

Abstract
Background
Many current gene prediction methods use only one model to represent protein-coding regions in a genome, and so are less likely to predict the location of genes that have an atypical sequence composition. It is likely that future improvements in gene finding will involve the development of methods that can adequately deal with intra-genomic compositional variation.
Results
This work explores a new approach to gene-prediction, based on the Self-Organizing Map, which has the ability to automatically identify multiple gene models within a genome. The current implementation, named RescueNet, uses relative synonymous codon usage as the indicator of protein-coding potential.
Conclusions
While its raw accuracy rate can be less than other methods, RescueNet consistently identifies some genes that other methods do not, and should therefore be of interest to gene-prediction software developers and genome annotation teams alike. RescueNet is recommended for use in conjunction with, or as a complement to, other gene prediction methods.
Background
Computational gene prediction methods have yet to achieve perfect accuracy, even in the relatively simple prokaryotic genomes. Problems in gene prediction centre on the fact that many protein families remain uncharacterised. As a result, it seems that only approximately half of an organism's genes can be confidently predicted on the basis of homology to other known genes [1-3], so ab initio prediction methods are usually employed to identify many protein-coding regions of DNA.
Currently, the most popular prokaryotic gene-prediction methods, such as GeneMark.hmm [4] and Glimmer2 [5], are based on probabilistic Markov models that aim to predict each base of a DNA sequence using a number of preceding bases in the sequence. These methods are undoubtedly very successful, with published sensitivity rates between 90% and 99% for most prokaryotic genomes. However, as the sensitivity rates of the methods rise, specificity generally tends to fall, and while the application of sophisticated post-processing rules can correct many false-positive predictions, no method has yet achieved 100% accuracy. This is especially the case in the more complex eukaryotic gene-finding problem, where less than 80% of exons in anonymous genomic sequences are correctly predicted by current methods [2,6-8].
For the foreseeable future it does not seem that the exact set of genes in any organism can be automatically predicted by any single computational method. In practice, this has meant that the best predictions are to be found by combining evidence from two or more independent methods [3,9]. Genome annotation teams often compare the evidence offered by multiple gene-finders in order to predict the gene complement of a given genome. Because of the degree of 'manual' annotation that now takes place in the major genome sequencing centres, a gene-prediction tool will be of practical use if it can exclusively predict genes that other gene-finders cannot.
Many ab initio gene-prediction methods are based on single models of protein-coding regions and therefore make the implicit assumption that all protein-coding regions within a particular genome will share similar statistical properties. However, evidence has mounted that single gene models of intrinsic coding measures are no longer fully satisfying [10,11]. The problem with single model methods centres on the degree of oligonucleotide composition variation that exists within most genomes. On the codon level, intra-genomic variation in codon bias has long been correlated with expression level [12]. Counterbalancing the translational selection theory of codon bias is the effect of mutational bias [13,14]. Many other, often more subtle levels of variation have been recognised over the years, with many disparate evolutionary pressures shown to be acting on codon usage bias [15]. For example, strand-specific codon usage biases have often been recognised [16-20], leading to more general studies of correlation between the location of the gene on the genome and codon bias [21], and the more specific discovery of a A+T skewed bias near the replication terminus of bacterial genomes [22]. Other effects shown to shape codon usage are gene length [23] and selection at the amino acid level [24]. It has also been suggested that content variation can occur at the exon level in eukaryotic genes, the possibility existing that some exons in a gene may have different codon usage patterns to others [1]. Given that some of the above pressures on codon usage have only recently been discovered, it is likely that some more subtle patterns have yet to be recognised, and therefore it is difficult to predict the level of compositional variation that will be present in an anonymous genomic sequence.
The need for gene-finding methods that can overcome the problems presented by intra-genomic variation was recognised and addressed in the case of prokaryotic genomes by GeneMark-Genesis [25], which derives two models for each genome according to typical and atypical codon usage clusters in that genome. This increase in the number of gene models led to an increase in accuracy of the GeneMark method. While Hayes & Borodovsky experimented with a third ('highly-typical') codon usage cluster and an associated model in some cases, they did not see the need to further sub-cluster the atypical codon usage set in order to make even more models. Overly sub-clustering the training data would not be useful in the case of Markov-based methods, as the data contained in each sub-cluster may not allow for a good estimation of model parameters. However, generating more specific models for subtle patterns found in the training set can only be advantageous if it can be done in a way that minimises loss of overall accuracy and produces no extra false-positive predictions.
This paper aims to show how the Self-Organizing Map neural network algorithm can be used to automatically identify the major trends in oligonucleotide variation in a genome, and in doing so provide multiple gene models for use in gene prediction. It will be explained that this approach is an effective solution to the problem of intra-genomic variation. Specific examples of genes predicted only by this method are offered, thus demonstrating the usefulness of the approach in genome annotation.
