

Abstract
Neurodegenerative diseases involve the progressive loss of neurons, and a pathological hallmark is the presence of abnormal inclusions containing misfolded proteins. Although the precise molecular mechanisms triggering neurodegeneration remain unclear, endoplasmic reticulum (ER) stress, elevated oxidative and nitrosative stress, and protein misfolding are important features in pathogenesis. Protein disulphide isomerase (PDI) is the prototype of a family of molecular chaperones and foldases upregulated during ER stress that are increasingly implicated in neurodegenerative diseases. PDI catalyzes the rearrangement and formation of disulphide bonds, thus facilitating protein folding, and in neurodegeneration may act to ameliorate the burden of protein misfolding. However, an aberrant posttranslational modification of PDI, S-nitrosylation, inhibits its protective function in these conditions. S-nitrosylation is a redox-mediated modification that regulates protein function by covalent addition of nitric oxide- (NO-) containing groups to cysteine residues. Here, we discuss the evidence for abnormal S-nitrosylation of PDI (SNO-PDI) in neurodegeneration and how this may be linked to another aberrant modification of PDI, S-glutathionylation. Understanding the role of aberrant S-nitrosylation/S-glutathionylation of PDI in the pathogenesis of neurodegenerative diseases may provide insights into novel therapeutic interventions in the future.
1. Introduction
Neurodegenerative diseases share several common pathological characteristics, including the aberrant aggregation of misfolded proteins, leading to the formation of abnormal protein inclusions [1]. These diseases are also frequently classified as protein conformational disorders in which protein aggregation occurs due to the exposure of hydrophobic regions [2]. The most common neurodegenerative diseases include Alzheimer's disease (AD), Parkinson's disease (PD), amyotrophic lateral sclerosis (ALS), Creutzfeldt-Jakob disease (CJD), and Huntington's disease (HD). These diseases differ according to the specific group of neurons targeted and the type of misfolded proteins that aggregate. In AD, the accumulation of aggregated proteins occurs in cortical regions and involves both β-amyloid (βA), which forms extracellular amyloid plaques, and tau, which is hyperphosphorylated and forms intracellular neurofibrillary tangles (NFT) [3, 4]. PD involves the formation of Lewy bodies (LB) containing misfolded α-synuclein [5], and in HD aggregated Huntington protein with expanded polyglutamine repeats forms inclusions in the nucleus [6]. Similarly, in ALS, cytoplasmic inclusions contain copper/zinc (CuZn) superoxide dismutase 1 (SOD1) [7–9], TAR DNA binding protein 43 (TDP-43) [10–13], or fused in sarcoma/translated in liposarcoma (FUS/TLS) [14]. Recently, a hexanucleotide repeat expansion in an intronic region of the chromosome 9 open reading frame 72 (C9orf72) gene, encoding a gene of unknown function, was linked to the greatest proportion of familial ALS cases [15, 16]. For AD, PD, and ALS, 90–95% of cases arise sporadically, while the remainder are familial in nature. Genetic mutations in Amyloid Precursor Protein (APP), leads to increased accumulation of A-β and fibril formation [17–20], and Presenilin 1, 2 (PS 1, 2), which regulates APP processing via gamma secretase [21–23], causes rare familial cases of AD [24]. Similarly, some forms of autosomal dominant familial PD is caused by α-synuclein mutations [25] leading to the aggregation of α-synuclein into insoluble fibrils, which are the primary components of LB [26], while mutations in PINK1, Parkin, and DJ-1 cause autosomal recessive PD cases [27]. However, in contrast to these conditions, HD is early onset and entirely genetic in nature.
The causal factors underlying the pathogenesis of sporadic neurodegenerative diseases remain poorly understood. However, due to the typical late onset of these disorders, neurodegeneration can be conceptualized as pathology that arises during the normal aging process, involving increases in oxidative stress and the production of free radicals which damage cells by decreasing antioxidant defenses. In AD, increased free radical accumulation and elevated levels of oxidative and nitrosative stress are associated with alterations in A-β metabolism [28, 29]. Meanwhile, in PD, nitrosative stress is associated with impairment of the mitochondrial respiratory chain, leading to energy deficiency and cell death [30]. In addition, oxidative and nitrosative stress are associated with endoplasmic reticulum (ER) stress, through the accumulation of misfolded proteins in the ER, and upregulation of molecular chaperones in the protein disulphide isomerase (PDI) family [31]. PDI possesses both general protein chaperone and disulphide interchange activity, thus facilitating the formation of native disulphide bonds in proteins. It also facilitates the degradation of these proteins via ER-associated degradation (ERAD), whereby irreparably misfolded proteins are targeted for retrotranslocation to the cytoplasm, where they undergo polyubiquitination and subsequent degradation by the proteasome [32–35]. There is now sufficient evidence that in conditions of elevated nitrosative stress, PDI undergoes an aberrant posttranslational modification known as S-nitrosylation, which inhibits its enzymatic activity [36]. Hence, in late onset neurodegenerative disease, there is a decrease in cellular defences and a corresponding increase in oxidative and nitrosative damage to lipids, proteins, DNA, and RNA [37, 38].
In this review, we will begin by examining the role of nitrosative stress, redox potential, and S-nitrosylation/S-glutathionylation of proteins linked to neurodegeneration. The structure and function of PDI family members will be discussed, and the importance of PDI in neurodegenerative disease will be highlighted. We will examine the evidence that PDI is aberrantly S-nitrosylated and discuss the functional significance of this modification in neurodegeneration. Finally, we speculate that PDI may also be S-glutathionylated in neurodegenerative disease.
2. Nitrosative Stress
Reactive nitrogen and oxygen species (RNS and ROS), primarily superoxide anion (O2 −), hydrogen peroxide (H2O2), or nitric oxide (NO), are highly reactive molecules that normally function at low levels as mediators of intracellular signalling processes in mammalian cells [36, 39]. However, RNS and ROS can accumulate in cells under pathological conditions, triggering nitrosative or oxidative stress. This leads to numerous detrimental effects on cellular function including posttranslational modifications of proteins, lipid peroxidation, DNA, damage, and dysregulation of redox signalling [28, 37, 38, 40]. Nitrosative or oxidative stress results when there is an imbalance between the production of RNS/ROS and cellular antioxidant defence mechanisms such ascorbic acid, glutathione (GSH), or enzymes including superoxide dismutases, catalases, and glutathione peroxidases. GSH is a particularly important antioxidant as it is the most abundant cellular thiol-containing molecule; the ratio of reduced GSH to its oxidized form (GSSG) makes a major contribution to cellular redox potential and homeostasis [28, 29, 41]. However, the thiol/disulfide systems, which include GSH/GSSG, and plasma cysteine/cystine (Cys/CySS) pools are not necessarily in equilibrium and may respond differentially to specific stressors [42]. Nitrosative or oxidative stress may be induced by familial mutations, exogenous toxins (xenobiotics, pesticides), or via normal aging processes such as alterations in mitochondrial respiration [31, 43]. Neurons are particularly vulnerable to the effects of RNS/ROS due to a relative deficiency in antioxidant enzymes glutathione peroxidase (GPx) and catalase (Cat), compared to other cell types, and their higher metabolic demands which generate RNS/ROS from mitochondrial metabolism [38, 39, 43, 44].
