, Neal ML, Gu Q, Bassingthwaighte JB, Omens JH, McCulloch AD. Coupling of a 3D finite element model of cardiac ventricular mechanics to lumped systems models of the systemic and pulmonic circulation. Ann Biomed Eng. 2007;35:1–18. doi: 10.1007/s10439-006-9212-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kono T, Sabbah HN, Rosman H, Alam M, Stein PD, Goldstein S. Left atrial contribution to ventricular filling during the course of evolving heart failure. Circulation. 1992;86:1317–1322. doi: 10.1161/01.cir.86.4.1317. [DOI] [PubMed] [Google Scholar]
- 24.Ramanathan C, Jia P, Ghanem R, Ryu K, Rudy Y. Activation and repolarization of the normal human heart under complete physiological conditions. Proc Natl Acad Sci U S A. 2006;103:6309–6314. doi: 10.1073/pnas.0601533103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nishimura RA, Hayes DL, Holmes DR, Jr, Tajik AJ. Mechanism of hemodynamic improvement by dual-chamber pacing for severe left ventricular dysfunction: an acute Doppler and catheterization hemodynamic study. J Am Coll Cardiol. 1995;25:281–288. doi: 10.1016/0735-1097(94)00419-q. [DOI] [PubMed] [Google Scholar]
- 26.Berko B, Gaasch WH, Tanigawa N, Smith D, Craige E. Disparity between ejection and end-systolic indexes of left ventricular contractility in mitral regurgitation. Circulation. 1987;75:1310–1319. doi: 10.1161/01.cir.75.6.1310. [DOI] [PubMed] [Google Scholar]
- 27.Auricchio A, Stellbrink C, Block M, et al. Effect of pacing chamber and atrioventricular delay on acute systolic function of paced patients with congestive heart failure. The Pacing Therapies for Congestive Heart Failure Study Group. The Guidant Congestive Heart Failure Research Group. Circulation. 1999;99:2993–3001. doi: 10.1161/01.cir.99.23.2993. [DOI] [PubMed] [Google Scholar]
- 28.Vernooy K, Verbeek XA, Cornelussen RN, et al. Calculation of effective VV interval facilitates optimization of AV delay and VV interval in cardiac resynchronization therapy. Heart Rhythm. 2007;4:75–82. doi: 10.1016/j.hrthm.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 29.Braunwald E, Welch GH, Jr, Sarnoff SJ. Hemodynamic effects of quantitatively varied experimental mitral regurgitation. Circ Res. 1957;5:539–545. doi: 10.1161/01.res.5.5.539. [DOI] [PubMed] [Google Scholar]
- 30.Breithardt OA, Stellbrink C, Herbots L, et al. Cardiac resynchronization therapy can reverse abnormal myocardial strain distribution in patients with heart failure and left bundle branch block. J Am Coll Cardiol. 2003;42:486–494. doi: 10.1016/s0735-1097(03)00709-5. [DOI] [PubMed] [Google Scholar]
- 31.Usyk TP, McCulloch AD. Electromechanical model of cardiac resynchronization in the dilated failing heart with left bundle branch block. J Electrocardiol. 2003;36 (Suppl):57–61. doi: 10.1016/j.jelectrocard.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 32.Leung DY, Armstrong G, Griffin BP, Thomas JD, Marwick TH. Latent left ventricular dysfunction in patients with mitral regurgitation: feasibility of measuring diminished contractile reserve from a simplified model of noninvasively derived left ventricular pressure-volume loops. Am Heart J. 1999;137:427–434. doi: 10.1016/s0002-8703(99)70487-4. [DOI] [PubMed] [Google Scholar]
- 33.Kennedy JW, Doces JG, Stewart DK. Left ventricular function before and following surgical treatment of mitral valve disease. Am Heart J. 1979;97:592–598. doi: 10.1016/0002-8703(79)90186-8. [DOI] [PubMed] [Google Scholar]
- 34.Grimes RY, Levine RA, Walker PG, Yoganathan AP. Dynamics of systolic pulmonary venous flow in mitral regurgitation: mathematical modeling of the pulmonary venous system and atrium. J Am Soc Echocardiogr. 1995;8:631–642. doi: 10.1016/s0894-7317(05)80376-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.