A further advantage of using the Self-Organizing Map for gene prediction is the ability of the algorithm to use complex descriptors as measures of gene coding potential. We demonstrate this ability using relative synonymous codon usage (RSCU) as our measure of gene coding potential. Unlike other gene coding measures, RSCU is not based on the absolute frequency of k-mers, but instead describes the codon choice for each amino acid. Markov chains based on the RSCU measure would have transition probabilities that are conditioned on the underlying amino-acids. Although theoretically possible [26], the practical computation of such Markov chains would give rise to major difficulties. Therefore, the ability of our approach to make use of a sophisticated gene coding descriptor such as RSCU is a distinct advantage of our approach over Markov model based methods.
Implementation
Coding measure
In this study, relative synonymous codon usage (RSCU) vectors are used as the measure of protein-coding potential for a given window of sequence. The RSCU value for a codon 'i' is defined as:
![]()
where Obsi is the observed number of occurrences of codon 'i', and Expi is the expected number of occurrences of the same codon (based on the number of times the relevant amino acid is present in the gene and the number of synonymous alternatives to 'i', assuming a uniform choice of synonymous codons). In order to make the data more compatible with the mathematical methods used, the log of each RSCUi value is found so that the resulting value is positive if the codon is used more than expected in that gene, and negative if the codon is used less than expected. Values were capped at ± 10, and set to 0 in the case of the non-occurrence of an amino acid in the sample. Taking the RSCU values for each of the codons with synonymous alternatives (and ignoring the 3 stop codons and the Trp and Met codons), each sample can be represented by a vector of 59 values.
Self-Organizing Map
The Self-Organizing Map (SOM) is based around the concept of a lattice of interconnected nodes, each of which contains a model. The models begin as random values, but during the iterative training process they are modified to represent different subsets of the training set. In this work for example, the training set and the lattice node models are 59-dimensional RSCU vectors, and the models change during training to become similar to common or repeated patterns in the training set. The algorithm is fully described elsewhere [27], but we briefly summarize for our context:
(1) A vector (Xi), corresponding to a gene's RSCU values, is loaded from the training dataset.
(2) The lattice node is found whose model vector most closely resembles the input pattern. This node is denoted the 'winning node'.
(3) The winning node's model, W (as well as a certain number of 'neighbourhood bubble' node models) is changed to be more similar to the input vector by the equation:
Wnew = Wold + η (Xi - Wold) (2)
(4) If all the vectors in the training dataset are processed, we say that an epoch has been completed. In this study, all SOMs are trained for 3000 epochs.
The 'neighbourhood bubble' mentioned in step 3 is a group of nodes centered at the winning node. The radius of this bubble is initialised to be large and is linearly decreased during training until only the winning node's model is changed. Changing the models on the winning node's neighbours allows the clustering of similar patterns. The learning rate (η) in step 3 is initialised close to 1 and is also linearly decreased during training until it is held constant at a predefined fraction. The linear decrease in learning rate means that each node's model will not get changed as much or as often as training progresses. Two recognised phases of training result; an ordering phase where the lattice takes its general shape, and a convergence phase where the nodes get more specialised to respond to specific patterns.
In this work, similarity between two vectors is measured by finding the cosine of the angle between them. A cosine of 1 represents exactly similar vectors while a cosine of 0 represents exactly dissimilar vectors.
The SOM is used mainly in data visualisation, as it can be effectively used to reduce high-dimensional data to a two dimensional map. One of the main strengths of the method is the ability to automatically cluster similar patterns in its training set. In the context of codon usage data, the SOM has been previously used to cluster genes on the basis of similar codon usage [28-30]. However, the previous studies have concentrated on identifying genes with atypical codon usage and hypothesising their origin as horizontally transferred genes. It has since been shown that atypical codon usage is not sufficient evidence to show that a gene has origins in horizontal transfer events [22,31]. In contrast, this study uses the fact that once a SOM has been trained using codon usage information, the nodes of the SOM encapsulate models that are representative of the major codon usage patterns within the training set.
If a new sequence is inputted to a trained SOM, we can easily be told which node's model is most similar to this new sequence, and most importantly, how similar. The similarity (cosine) score is then converted to the probability that the sequence is protein-coding. This is achieved by finding the mean cosine score received by a set of random length, random sequence genes that are generated using the same nucleotide bias as the mutational bias found in the genome. Using the mean score, each similarity score can be converted to a z-score, which is in effect the probability that the sequence is not a random sequence.
Using the SOM to find genes
Separate SOMs are trained for each of the 15 genomes under test. The SOMs are each 15×15 nodes in size and trained for 3000 cycles. Finding genes via homology search is usually the first step to be carried out in a genome annotation process, so our training sets consist of all genes in the relevant organism that were previously confirmed by homology searches and are also at least 750 bp long. Note that unlike other gene-finding methods, no statistical knowledge of non-coding DNA is necessary as part of the SOM's training.
In analysing an entire genome sequence, a sliding window is used to split each of the six reading frames into small samples. The default window size is 110 triplets, which has been chosen as a balance between having a window size long enough to evaluate a meaningful RSCU vector, and short enough to predict short genes. Each window is offset from the next by 10 triplets. An arbitrary probability score of 0.1 is used as the threshold for deciding if a sequence was protein-coding, and all samples that scored higher than 0.1 are recorded as predictions. If a stop codon lies in the sample, the gene prediction is annotated as having ended at that point. Note, however, that no effort is made to find stop codons if they are not within the prediction, and no effort whatsoever is made to find any start codons in the prediction.