RNS are derived primarily from O2 − and NO, a small, diffusible inter- and intracellular messenger that normally mediates many intracellular signalling pathways [29, 31, 45, 46]. NO is generated by NO synthases (NOS) that use oxygen (O2) and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase to convert L-arginine to L-citrulline [47]. NOS is constitutively expressed in several isoforms in the central nervous system (CNS): endothelial NOS (eNOS), inducible NOS (iNOS), neuronal NOS (nNOS), and an isoform expressed in the inner mitochondrial membrane (mtNOS) [48–50]. The covalent addition of NO to a cysteine thiol or thiolate anion on specific proteins, to form an S-nitrosothiol (SNO) group, is a process termed “S-nitrosylation” [36, 51–56].
3. S-Nitrosylation
In recent years, S-nitrosylation has been increasingly implicated in many physiological and pathological conditions [36]. Under normal conditions, S-nitrosylation is a reversible posttranslational modification analogous to acetylation and phosphorylation that regulates protein activity [55, 57]. The SNO-group can be removed in these situations by denitrosylation enzymes, primarily S-glutathione reductase (GSNOR; alcohol dehydrogenase III) in conjunction with GSH and NADH as an electron donor [58, 59]. However, reduced oxidoreductase thioredoxin (TRX) [60, 61] can oxidize S-nitrosoglutathione (GSNO) to release GSH and NO [62, 63]. Recombinant human PDI can denitrosylate GSNO [64] and in vitro SOD1 can modify the stability of S-nitrosothiols by enhancing the decomposition of GSNO, resulting in production of NO [65], possibly by its reduced metal ions [66].
S-nitrosylation is both a reversible and irreversible process [67]. Under pathological conditions, S-nitrosylation of specific proteins is an abnormal, irreversible process and is linked to protein misfolding, ER stress, mitochondrial dysfunction, synaptic degeneration, and cell death [36]. A well-recognized mechanism for NO production in neurodegenerative diseases is activation of N-methyl-D-Aspartate receptors (NMDAr) [68, 69]. Activation of NMDArs generates ROS and results in calcium (Ca2+) influx into the cell [31, 70–72], which in turn activates nNOS to produce NO [50]. S-nitrosylation may also lead to NO-independent oxidation of proteins via ROS, producing reversible modifications in the form of intramolecular/mixed disulphide bonds. One of the proposed pathways for the further oxidation of cysteines is through the hydrolysis of sulfenic acid (SOH), which then may be susceptible to irreversible oxidation from accumulating ROS leading to stable sulfinic (–SO2H) or sulfonic (–SO3H) acid formation [73–75]. However, –SO2H can be reduced back to the free thiol group if the enzyme sulfiredoxin is induced and this can occur in neurons due to activation of NMDAr by increased synaptic activity [76]. In addition, S-nitrosylation can reversibly influence further posttranslation modifications of cysteine residues. When there are two proximal cysteine residues, S-nitrosylation of one of these can facilitate disulphide bond formation [77–79]. Under conditions of excessive nitrosative stress, however, S-nitrosylation inhibits the formation of disulphide bonds [67, 75]. Another pathological mechanism linked to S-nitrosylation has also been implicated in ALS. Cells expressing familial ALS mutants, SODA4V and SODG37R, have increased denitrosylation activity of GSNO in comparison to wild type (WT) SOD1 [80]. This deficiency in S-nitrosylation is especially elevated in mitochondria of mutant SOD1 cells [81].
Whilst most proteins contain multiple cysteine residues, the features underlying the specificity for S-nitrosylation are not fully defined, but appear to rely on tertiary rather than primary structure. Previous studies have suggested that the formation of S-nitrosylated proteins (SNO proteins) requires a cysteine flanked by a proximal acid-base motif, hydrophobic content, low pKa, and high exposure of sulfur atoms [67, 82]. However, a recent bioinformatics study predicted that the known SNO-Cys sites in proteins are more heterogenous than this, although the presence of a charged residue in close proximity to NO-Cys and another oppositely charged residue within a larger region was a common feature [82]. The stability of the resulting SNO-group depends upon the local environment of the cysteine residues, but studies of the dissociation energies of the S–N bond suggest that there is a wide variation, with this bond remaining stable theoretically from seconds to years [83, 84].
Up to one thousand SNO proteins have now been identified [85] including many proteins linked to neurodegenerative diseases [36, 77, 86–89]. For instance, S-nitrosylation of dynamin-related protein (Drp1) (SNO-Drp), found in post-mortem brains of AD cases, is associated with β-A for-mation and subsequent activation of mitochondrial fission [77, 87]. In sporadic and familial PD, S-nitrosylated Parkin (SNO-Parkin) has reduced E3 ligase function, leading to proteasomal dysfunction [90]. Similarly, proteins involved in apoptosis (XIAP/Caspase 3, GAPDH-Siah), antioxidant activity (Prx2), the phosphatase pathway (PTEN), neuroinflammation (COX2), and autophagy (JNK1 and IKKβ) are also S-nitrosylated (for comprehensive review see [36]). Furthermore, SNO-proteins may alter cellular redox homeostasis through an interaction with GSH and therefore may influence other post-translational modifications, such as S-glutathionylation [36, 41]. Some proteins, such as NMDAr, are S-nitrosylated under both normal and pathological conditions [36]. S-nitrosylation/denitrosylation of NMDAr is important in physiological cellular signalling processes [52, 53, 91], but overactivation is associated with an increased production of SNO-proteins and neurodegeneration [31]. However, it should be noted that S-nitrosylation of NMDAr at Cys399 is protective by deactivation of the receptor, thus preventing glutamate excitotoxicity [53, 67, 78, 91].