Post-processing the predictions
Once all the samples are processed, some simple post-processing is carried out. Naturally, all same-frame concurrent predictions are merged. Predictions that are totally overlapped by another prediction are deleted if they are less than 75% the length of the other. Similarly, any prediction in which more than half its length is overlapped is deleted if it is less than half the length of the other prediction. Alternatively, any prediction that is less than 90% as long as the overlapping prediction and receiving a lower score is deleted. Finally, any prediction that is overlapped on both ends to a total overlap of at least 70% is also deleted. A prediction size of 75 codons was found by trial and error to be the smallest gene-coding region that could accurately be found using RescueNet.
While the above rules aim to delete smaller erroneous predictions, it is recognised that the loose nature of the rules leave room for many other overlapping predictions. However, it was found that in many overlapping cases it was difficult to decide which prediction to delete. Therefore, the best solution is to leave both predictions rather than misleading an annotator by giving only one, possibly erroneous, prediction.
In assessing the accuracy of our method, we had to take into account that our method will not predict most start sites, and some stop sites, exactly. We assume that our method will be of most use to annotation teams who rigorously inspect the results of our method in conjunction with the results of other gene prediction programs. Such annotation teams base their final genome annotation on widespread evidence, so the fact that our method may produce inexact start and stop sites will not be a major disadvantage. Therefore, a correct prediction is defined here as one that predicts more than 50% of an annotated gene in the correct frame. This criterion means that only predictions that are useful to annotators are considered to be correct.
Results and discussion
Evaluating accuracy rates
Previous studies discuss the possibility that the GenBank annotation of various genomes may be incomplete or incorrect in some cases [5,32]. Since many GenBank annotations are not experimentally corroborated, this possibility remains strong. Large-scale benchmarking of gene-prediction algorithms is therefore difficult, because few 'gold standard' annotations exist for prokaryotic genomes. Also, in most cases hypothetical gene annotations in the public databases have their roots in the predictions of an ab initio method, thus biasing any comparison of accuracy in favour of the particular method used in the annotation of that genome. However, for the purpose of defining accuracy in this study we must assume that all GenBank annotations are correct and complete. Sensitivity (Sn) is defined here as the percentage of GenBank gene records that are predicted correctly by our method. Specificity (Sp) is defined as the percentage of total RescueNet gene predictions that are correct.
Table 1 shows the results of RescueNet's predictions in 15 genomes. All results were generated using the default settings described above. Sensitivity and specificity values for each genome are shown, along with sensitivity values for those genes that are above the prediction length threshold of 75 codons (225 bp) and sensitivity values for those genes that have database matches.
Table 1.
Accuracy of RescueNet in 15 bacterial genomes.
| Organism | GC % | Number of Genes Annotated | Training Set Size | Sn. (%) | Sn. >225 bp (%) | Sn. Conserved (%) | Sp. (%) |
| Buchnera | 26.2 | 564 | 292 | 88.65 | 91.24 | 89.97 | 96.18 |
| B. burgdorferi | 28.6 | 857 | 403 | 90.54 | 96.39 | 95.66 | 98.02 |
| C. jejuni | 30.6 | 1654 | 673 | 90.14 | 95.08 | 92.14 | 99.23 |
| M. jannaschii | 31.4 | 1715 | 692 | 88.39 | 91.82 | 91.02 | 96.50 |
| M. genitalium | 31.7 | 483 | 301 | 89.44 | 91.52 | 89.89 | 92.32 |
| H. influenzae | 38.0 | 1754 | 885 | 91.56 | 96.34 | 93.10 | 98.01 |
| H. pylori | 38.9 | 1593 | 712 | 91.39 | 96.80 | 95.70 | 95.49 |
| A. aeolicus | 43.3 | 1517 | 723 | 95.78 | 96.54 | 95.57 | 87.80 |
| B. subtilis | 43.5 | 4220 | 1832 | 87.93 | 94.95 | 89.86 | 89.47 |
| Synechocystis | 47.6 | 3169 | 954 | 93.18 | 96.53 | 91.55 | 90.95 |
| Y. pestis | 47.6 | 4043 | 1640 | 91.04 | 94.84 | 93.66 | 88.29 |
| E. coli | 50.8 | 4290 | 1983 | 89.39 | 92.85 | 92.54 | 89.04 |
| D. radiodurans | 67.0 | 2622 | 1436 | 84.28 | 85.65 | 92.61 | 95.50 |
| R. solanacearum | 67.0 | 3442 | 1748 | 84.74 | 88.60 | 89.82 | 93.20 |
| S. coelicolor | 72.1 | 7851 | 956 | 88.35 | 91.55 | 91.55 | 90.10 |
The genomes are listed according to ascending G+C content. For each genome, the table shows: Genome GC content (GC %), the number of genes annotated in GenBank for that genome, the number of genes in the RescueNet training set, overall RescueNet sensitivity (Sn.), the sensitivity of RescueNet in finding genes longer than the 225 bp minimum prediction size (Sn. >225 bp), the sensitivity of RescueNet in finding genes that have been confirmed by homology with other genes in GenBank (Sn. Conserved), and finally, overall RescueNet specificity (Sp.)
Sequence data used in this study include the following 15 genomes and associated published genes available from the GenBank database: A. aeolicus [33], B. subtilis [34], Buchnera sp. [35], B. burgdorferi [36], C. jejuni [37], D. radiodurans (chromosome 1) [38], E. coli [39], H. influenzae [40], H. pylori [41], M. genitalium [42], M. jannaschii [43], R. solanacearum [44], S. coelicolor [45], Synechocystis sp. [46], and Y. pestis [47]. These genomes were chosen to be representative of a wide range of GC content.
High G+C content genomes
Three of the genomes tested have very high G+C content (D. radiodurans, R. solanacearum and S. coelicolor). High G+C content genomes present a problem to many gene-finding methods because of the relative infrequency of randomly occurring stop codons. The scarcity of stop codons has the effect of a large number of long, overlapping ORFs occurring in the sequence, relatively few of which are actually protein-coding. Many of the current gene-finders fail to discriminate accurately between coding and non-coding ORFs in this type of situation.
In our method, the relatively high specificity in each high G+C content genome suggests that RescueNet may have advantages in their annotation (see Table 1). To illustrate a case where RescueNet may be of practical use, we can consider the ORF annotated as DR1142 (see Figure 1) from D. radiodurans. This ORF is annotated to be protein-coding on the basis of the Glimmer2 prediction only. The RescueNet prediction in this area overlaps DR1142, but on the opposite strand. This type of situation, where a RescueNet prediction directly contradicts a GenBank/Glimmer2 annotation, occurs at least 23 times in the D. radiodurans genome. It is entirely possible that the Glimmer2 predictions are wrong in some of these cases, and the RescueNet predictions correct, but this cannot be proven without biochemical characterisation of the relevant gene. However, in the specific case of the DR1142 annotation, the RescueNet prediction has a much stronger database match than the GenBank annotation, and so has a high possibility of being correct.
Figure 1.

Screenshot from the Artemis sequence viewer [49] showing a sample region of D. radiodurans and accompanying RescueNet predictions. Annotated genes are shown as white blocks, and predictions are shown in-frame as shaded blocks. Note the relative infrequency of stop codons (vertical lines in each frame) and the many ORFs that are not protein-coding regions. Note also the selected gene DR1142 and the contradicting RescueNet prediction. DR1142 is a hypothetical gene, predicted to be so by Glimmer2, and there is a strong possibility that the CDS marked by RescueNet is the correct prediction. The possibility is also raised by RescueNet that the gene DR1143 may be longer than previously annotated and contains a frameshift.
Another interesting pointer to the advantages of RescueNet in high G+C content genomes is the substantially higher percentage of genes with database matches that are correctly predicted by RescueNet (see Table 1). In D. radiodurans, for example, 92.54% of genes with database matches are correctly predicted by RescueNet compared with only 84.28% of the total GenBank gene annotations. These figures suggest that hypothetical genes that are predicted only by Markov-based methods are poorly recognised by RescueNet, possibly because many hypothetical genes in high G+C content genomes may in fact be false gene predictions.
Predicting the location of frameshifts
The general location of frameshifts within a gene sequence can be found by our method. Two features of our approach facilitate this. Firstly, even though the overall codon usage of a frameshifted gene could seem unusual, the two coding sections of the gene should each retain the organism's native codon usage. Secondly, our approach does not require that a prediction be bounded by a start and a stop codon. The sliding window used in our algorithm can therefore predict the correct coding frames each side of the frameshift.
In an interesting example in Figure 1, two RescueNet predictions overlap the D. radiodurans gene DR1143 in such a way that it seems that there may be a frameshift that extends the protein-coding region of the gene past the annotated stop codon. In fact, combining the two RescueNet predictions offers a better database match to the same genes that the original annotation matches. This increases the possibility that the actual gene contains an authentic frameshift or at least that the extra RescueNet prediction is an evolutionary artefact.
Figure 2 shows another example of frameshifts which are detected by RescueNet. In this case, the H. influenzae genes HI0218 and HI0220 both contain frameshifts, but both are handled by RescueNet's predictions. Note that the GeneMark algorithms are known to show the location of frameshifted regions in much the same manner as we have described, but our approach has required no modification to our basic algorithm in order to facilitate the prediction of frameshifted genes.
Figure 2.
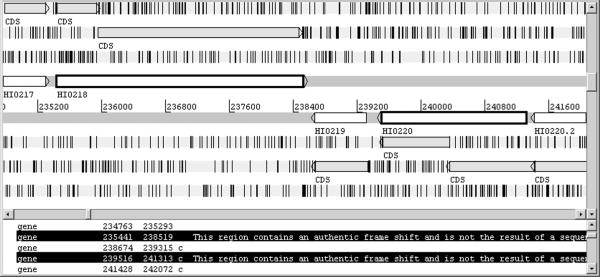
Artemis screenshot showing a sample region of the H. influenzae genome and associated RescueNet predictions. As in Fig. 1, the annotated genes are shown as white blocks and the RescueNet predictions are shown in-frame as shaded blocks. Note that genes HI0218 and HI0220 contain authentic frameshifts. RescueNet gives two predictions that overlap each of these gene, and they meet near the frameshift point.