4. S-Glutathionylation
S-glutathionylation is another posttranslational modification that has been implicated in the regulation of diverse proteins involved in energy metabolism, signalling pathways, Ca2+ homeostasis, antioxidant enzymatic activity, and protein folding [92] (for a comprehensive review see [41]). S-glutathionylation is induced by RNS/ROS and involves the formation of a disulfide between GSH and a cysteine residue [41]. As reduced GSH is the most abundant cellular thiol, it plays an important role in S-glutathionylation [41], although protein thiols represent a similar redox pool, and therefore may also be critical in providing antioxidant protection against oxidative stress [93]. S-nitrosylated cysteines can be converted to S-glutathionylated cysteines, supporting the premise that products of nitrosative stress induce S-glutathionylation [41]. However, the exact identity of the metabolites that act as proximal donors in this reaction remain to be elucidated [41] and it is unclear whether SNO proteins are intermediates for S-glutathionylation in vivo. Under oxidizing conditions, S-glutathionylation is reversible via the release of GSH from cysteine residues by thiol-disulphide oxidoreductase enzymes (TDOR). TDOR enzymes include TRX, which reduces intra- and intermolecular disulphide bonds, and glutaredoxin (GRX) which reduces protein-GSH bonds [94–96]. TRX and GRX catalyze the reduction of disulphide bonds and reactivate proteins that have undergone oxidation from sulfhydryl groups [95, 96]. Alterations in the ratio of GSH/GSSG and conditions that promote RNS/ROS production result in cysteine modifications that are precursors to the formation of mixed disulphides with GSH [95, 97, 98]. However, the role of S-glutathionylation during nitrosative and oxidative stress has not been completely defined. Glutathionylation at physiological levels may therefore represent a mechanism whereby cysteine residues faced with oxidation are protected from irreversible damage. The reduction of GSH-protein disulphide by GRX is essential in this process as it maintains the cellular availability of GSH and acts in concert with TRX to maintain the cellular thiol status [95].
S-glutathionylation has been implicated in neurodegeneration [95, 99–101]. The ratio of GSH/GSSG decreases in brains of aged rats [102], and accumulation of S-glutathionylated p53 in the inferior parietal lobule of AD patients has also been reported [101]. In PD models, administration of the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), which causes damage to dopaminergic neurons, caused an early decrease in the levels of GSH, inhibition of mitochondrial complex 1, and dopaminergic cell loss [103]. Furthermore, increases in GSH, GRX, and GSH reductase were detected in the brains of transgenic HD mice models (R6) [104, 105]. S-glutathionylation of SOD1 isolated from human erythrocytes at Cys111 promoted SOD1 monomer formation and subsequent aggregation [106]. Hence, alterations in S-glutathionylation and redox potential are important mediators of protein misfolding, and aberrant disulphide bond formation is implicated in this process.
5. ER Stress and Neurodegeneration
The major cellular location for protein disulphide bond formation is the ER. The highly oxidizing environment of this compartment (GSH : GSSG ratio~3 : 1) is necessary for formation of disulphide bonds and is in stark contrast to the reducing environment of the cytosol (GSH : GSSG ratio~100 : 1) [41, 92, 107]. The ER environment, therefore, is highly sensitive to changes in nitrosative and oxidative stress [31, 36].
ER stress is increasingly implicated as a pathogenic mechanism in neurodegenerative diseases [108–114]. ER stress occurs when misfolded proteins accumulate within the ER lumen, triggering the unfolded protein response (UPR) [115]. The UPR is a set of signalling pathways that initially aim to restore homeostasis by: (1) reducing protein synthesis and translocation, attenuating further accumulation of unfolded proteins in the ER, (2) activation of ER-resident chaperones including PDI to increase the protein folding capacity of the ER, and (3) induction of ERAD. The UPR activates three ER stress sensor proteins: inositol requiring kinase 1 (IRE1 α/β), double-stranded RNA-activated protein kinase- (PKR-) like ER kinase (PERK), and activating transcription factor 6 (ATF6), which transduce signals to the nucleus and cytosol [115, 116]. However, if homeostasis cannot be restored, apoptosis is triggered [115, 117]. Prolonged UPR activation linked to RNS or ROS triggers apoptosis through C/EBP homologous protein (CHOP), caspase 4, c-Jun, and c-Jun N-terminal kinase (JNK) [41, 118, 119].
PDI family members fulfil crucial roles in regulating ER stress by maintaining native protein conformation and facilitating protein degradation [120]. The remainder of this review will focus on the PDI family and the effect of S-nitrosylation/S-glutathionylation on PDI and its functional role in neurodegeneration.
6. PDI Family Members
There are currently 21 identified members of the PDI family [32, 120–125], which share several features in common; at least one domain with a TRX fold, the presence of a signal sequence, and ER localization due to the presence of an KDEL or other ER retention signal [32, 120, 126]. Whilst PDI family members contain a TRX domain, they essentially differ from TRX due to their higher redox potentials, substrate binding domains, and their ability to display both isomerase and chaperone activities, which renders them more efficient than TRX at forming/reforming disulphide bonds [127, 128]. Whilst PDI family members primarily mediate protein folding, other functions have also been ascribed to them, including regulation of Ca2+ homeostasis [129, 130] and ERAD, thus ameliorating protein misfolding within the ER [33–35].
PDI disulphide-isomerase activity catalyzes the rearrangement of nonnative (incorrectly formed) disulphide bonds on nascent proteins, which would otherwise result in the formation of a misfolded structure. This activity is mediated through catalysis of thiol disulphide exchange (isomerization), whereby non-native disulphide bonds are initially reduced, and then oxidized to form the native structure [131–133]. Disulphide formation and stability are facilitated by the redox conditions of the ER [31]. Thus, active-site cysteines shift between two redox states: oxidation and the formation of disulphide bonds and reduction leading to the formation of thiols with free sulfhydryls [134]. In addition, PDI also has general chaperone activity which is independent of its disulphide interchange function [135–137]. This chaperone activity does not require its catalytic domains or active sites [138, 139].
PDI (PDIA1), the prototype of the PDI family, is a 55 kDa protein with two catalytically inactive TRX domains (b and b′), inserted between two TRX-like catalytic domains (a and a′), and an acidic C terminal domain with an ER-retention motif (KDEL). PDIA1 contains a CXXC catalytically active motif (Figure 1). All domains of PDI are required for efficient catalysis of disulphide bond formation and rearrangement [32, 120, 140]. The structure of yeast PDI has revealed that the binding of PDI to misfolded protein substrates is facilitated by two of the active cysteines positioned on opposite sides of the molecule [140, 141]. The noncatalytic b′ domain is situated on the base and is the major site for binding of substrates [141], although other domains also contribute to this process. The b-b combination of noncatalytic domains is present only in PDIA1, PDIA2 (PDIp), PDIA3 (ERp57), and PDIA4 (ERp72) family members [142–146]. PDIA1 has the broadest substrate specificity of the PDI family members examined to date [144].
Figure 1.