Comparison with a Markov-based method
There may be a perception that any method using codon usage as the coding measure will only give predictions that are a subset of the predictions given by a Markov-based method that uses a 4th or 5th order model. To counter this argument, we compared the predictions of our method in two genomes (H. influenzae and H. pylori) to those of the web-based version of GeneMark.hmm 2.1 for prokaryotes http://opal.biology.gatech.edu/GeneMark/gmhmm2_prok.cgi, which generated results using two models; the 'typical' and 'atypical' models.
The published sensitivity of GeneMark.hmm in the H. influenzae genome (96.2%, see [4]) is higher than that of our method, and the published specificity (89.8%) is lower, so GeneMark.hmm should give more predictions overall for this genome. However, 11 H. influenzae genes are predicted correctly by our method which are not predicted by GeneMark.hmm using 5th order models, and 14 genes are predicted correctly by our method which are not predicted by GeneMark.hmm using 4th order models. In the H. pylori genome, GeneMark.hmm has again a higher sensitivity and a lower specificity (94.0% & 91.3% respectively), but even more genes are exclusively predicted by our method; 25 genes as compared to the 5th order GeneMark.hmm models and 30 genes as compared to the 4th order models. Although these genes represent a small proportion of the total number of genes in the respective organisms, the fact that they are only predicted by RescueNet gives some indication of the advantage of using RescueNet in conjunction with other gene prediction methods.
Possible future improvements
RSCU is only one of many possible criterion with which to measure coding potential (see [48] for a review of others). In-phase hexamers are accepted as the most accurate k-mer frequency based measure of coding potential, and so their use as the coding measure in a Self-Organizing Map may offer improvement in accuracy over RescueNet. However, the larger space dimension of the hexamer coding measure may force a larger sliding window to be used and therefore the use of hexamers could actually decrease the precision of gene prediction.
The future use of alternative coding measures with our approach may also help to overcome difficulties in recognising genes that are reputed to be horizontally transferred in origin. Horizontally transferred genes would be more likely to have dissimilar codon usage patterns to other genes in the genome. Since our approach currently relies on the codon usage patterns it finds in the training set, it is unlikely to mark areas of unseen codon usage as protein-coding regions. Note, however, we are not suggesting all genes that were not recognised by our approach are of horizontally transferred origin. There are many explanations for a gene displaying atypical codon usage, and codon usage cannot be used as an accurate indicator of horizontal transfer.
There may be other ways to improve the accuracy of our method. The current implementation has a rather simple post-processing step that does not rely on modifying the prediction in order to include start or stop codons. While the practise of not constraining a prediction to be bound by a start and stop codon stands in stark contrast to other methods, we did not wish to lengthen or shorten any predictions artificially, since doing so can mislead annotation teams (especially in start site annotation). Relatively simple post-processing steps may, in fact, be advantageous. Our predictions represent a raw account of regions of the genome that display typical or native codon usage patterns, and this in itself may be of interest to annotation teams who use codon usage plots as the basis for some genomic feature annotations.
Conclusion
Gene-finding in prokaryotic genomes is still not a completely solved problem, partly because current methods use a limited number of models to represent the training data. In this paper, we have introduced an alternative, independent approach to the problem. The Self-Organizing Map approach has the potential to overcome the issue of variation in the statistical properties of the training set data, and can automatically train a representative number of gene-models, depending on the degree of variation within the training data.
While the current implementation of our approach produces lower raw sensitivity scores in comparison to established Markov-based techniques, we have clearly shown that our method can predict some genes that other methods cannot. We have also demonstrated advantages in annotating the traditionally 'difficult' high G+C content genomes. Annotation teams who are concerned with the complete and accurate annotation of a sequenced genome should find our method useful when used alongside other gene-finding methods. The relatively high specificity of our method, coupled with the independent nature of the algorithm, should make it a useful tool in confirming the predictions of other software programs and in some cases pointing out areas of conflicting or contradictory predictions that are worthy of further examination.
Availability
Project name: RescueNet
Project home page: http://bioinf.nuigalway.ie/RescueNet/
Operating systems: Windows, Linux, IRIX, Digital Unix. Source code also available.
Programming Language: C++
Licence: GNU GPL
Restrictions to use by non-academics: Please contact the authors.
Authors' contributions
SM conceived of the study, and designed, implemented and tested the RescueNet software. JMcI, TS and AG supervised, and participated in the design of, the study. All authors read and approved the final manuscript.
Acknowledgments
Acknowledgements
The authors would like to thank Kim Rutherford of the Welcome Trust Sanger Institute Pathogen Sequencing Group for his help and suggestions. We also thank the two anonymous reviewers for their useful comments. S.M. is funded by an EMBARK postgraduate fellowship from the Irish Research Council for Science, Engineering & Technology.
Contributor Information
Shaun Mahony, Email: shaun.mahony@nuigalway.ie.
James O McInerney, Email: james.o.mcinerney@may.ie.
Terry J Smith, Email: terry.smith@nuigalway.ie.
Aaron Golden, Email: aaron.golden@nuigalway.ie.
References
- Mathe C, Sagot MF, Schiex T, Rouze P. Current methods of gene prediction, their strengths and weaknesses. Nucleic Acids Res. 2002;30:4103–4117. doi: 10.1093/nar/gkf543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claverie JM. Computational methods for the identification of genes in vertebrate genomic sequences. Hum Mol Genet. 1997;6:1735–1744. doi: 10.1093/hmg/6.10.1735. [DOI] [PubMed] [Google Scholar]
- Fickett JW. Finding genes by computer: the state of the art. Trends Genet. 1996;12:316–320. doi: 10.1016/0168-9525(96)10038-X. [DOI] [PubMed] [Google Scholar]
- Lukashin AV, Borodovsky M. GeneMark.hmm: new solutions for gene finding. Nucleic Acids Res. 1998;26:1107–1115. doi: 10.1093/nar/26.4.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcher AL, Harmon D, Kasif S, White O, Salzberg SL. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999;27:4636–4641. doi: 10.1093/nar/27.23.4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guigo R, Agarwal P, Abril JF, Burset M, Fickett JW. An assessment of gene prediction accuracy in large DNA sequences. Genome Res. 2000;10:1631–1642. doi: 10.1101/gr.122800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bork P. Powers and pitfalls in sequence analysis: the 70% hurdle. Genome Res. 2000;10:398–400. doi: 10.1101/gr.10.4.398. [DOI] [PubMed] [Google Scholar]
- Reese MG, Hartzell G, Harris NL, Ohler U, Abril JF, Lewis SE. Genome annotation assessment in Drosophila melanogaster. Genome Res. 2000;10:483–501. doi: 10.1101/gr.10.4.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogic S, Ouellette BF, Mackworth AK. Improving gene recognition accuracy by combining predictions from two gene-finding programs. Bioinformatics. 2002;18:1034–1045. doi: 10.1093/bioinformatics/18.8.1034. [DOI] [PubMed] [Google Scholar]
- Borodovsky M, McIninch JD, Koonin EV, Rudd KE, Medigue C, Danchin A. Detection of new genes in a bacterial genome using Markov models for three gene classes. Nucleic Acids Res. 1995;23:3554–3562. doi: 10.1093/nar/23.17.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathe C, Dehais P, Pavy N, Rombauts S, Van Montagu M, Rouze P. Gene prediction and gene classes in Arabidopsis thaliana. J Biotechnol. 2000;78:293–299. doi: 10.1016/S0168-1656(00)00196-6. [DOI] [PubMed] [Google Scholar]
- Ikemura Toshimichi. Correlation between the Abundance of Escherichia coli Transfer RNAs and the Occurance of the Respective Codons in its Protein Genes: A Proposal for a Synonymous Codon Choice that is Optimal for the E. coli Translational System. J Mol Biol. 1981;151:389–409. doi: 10.1016/0022-2836(81)90003-6. [DOI] [PubMed] [Google Scholar]
- Bulmer M. The selection-mutation-drift theory of synonymous codon usage. Genetics. 1991;129:897–907. doi: 10.1093/genetics/129.3.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp Paul M., Stenico Michele, Peden John F., Lloyd Andrew T. Codon usage: mutational bias, translational selection, or both? Biochem Soc Trans. 1993;21:835–841. doi: 10.1042/bst0210835. [DOI] [PubMed] [Google Scholar]
- Duret L. Evolution of synonymous codon usage in metazoans. Curr Opin Genet Dev. 2002;12:640–649. doi: 10.1016/S0959-437X(02)00353-2. [DOI] [PubMed] [Google Scholar]
- Lobry JR. Asymmetric substitution patterns in the two DNA strands of bacteria. Mol Biol Evol. 1996;13:660–665. doi: 10.1093/oxfordjournals.molbev.a025626. [DOI] [PubMed] [Google Scholar]
- Francino MP, Ochman H. Strand asymmetries in DNA evolution. Trends Genet. 1997;13:240–245. doi: 10.1016/S0168-9525(97)01118-9. [DOI] [PubMed] [Google Scholar]
- McInerney James O. Replicational and transcriptional selection on codon usage in Borrelia burgdorferi. Proc Natl Acad Sci. 1998;95:10698–10703. doi: 10.1073/pnas.95.18.10698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrazek J, Karlin S. Strand compositional asymmetry in bacterial and large viral genomes. Proc Natl Acad Sci U S A. 1998;95:3720–3725. doi: 10.1073/pnas.95.7.3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafay B, Lloyd AT, McLean MJ, Devine KM, Sharp PM, Wolfe KH. Proteome composition and codon usage in spirochaetes: species-specific and DNA strand-specific mutational biases. Nucleic Acids Res. 1999;27:1642–1649. doi: 10.1093/nar/27.7.1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean Michael J., Wolfe Kenneth H., Devine Kevin M. Base Composition Skews, Replication Orientation, and Gene Orientation in 12 Prokaryote Genomes. J Mol Evol. 1998;47:691–696. doi: 10.1007/pl00006428. [DOI] [PubMed] [Google Scholar]