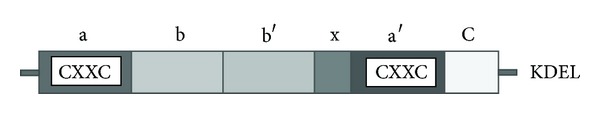
Domains of PDIA1. TRX-like domains representing catalytic active domains a a′. The b domain and b′ are catalytically inactive. The linker region is responsible for binding to the substrate. The C terminal is followed by an ER retrieval signal KDEL.
PDIA2 is primarily expressed in pancreatic cells and dopaminergic neurons [146–148]. The domain structure of PDIA2 is similar to PDIA1, with a CXXC motif in the homologous a, a′ domains, intervening b, b′ domains, a x-linker region, and an N-terminal ER sequence [149]. PDIA2 also contains a KEEL motif at the C-terminus, an ER retention signal analogous to KDEL [150]. Similar to PDI, PDIA2 can interact with protein substrates with and without cysteine residues [148, 151], suggesting that PDIA2 may act as a chaperone independent of catalyzing disulphide bond formation [147]. However, although its domain organization is similar to PDI, its physiological role remains unclear.
PDIA3 is the second most abundant soluble protein after PDIA1 found in the ER [120]. It contains a protein sequence homologous to PDIA1, with similarities in domain architecture but differences in substrate binding [152]. Whilst PDIA3 is an oxidoreductase with thiol-dependent reductase activity [153], it is different to the other PDI family members in that it acts primarily on glycosylated proteins by associating noncovalently with the lectin chaperones calnexin and calreticulin [154]. The catalytic properties differ from PDIA1 and the redox potential of PDIA3 is also lower than PDIA1 [155, 156]. PDIA3, like PDIA1, has two CXXC motifs at the conserved active sites and four similar TRX-like domains (a-b-b′-a′) [153, 156]. The C-terminus of PDIA3 has an ER retention signal with a sequence similar to that of PDIA1 [153] and a nuclear localisation signal near the C terminal with a high affinity for importin [128, 157, 158]. In addition, PDIA3 and PDIA1 differ in terms of substrate binding specificity due to differences in homology in their b′ domains. The binding domain of PDIA3 is enriched in lysine and arginine residues, so that PDIA3 binds to proteins containing negatively charged P domains, such as those found in calreticulin [142, 158]. The oxidative and catalytic property of PDIA3 and PDIA1 both rely on a charged glutamic acid and a pair of lysine residues found behind the active CXXC site [120].
Some PDI family members possess more than two CXXC active sites. PDIr, Erp46, and PDIA4, also known as Ca2+ binding protein (CaBP2) [159], all have three active sites [121, 160–163], and ERdJ5 contains four active sites [164]. PDIA4 is similar to PDIA1 in its catalytic domains but has lower sequence similarity in the other domains. It can also act as a substitute for PDIA3 in folding specific proteins, but it does not bind to glycoproteins [165]. Other PDI gene family members include DNAJC10, ERP27, ERP29 (ERP28), ERP44, PDIA5, PDIA6, PDILT, and TXNDC5 (for comprehensive review please refer to [125]). However, this review will focus on PDIA1, PDIA2, PDIA3, and PDIA4 as these are the only PDI family members to date that are reported to undergo S-nitrosylation.
7. The Presence of PDI in Non-ER Compartments
Whilst PDI family members are primarily considered to be ER-localized, they are also present in other cellular locations, including the nucleus, cytoplasm, cell surface, and extracellular space [128]. Few proteins linked to neurodegeneration are present in the ER, so it is possible that PDI plays an important role in these locations. In the ER, PDI must be maintained in a balance between its oxidized and reduced states to facilitate disulphide bond formation [166, 167]. However, in non-ER compartments, PDI family members have an increased ability to catalyze the reduction of disulphide bonds compared to the ER [168]. The mechanism of transit of PDI from the ER remains unknown, and because of the presence of the KDEL retention signal, observations of non-ER localized PDI have previously been questioned [128]. However, other primarily ER-localized proteins that possess a KDEL motif, such as calreticulin and binding immunoglobulin protein (BiP), are also secreted and located in the nucleus, cytoplasm and cell surface [169–176].
PDI in the cytosol has been postulated to act as a cofactor with insulin-degrading enzyme (IDE) during insulin metabolism, while acting in concert with reduced GSH to catalyze disulphide bond cleavage [177]. There is also evidence that PDI redistributes away from its ER location into the cytoplasm in pathological conditions. ER stress causes the redistribution of PDIA1 and PDIA3 from the ER to the cytosol [178], consistent with the notion that PDI in locations other than the ER is neuroprotective. Furthermore, one study demonstrated that overexpression of reticulon-4A (NOGO A) triggered the redistribution of PDI from the ER into vesicular-type structures localized in an undefined cellular compartment, both in vitro and in vivo, which occurred in the absence of the UPR [179]. Deletion of NOGO A, B from ALS mouse models, involving transgenic overexpression of mutant SOD1G93A, led to earlier onset and increased disease progression, indicating that reticulons mediate PDI function and redistribution in neurodegeneration [179]. A more recent study, using human neuroblastoma SH-SY5Y cells overexpressing reticulon protein 1C (RTN-1C), demonstrated that redistribution of PDI away from the ER into vesicular structures led to a consequent increase in the enzymatic activity of PDI and a decrease in S-nitrosylation [180].
PDI has also been detected at the cell membrane, where a role in NO signalling has been described. S-nitrosylated extracellular proteins transfer NO to the cytosol via the reducing activity of cell surface PDI [181, 182]. During this process, cell-surface PDI also undergoes thiol modification [183]. Furthermore, PDIA3 interacts with prion proteins (PrP) at the cell surface and may play a key role in PrP accumulation [184]. In addition, PDIA1 and PDIA3 have been detected in the nucleus, where they are posited to anchor DNA loops to the nuclear matrix [128, 185, 186]. PDI-like activity has also been detected in mitochondria, although PDIA1 has not been identified in this compartment [187], and it is possible that Mia 40 contributes to this activity [188, 189].
PDIA1 and PDIA3 have also been detected at mitochondrial-associated ER membranes, where, remarkably, they may regulate apoptosis signalling [190]. The expression of polyglutamine expanded Huntington protein led to PDIA1 and PDIA3 accumulation in this location, where it triggered mitochondrial outer membrane permeabilization through activation of proapoptotic BCL-2 family members, triggering apoptosis [190]. Hence, whilst PDI functions protectively through its chaperone and isomerase activities [191], it can also trigger pro-apoptotic mechanisms [190]. While this process has not yet been fully defined, the novel proapoptotic function of PDI may represent a new link between protein misfolding and cell death.