- Guindon S, Perriere G. Intragenomic base content variation is a potential source of biases when searching for horizontally transferred genes. Mol Biol Evol. 2001;18:1838–1840. doi: 10.1093/oxfordjournals.molbev.a003972. [DOI] [PubMed] [Google Scholar]
- Duret L, Mouchiroud D. Expression pattern and, surprisingly, gene length shape codon usage in Caenorhabditis, Drosophila, and Arabidopsis. Proc Natl Acad Sci U S A. 1999;96:4482–4487. doi: 10.1073/pnas.96.8.4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton BR. Selection at the amino acid level can influence synonymous codon usage: implications for the study of codon adaptation in plastid genes. Genetics. 2001;159:347–358. doi: 10.1093/genetics/159.1.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes WS, Borodovsky M. How to interpret an anonymous bacterial genome: machine learning approach to gene identification. Genome Res. 1998;8:1154–1171. doi: 10.1101/gr.8.11.1154. [DOI] [PubMed] [Google Scholar]
- Rodolphe F, Mathe C. Translation conditional models for protein coding sequences. J Comput Biol. 2000;7:249–260. doi: 10.1089/10665270050081504. [DOI] [PubMed] [Google Scholar]
- Kohonen T. Self-Organizing Maps. Berlin, Springer-Verlag; 1995. [Google Scholar]
- Kanaya S, Kinouchi M, Abe T, Kudo Y, Yamada Y, Nishi T, Mori H, Ikemura T. Analysis of codon usage diversity of bacterial genes with a self-organizing map (SOM): characterization of horizontally transferred genes with emphasis on the E. coli O157 genome. Gene. 2001;276:89–99. doi: 10.1016/S0378-1119(01)00673-4. [DOI] [PubMed] [Google Scholar]
- Wang HC, Badger J, Kearney P, Li M. Analysis of codon usage patterns of bacterial genomes using the self-organizing map. Mol Biol Evol. 2001;18:792–800. doi: 10.1093/oxfordjournals.molbev.a003861. [DOI] [PubMed] [Google Scholar]
- Abe T, Kanaya S, Kinouchi M, Ichiba Y, Kozuki T, Ikemura T. Informatics for unveiling hidden genome signatures. Genome Res. 2003;13:693–702. doi: 10.1101/gr.634603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koski Liisa B., Morton Richard A., Golding G. Brian. Codon Bias and Base Composition Are Poor Indicators of Horizontally Transferred Genes. Mol Biol Evol. 2001;18:404–412. doi: 10.1093/oxfordjournals.molbev.a003816. [DOI] [PubMed] [Google Scholar]
- Besemer J, Lomsadze A, Borodovsky M. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001;29:2607–2618. doi: 10.1093/nar/29.12.2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckert G, Warren PV, Gaasterland T, Young WG, Lenox AL, Graham DE, Overbeek R, Snead MA, Keller M, Aujay M, Huber R, Feldman RA, Short JM, Olsen GJ, Swanson RV. The complete genome of the hyperthermophilic bacterium Aquifex aeolicus. Nature. 1998;392:353–358. doi: 10.1038/32831. [DOI] [PubMed] [Google Scholar]
- Kunst F, Ogasawara N, Moszer I, Albertini AM, Alloni G, Azevedo V, Bertero MG, Bessieres P, Bolotin A, Borchert S, Borriss R, Boursier L, Brans A, Braun M, Brignell SC, Bron S, Brouillet S, Bruschi CV, Caldwell B, Capuano V, Carter NM, Choi SK, Codani JJ, Connerton IF, Danchin A. The complete genome sequence of the gram-positive bacterium Bacillus subtilis. Nature. 1997;390:249–256. doi: 10.1038/36786. [DOI] [PubMed] [Google Scholar]
- Shigenobu S, Watanabe H, Hattori M, Sakaki Y, Ishikawa H. Genome sequence of the endocellular bacterial symbiont of aphids Buchnera sp. APS. Nature. 2000;407:81–86. doi: 10.1038/35024074. [DOI] [PubMed] [Google Scholar]
- Fraser CM, Casjens S, Huang WM, Sutton GG, Clayton R, Lathigra R, White O, Ketchum KA, Dodson R, Hickey EK, Gwinn M, Dougherty B, Tomb JF, Fleischmann RD, Richardson D, Peterson J, Kerlavage AR, Quackenbush J, Salzberg S, Hanson M, van Vugt R, Palmer N, Adams MD, Gocayne J, Venter JC. Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature. 1997;390:580–586. doi: 10.1038/37551. [DOI] [PubMed] [Google Scholar]
- Parkhill J, Wren BW, Mungall K, Ketley JM, Churcher C, Basham D, Chillingworth T, Davies RM, Feltwell T, Holroyd S, Jagels K, Karlyshev AV, Moule S, Pallen MJ, Penn CW, Quail MA, Rajandream MA, Rutherford KM, van Vliet AH, Whitehead S, Barrell BG. The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature. 2000;403:665–668. doi: 10.1038/35001088. [DOI] [PubMed] [Google Scholar]
- White O, Eisen JA, Heidelberg JF, Hickey EK, Peterson JD, Dodson RJ, Haft DH, Gwinn ML, Nelson WC, Richardson DL, Moffat KS, Qin H, Jiang L, Pamphile W, Crosby M, Shen M, Vamathevan JJ, Lam P, McDonald L, Utterback T, Zalewski C, Makarova KS, Aravind L, Daly MJ, Fraser CM. Genome sequence of the radioresistant bacterium Deinococcus radiodurans R1. Science. 1999;286:1571–1577. doi: 10.1126/science.286.5444.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blattner FR, Plunkett G., 3rd, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1474. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, Bult CJ, Tomb JF, Dougherty BA, Merrick JM. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 1995;269:496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, Ketchum KA, Klenk HP, Gill S, Dougherty BA, Nelson K, Quackenbush J, Zhou L, Kirkness EF, Peterson S, Loftus B, Richardson D, Dodson R, Khalak HG, Glodek A, McKenney K, Fitzegerald LM, Lee N, Adams MD, Venter JC. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature. 1997;388:539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- Fraser CM, Gocayne JD, White O, Adams MD, Clayton RA, Fleischmann RD, Bult CJ, Kerlavage AR, Sutton G, Kelley JM. The minimal gene complement of Mycoplasma genitalium. Science. 1995;270:397–403. doi: 10.1126/science.270.5235.397. [DOI] [PubMed] [Google Scholar]
- Bult CJ, White O, Olsen GJ, Zhou L, Fleischmann RD, Sutton GG, Blake JA, FitzGerald LM, Clayton RA, Gocayne JD, Kerlavage AR, Dougherty BA, Tomb JF, Adams MD, Reich CI, Overbeek R, Kirkness EF, Weinstock KG, Merrick JM, Glodek A, Scott JL, Geoghagen NS, Venter JC. Complete genome sequence of the methanogenic archaeon, Methanococcus jannaschii. Science. 1996;273:1058–1073. doi: 10.1126/science.273.5278.1058. [DOI] [PubMed] [Google Scholar]
- Salanoubat M, Genin S, Artiguenave F, Gouzy J, Mangenot S, Arlat M, Billault A, Brottier P, Camus JC, Cattolico L, Chandler M, Choisne N, Claudel-Renard C, Cunnac S, Demange N, Gaspin C, Lavie M, Moisan A, Robert C, Saurin W, Schiex T, Siguier P, Thebault P, Whalen M, Wincker P, Levy M, Weissenbach J, Boucher CA. Genome sequence of the plant pathogen Ralstonia solanacearum. Nature. 2002;415:497–502. doi: 10.1038/415497a. [DOI] [PubMed] [Google Scholar]
- Bentley SD, Chater KF, Cerdeno-Tarraga AM, Challis GL, Thomson NR, James KD, Harris DE, Quail MA, Kieser H, Harper D, Bateman A, Brown S, Chandra G, Chen CW, Collins M, Cronin A, Fraser A, Goble A, Hidalgo J, Hornsby T, Howarth S, Huang CH, Kieser T, Larke L, Murphy L, Oliver K, O'Neil S, Rabbinowitsch E, Rajandream MA, Rutherford K, Rutter S, Seeger K, Saunders D, Sharp S, Squares R, Squares S, Taylor K, Warren T, Wietzorrek A, Woodward J, Barrell BG, Parkhill J, Hopwood DA. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2) Nature. 2002;417:141–147. doi: 10.1038/417141a. [DOI] [PubMed] [Google Scholar]
- Kaneko T, Sato S, Kotani H, Tanaka A, Asamizu E, Nakamura Y, Miyajima N, Hirosawa M, Sugiura M, Sasamoto S, Kimura T, Hosouchi T, Matsuno A, Muraki A, Nakazaki N, Naruo K, Okumura S, Shimpo S, Takeuchi C, Wada T, Watanabe A, Yamada M, Yasuda M, Tabata S. Sequence analysis of the genome of the unicellular cyanobacterium Synechocystis sp. strain PCC6803. II. Sequence determination of the entire genome and assignment of potential protein-coding regions. DNA Res. 1996;3:109–136. doi: 10.1093/dnares/3.3.109. [DOI] [PubMed] [Google Scholar]
- Parkhill J, Wren BW, Thomson NR, Titball RW, Holden MT, Prentice MB, Sebaihia M, James KD, Churcher C, Mungall KL, Baker S, Basham D, Bentley SD, Brooks K, Cerdeno-Tarraga AM, Chillingworth T, Cronin A, Davies RM, Davis P, Dougan G, Feltwell T, Hamlin N, Holroyd S, Jagels K, Karlyshev AV, Leather S, Moule S, Oyston PC, Quail M, Rutherford K, Simmonds M, Skelton J, Stevens K, Whitehead S, Barrell BG. Genome sequence of Yersinia pestis, the causative agent of plague. Nature. 2001;413:523–527. doi: 10.1038/35097083. [DOI] [PubMed] [Google Scholar]
- Fickett JW, Tung CS. Assessment of protein coding measures. Nucleic Acids Res. 1992;20:6441–6450. doi: 10.1093/nar/20.24.6441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B. Artemis: sequence visualization and annotation. Bioinformatics. 2000;16:944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]