8. Role of PDI in Neurodegeneration
There is now substantial evidence linking PDI family members to protein misfolding in neurodegeneration. PDIA1 is upregulated in AD brain tissues [192], PDIA3 forms a complex with calreticulin and A-β peptides in patients' CSF [193], and NFTs are immunopositive for PDI [194, 195]. Similarly, in cellular models of PD, treatment of dopaminergic neurons with 6-hydroxydopamine (6-OHDA) induces ER stress, oxidation, and aggregation of PDIA3 [196]. PDIA2 is upregulated in SH-SY5Y human neuroblastoma treated with either 1-methy-4-phenyl-pyridinium (MPP+) or proteasome inhibitor lactacystin while immunoreactivity to PDIA2 has also been detected in LB in postmortem brains of PD patients [146]. Furthermore, the a′ domain of PDIA1 inhibits α-synuclein fibril formation [197], and coexpression of PDIA1 decreased synphilin-1 positive LB formation in the cytoplasm [75]. PDIA1 was upregulated in the brains of Creutzfeldt-Jakob disease (CJD) patients [198], while PDIA1 and PDIA3 were upregulated in prion disease in scrapie infected rodents [199]. Pharmacological inhibition of PDIA3 using bacitracin increased the accumulation of aggregated PrP, also suggesting that PDI is not functional in prion disease [184]. Furthermore, upregulation of PDIA1 and PDIA3 was associated with mitochondrial dysfunction in cells expressing misfolded PrP [199]. The detection of mitochondrial apoptosis triggered by PDIA1 and PDIA3 in HD models [190] also highlights the intrinsic link between PDI upregulation and mitochondrial dysregulation in neurodegeneration [199].
There is also increasing evidence for an important role for PDI in ALS. PDIA1 is upregulated and is a component of TDP-43 and FUS-positive cytoplasmic inclusions in motor neurons of sporadic ALS patients [200, 201]. Additionally, PDIA1 is a risk factor for the development of ALS [202]. PDIA1 also colocalizes with mutant SOD1-positive inclusions in cell culture and transgenic SOD1 rodents [89, 203, 204]. Overexpression of PDIA1 decreases the formation of mutant SOD1 inclusions whereas knockdown of PDI using siRNA increases the proportion of inclusions [89]. Furthermore, a synthetic mimic of the PDIA1 active site; (±)-trans-1,2-bis (mercaptoacetamido)cyclohexane (BMC), is protective against mutant SOD1 aggregation in cell culture [89]. SOD1 contains four cysteine residues, and non-native disulphide bonds between Cys6 and Cys111 have been implicated in mutant SOD1 aggregation [205]. Conversely, upregulation of PDIA1 in microglia in SOD1G93A mice was associated with increased levels of NADPH oxidase (NOX), superoxide, and tumour necrosis factor-α. Pharmacological inhibition and knockdown of PDIA1 using siRNA decreased superoxide and NOX activation in microglia, therefore providing further evidence for a potential neurotoxic role of PDIA1 [206].
PDI is therefore upregulated during UPR activation and is part of a cellular protective mechanism that prevents protein misfolding and aggregation in neurodegeneration. PDI family members are especially vulnerable to oxidative and nitrosative-linked posttranslational modifications due to the highly oxidizing environment of the ER and the presence of cysteine residues in the PDI catalytic regions. Irreversible S-nitrosylation of PDI (SNO-PDI) may therefore ameliorate its protective function in neurodegenerative disorders and thus contribute to disease.
9. SNO-PDI and Neurodegeneration
PDI is S-nitrosylated by endogenous nNOS in both its TRX domains leading to a significant reduction in isomerase and chaperone activity [75]. Also, induction of SNO-PDI using NO donor S-nitrosocysteine (SNOC) completely abrogates the catalytic activity of PDI, resulting in neuronal cell death [207].
SNO-PDI has been detected in postmortem brain tissue of sporadic PD and AD patients [75] and lumbar spinal cord tissues of ALS patients and SOD1G93A mice [89]. This was linked to excessive production of NO or exposure to exogenous agents such as rotenone [75]. PDI was shown to be modified in the cysteine thiol groups in the C-terminal CXXC motif, leading to the accumulation of polyubiquitinated proteins and activation of the UPR [75]. SNO-PDI formation is associated with synphilin misfolding in PD [31] and mitochondrial mediated apoptosis in prion infection [199]. SNO-PDI is also found in cultured astrocytes after ischemia/reperfusion-induced iNOS production, leading to increases in ubiquitinated aggregates that colocalize with SOD1 [7].
One potential physiological mechanism of SNO-PDI production involves pathological hyperactivation of NMDAr [31] and inhibition of mitochondria leading to the generation of ROS, nNOS, and NO [31, 70, 71]. Exposure of cortical neurons to NMDA produces SNO-PDI, leading to an increase in polyubiquitinated proteins and apoptosis after 24 hrs of treatment. Furthermore, overexpression of WT PDI leads to a decrease in polyubiquitination and apoptosis, suggesting that PDI may provide protection against excitotoxicity from excessive stimulation of NMDA receptors [75]. Additionally, treatment with Rotenone, a mitochondrial complex inhibitor, produces elevated levels of SNO-PDI [75], suggesting that mitochondria are another source of NO or cytosolic nNOS [31]. NO disrupts Ca2+ homeostasis, potentially via S-nitrosylation of the ER Ca2+ channel ryanodine receptor, and induction of ER stress [57, 208]. ER-resident proteins are particularly vulnerable to S-nitrosylation and as such a positive feedback mechanism would create a scenario whereby excessive RNS/ROS increasingly deactivates protective ER-resident chaperones such as PDI, prolonging UPR activation, leading to increases in ROS/RNS generation eventually resulting in cell death [31]. ER dysfunction due to excessive oxidative/nitrosative stress may, thus, lead to the S-nitrosylation of PDI in neurodegenerative disease [31]. However, PDI family members PDIA1, PDIA3, and PDIA4 can be S-nitrosylated independently of UPR induction [209]. Alternatively, PDI located at the cell surface may also promote production of SNO proteins. It has been previously suggested that extracellular SNO proteins may transfer NO to the cytoplasm via the reducing activity of cell surface PDI [181, 182]. According to this theory, reduced NO may readily penetrate the plasma membrane, leading to SNO production [128] (Figure 2). Hence, the formation of SNO-PDI results in the abrogation of the normally protective isomerase/chaperone activity of PDI, which may contribute to protein misfolding and production of SNO proteins. This suggests that SNO-PDI may be a common pathological mechanism contributing to neurodegenerative diseases.
Figure 2.
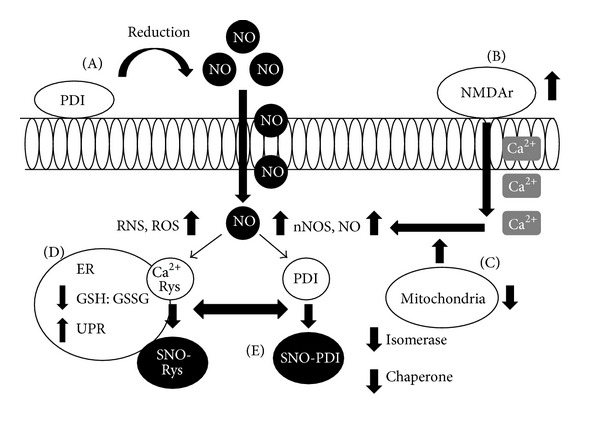
Cell surface PDI, NO, and SNO-PDI. (A) Cell surface PDI reduces NO from extracellular SNO proteins (SNO-P) and in the process undergoes thiol modification. (B) Hyperactivation of the NMDAr leads to an intracellular influx of Ca2+ ions (NMDAr may also undergo reversible S-nitrosylation to ameliorate excessive activity). (C) Inhibition of mitochondria contributes to an increase in intracellular NO which is potentially oxidized by O2 leading to an increase in NO, nNOS, ROS, and RNS. (D) Increases in RNS/ROS alters the ER redox environment, and NO S-nitrosylates Ca2+ ryanodine (Ryn) receptor leading to a disruption in Ca2+ homeostasis. (E) ER-resident proteins such as PDI are vulnerable to S-nitrosylation, deactivating its isomerase and chaperone activity, leading to accumulation of misfolded proteins, ER stress, and UPR induction.
10. S-Glutathionylation and PDI
A link between S-glutathionylated PDI and neurodegenerative disease has not yet been established [210]. However, cysteine residues in the a and a′ domains of PDI make it a potential target for S-glutathionylation [211].
PDI has been shown to be S-glutathionylated at two of its four active cysteine sites (Cys53, Cys56 or Cys397, Cys400) [92]. S-glutathionylation was induced in these cells by treatment with anticancer agent O2–[2,4-dinitro-5-(N-methyl-N-4-carboxyphenylamino) phenyl]1–(N,N dimethylamino)diazen-1-ium-1,2-diolate (PABA/NO), which led to a dose-dependent increase in intracellular NO [210], triggering UPR induction and cell death [92]. S-glutathionylation of PDI has been demonstrated in human leukemia (HL60) and ovarian cancer cells (SKOV3) inhibiting its isomerase function [205]. In addition, S-glutathionylation of PDI abrogates its chaperone activity and prevents binding to oestrogen receptor alpha (ERα) [212]. The PDI-ERα interaction may protect ERα from oxidation and ensure its native protein conformation [213]. However, aberrant S-glutathionylation of PDI leads to destabilisation of the receptor and dysregulatation of ERα signaling. This may subsequently mediate cell death via activation of the UPR and reduced ERα stability [212]. However, although PABA/NO treatment increased levels of intracellular NO, it did not lead to S-nitrosylation of PDI [210]. There are two pools of S-nitrosylated proteins, GSH stable and GSH labile proteins, with the latter pool being readily subject to conversion to S-glutathionylated products [41]. Therefore, the lack of SNO proteins after PABA/NO treatment may be due to conversion of SNO proteins to S-glutathionylated proteins [210] (Figure 3). This notion therefore provides a link between S-nitrosylation and S-glutathionylation, although the exact relationship between these modifications remains unknown [41].
Figure 3.
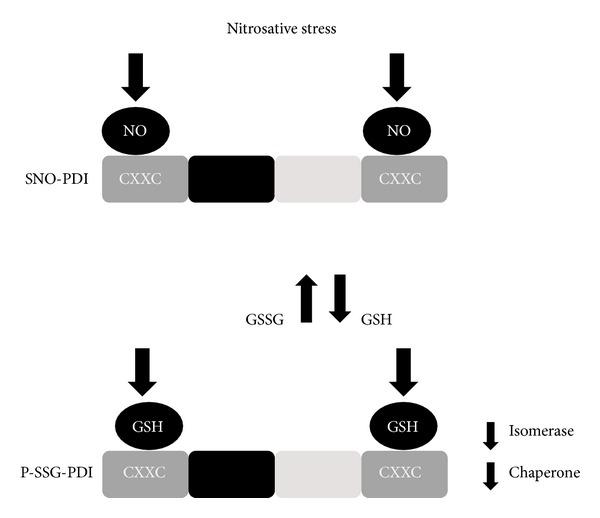
S-glutathionylation of PDI. Nitrosative stress from an exogenous agent (PABA/NO) increases intracellular NO and leads to the production of SNO-PDI. However, this may result in a decrease in GSSG/GSH ratio and increases in the free cellular pool of GSH. GSH then binds to the catalytic (a, a′) domains of PDI, resulting in S-glutathionylation (P-SSG) of its cysteine residues and attenuation of its protective isomerase and chaperone activity.
S-glutathionylation of PDI was proposed to be an upstream signalling event triggering misfolded protein accumulation and UPR induction [210, 211]. As PDI may regulate redox potential at the cell surface [182, 214], it therefore may facilitate cell adhesion [215], antigen processing [216], and glioma cell invasion [217]. S-glutathionylation of cell surface proteins alters extracellular and intracellular redox homeostasis [210]. Hence, irreversible S-glutathionylation/S-nitrosylation of cell surface PDI could alter redox potential, leading to amelioration of the protective chaperone/isomerase functions of PDI. This mechanism may therefore contribute to the excessive production of SNO and S-glutathionylated proteins observed in neurodegenerative disease.
11. Conclusion
PDIs are a large family of chaperones and foldases which have complex yet still inadequately described functions with emerging roles in neurodegenerative diseases. Whilst S-nitrosylation plays a normal physiological role in signalling pathways, aberrant modification is triggered during conditions of elevated nitrosative and oxidative stress. Accumulating evidence suggests that SNO-PDI plays a role in the pathogenesis of neurodegenerative diseases such as AD, PD, and ALS, and this may exacerbate neurodegeneration via a number of mechanisms. However, most of the available reports are correlative in nature and therefore more direct approaches examining the contribution of S-nitrosylation of PDI family members to neurodegeneration are warranted. S-nitrosylation is also linked to another previously described modification of PDI, S-glutathionylation, although the S-glutathionylation of PDI and its role in neurodegenerative diseases have not been elucidated. Whilst PDI family members are conventionally regarded as being ER localized, they are also present and catalytically active in several other cellular locations, which is likely to be particularly important in disease as few proteins associated with neurodegeneration are found in the ER. Finally, cell surface PDI, which reduces NO allowing it to pass through the plasma membrane, may lead to the production of SNO proteins and therefore also contribute to the pathogenesis of neurodegenerative diseases. The broad involvement of PDIs in human neurodegenerative diseases highlights the need for a better understanding of how they become inactivated by posttranslational modification, which is crucial to evaluate their use as possible targets for disease intervention.
References
- 1.Takalo M, Salminen A, Soininen H, Hiltunen M, Haapasalo A. Protein aggregation and degradation mechanisms in neurodegenerative diseases. American Journal of Neurodegenerative Disease. 2013;2(1):1–14. [PMC free article] [PubMed] [Google Scholar]
- 2.Soto C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nature Reviews Neuroscience. 2003;4(1):49–60. doi: 10.1038/nrn1007. [DOI] [PubMed] [Google Scholar]
- 3.Glenner GG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. 1984. Biochemical and Biophysical Research Communications. 2012;425(3):534–539. doi: 10.1016/j.bbrc.2012.08.020. [DOI] [PubMed] [Google Scholar]
- 4.Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein τ (tau) in Alzheimer cytoskeletal pathology. Proceedings of the National Academy of Sciences of the United States of America. 1986;83(13):44913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spillantini MG, Schmidt ML, Lee VM-Y, Trojanowski JQ, Jakes R, Goedert M. α-synuclein in Lewy bodies. Nature. 1997;388(6645):839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 6.DiFiglia M, Sapp E, Chase KO, et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277(5334):1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 7.Chen X, Guan T, Li C, et al. SOD1 aggregation in astrocytes following ischemia/reperfusion injury: a role of NO-mediated S-nitrosylation of protein disulfide isomerase (PDI) Journal of Neuroinflammation. 2012;9, article 237 doi: 10.1186/1742-2094-9-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mulligan VK, Kerman A, Laister RC, Sharda PR, Arslan PE, Chakrabartty A. Early steps in oxidation-induced SOD1 misfolding: implications for non-amyloid protein aggregation in familial ALS. Journal of Molecular Biology. 2012;421(4-5):631–652. doi: 10.1016/j.jmb.2012.04.016. [DOI] [PubMed] [Google Scholar]
- 9.Sheng Y, Chattopadhyay M, Whitelegge J, Valentine JS. SOD1 aggregation and ALS: role of metallation states and disulfide status. Current Topics in Medicinal Chemistry. 2012;12(22):2560–2572. doi: 10.2174/1568026611212220010. [DOI] [PubMed] [Google Scholar]
- 10.Arai T, Hasegawa M, Akiyama H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochemical and Biophysical Research Communications. 2006;351(3):602–611. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- 11.Cairns NJ, Neumann M, Bigio EH, et al. TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. American Journal of Pathology. 2007;171(1):227–240. doi: 10.2353/ajpath.2007.070182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mackenzie IRA, Bigio EH, Ince PG, et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Annals of Neurology. 2007;61(5):427–434. doi: 10.1002/ana.21147. [DOI] [PubMed] [Google Scholar]
- 13.Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 14.Deng H-X, Zhai H, Bigio EH, et al. FUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis. Annals of Neurology. 2010;67(6):739–748. doi: 10.1002/ana.22051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Musardo S, Saraceno C, Pelucchi S, Marcello E. Trafficking in neurons: searching for new targets for Alzheimer’s disease future therapies. European Journal of Pharmacology. 2013 doi: 10.1016/j.ejphar.2013.07.019. [DOI] [PubMed] [Google Scholar]
- 18.Goate A, Chartier-Harlin M-C, Mullan M, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349(6311):704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 19.Murrell J, Farlow M, Ghetti B, Benson MD. A mutation in the amyloid precursor protein associated with hereditary Alzheimer’s disease. Science. 1991;254(5028):97–99. doi: 10.1126/science.1925564. [DOI] [PubMed] [Google Scholar]
- 20.Chartier-Harlin M-C, Crawford F, Houlden H, et al. Early-onset Alzheimer’s disease caused by mutations at codon 717 of the β-amyloid precursor protein gene. Nature. 1991;353(6347):844–846. doi: 10.1038/353844a0. [DOI] [PubMed] [Google Scholar]
- 21.de Strooper B, Vassar R, Golde T. The secretases: enzymes with therapeutic potential in Alzheimer disease. Nature Reviews Neurology. 2010;6(2):99–107. doi: 10.1038/nrneurol.2009.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiological Reviews. 2001;81(2):741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 23.Wahlster L, Arimon M, Nasser-Ghodsi N, et al. Presenilin-1 adopts pathogenic conformation in normal aging and in sporadic Alzheimer’s disease. Acta Neuropathologica. 2013;125(2):187–199. doi: 10.1007/s00401-012-1065-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cruchaga C, Chakraverty S, Mayo K, et al. Rare variants in APP, PSEN1 and PSEN2 increase risk for AD in late-onset Alzheimer’s disease families. PLoS ONE. 2012;7(2) doi: 10.1371/journal.pone.0031039.e31039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276(5321):2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 26.Arima K, Uéda K, Sunohara N, et al. Immunoelectron-microscopic demonstration of NACP/α-synuclein-epitopes on the filamentous component of Lewy bodies in Parkinson’s disease and in dementia with Lewy bodies. Brain Research. 1998;808(1):93–100. doi: 10.1016/s0006-8993(98)00734-3. [DOI] [PubMed] [Google Scholar]
- 27.Bonifati V. Autosomal recessive parkinsonism. Parkinsonism and Related Disorders. 2012;18(supplement 1):S4–S6. doi: 10.1016/S1353-8020(11)70004-9. [DOI] [PubMed] [Google Scholar]
- 28.Dalle-Donne I, Rossi R, Colombo R, Giustarini D, Milzani A. Biomarkers of oxidative damage in human disease. Clinical Chemistry. 2006;52(4):601–623. doi: 10.1373/clinchem.2005.061408. [DOI] [PubMed] [Google Scholar]
- 29.Mangialasche F, Polidori MC, Monastero R, et al. Biomarkers of oxidative and nitrosative damage in Alzheimer’s disease and mild cognitive impairment. Ageing Research Reviews. 2009;8(4):285–305. doi: 10.1016/j.arr.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 30.Schapira AHV, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD. Mitochondrial complex I deficiency in Parkinson’s disease. Journal of Neurochemistry. 1990;54(3):823–827. doi: 10.1111/j.1471-4159.1990.tb02325.x. [DOI] [PubMed] [Google Scholar]
- 31.Benhar M, Forrester MT, Stamler JS. Nitrosative stress in the ER: a new role for S-nitrosylation in neurodegenerative diseases. ACS Chemical Biology. 2006;1(6):355–358. doi: 10.1021/cb600244c. [DOI] [PubMed] [Google Scholar]
- 32.Appenzeller-Herzog C, Ellgaard L. The human PDI family: versatility packed into a single fold. Biochimica et Biophysica Acta. 2008;1783(4):535–548. doi: 10.1016/j.bbamcr.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 33.Gillece P, Luz JM, Lennarz WJ, de la Cruz FJ, Römisch K. Export of a cysteine-free misfolded secretory protein from the endoplasmic reticulum for degradation requires interaction with protein disulfide isomerase. Journal of Cell Biology. 1999;147(7):1443–1456. doi: 10.1083/jcb.147.7.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Molinari M, Galli C, Piccaluga V, Pieren M, Paganetti P. Sequential assistance of molecular chaperones and transient formation of covalent complexes during protein degradation from the ER. Journal of Cell Biology. 2002;158(2):247–257. doi: 10.1083/jcb.200204122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsai B, Rodighiero C, Lencer WI, Rapoport TA. Protein disulfide isomerase acts as a redox-dependent chaperone to unfold cholera toxin. Cell. 2001;104(6):937–948. doi: 10.1016/s0092-8674(01)00289-6. [DOI] [PubMed] [Google Scholar]
- 36.Nakamura T, Tu S, Akhtar MW, Sunico CR, Okamoto S, Lipton SA. Aberrant protein S-nitrosylation in neurodegenerative diseases. Neuron. 2013;78(4):596–614. doi: 10.1016/j.neuron.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chakravarti B, Chakravarti DN. Oxidative modification of proteins: age-related changes. Gerontology. 2007;53(3):128–139. doi: 10.1159/000097865. [DOI] [PubMed] [Google Scholar]
- 38.Mariani E, Polidori MC, Cherubini A, Mecocci P. Oxidative stress in brain aging, neurodegenerative and vascular diseases: an overview. Journal of Chromatography B. 2005;827(1):65–75. doi: 10.1016/j.jchromb.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 39.Finkel T. Signal transduction by reactive oxygen species. Journal of Cell Biology. 2011;194(1):7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cutler RG, Pedersen WA, Camandola S, Rothstein JD, Mattson MP. Evidence that accumulation of ceramides and cholesterol esters mediates oxidative stress—induced death of motor neurons in amyotrophic lateral sclerosis. Annals of Neurology. 2002;52(4):448–457. doi: 10.1002/ana.10312. [DOI] [PubMed] [Google Scholar]
- 41.Xiong Y, Uys JD, Tew KD, Townsend DM. S-glutathionylation: from molecular mechanisms to health outcomes. Antioxidants and Redox Signaling. 2011;15(1):233–270. doi: 10.1089/ars.2010.3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jones DP. Redefining oxidative stress. Antioxidants and Redox Signaling. 2006;8(9-10):1865–1879. doi: 10.1089/ars.2006.8.1865. [DOI] [PubMed] [Google Scholar]
- 43.Calabrese V, Boyd-Kimball D, Scapagnini G, Butterfield DA. Nitric oxide and cellular stress response in brain aging and neurodegenerative disorders: the role of vitagenes. In Vivo. 2004;18(3):245–268. [PubMed] [Google Scholar]
- 44.Kedar NP. Can we prevent Parkinson’s and Alzheimer’s disease? Journal of Postgraduate Medicine. 2003;49(3):236–245. [PubMed] [Google Scholar]
- 45.Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiological Reviews. 1979;59(3):527–605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- 46.Garthwaite J, Boulton CL. Nitric oxide signaling in the central nervous system. Annual Review of Physiology. 1995;57:683–706. doi: 10.1146/annurev.ph.57.030195.003343. [DOI] [PubMed] [Google Scholar]
- 47.Bredt DS, Hwang PM, Glatt CE, Lowenstein C, Reed RR, Snyder SH. Cloned and expressed nitric oxide synthase structurally resembles cytochrome P-450 reductase. Nature. 1991;351(6329):714–718. doi: 10.1038/351714a0. [DOI] [PubMed] [Google Scholar]
- 48.Bustamante J, Czerniczyniec A, Lores-Arnaiz S. Brain nitric oxide synthases and mitochondrial function. Frontiers in Bioscience. 2007;12(3):1034–1040. doi: 10.2741/2123. [DOI] [PubMed] [Google Scholar]
- 49.Elfering SL, Sarkela TM, Giulivi C. Biochemistry of mitochondrial nitric-oxide synthase. The Journal of Biological Chemistry. 2002;277(41):38079–38086. doi: 10.1074/jbc.M205256200. [DOI] [PubMed] [Google Scholar]
- 50.Forstermann U, Schmidt HHHW, Pollock JS, et al. Isoforms of nitric oxide synthase. Characterization and purification from different cell types. Biochemical Pharmacology. 1991;42(10):1849–1857. doi: 10.1016/0006-2952(91)90581-o. [DOI] [PubMed] [Google Scholar]
- 51.Broillet M-C. S-nitrosylation of proteins. Cellular and Molecular Life Sciences. 1999;55(8-9):1036–1042. doi: 10.1007/s000180050354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lei SZ, Pan Z-H, Aggarwal SK, et al. Effect of nitric oxide production on the redox modulatory site of the NMDA receptor-channel complex. Neuron. 1992;8(6):1087–1099. doi: 10.1016/0896-6273(92)90130-6. [DOI] [PubMed] [Google Scholar]
- 53.Lipton SA, Choi Y-B, Pan Z-H, et al. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature. 1993;364(6438):626–632. doi: 10.1038/364626a0. [DOI] [PubMed] [Google Scholar]
- 54.Shahani N, Sawa A. Protein S-nitrosylation: role for nitric oxide signaling in neuronal death. Biochimica et Biophysica Acta. 2012;1820(6):736–742. doi: 10.1016/j.bbagen.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stamler JS, Simon DI, Osborne JA, et al. S-nitrosylation of proteins with nitric oxide: synthesis and characterization of biologically active compounds. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(1):444–448. doi: 10.1073/pnas.89.1.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stamler JS, Lamas S, Fang FC. Nitrosylation: the prototypic redox-based signaling mechanism. Cell. 2001;106(6):675–683. doi: 10.1016/s0092-8674(01)00495-0. [DOI] [PubMed] [Google Scholar]
- 57.Nakamura T, Lipton SA. S-nitrosylation of critical protein thiols mediates protein misfolding and mitochondrial dysfunction in neurodegenerative diseases. Antioxidants and Redox Signaling. 2011;14(8):1479–1492. doi: 10.1089/ars.2010.3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Staab CA, Hellgren M, Höög J-O. Medium- and short-chain dehydrogenase/reductase gene and protein families: dual functions of alcohol dehydrogenase 3: implications with focus on formaldehyde dehydrogenase and S-nitrosoglutathione reductase activities. Cellular and Molecular Life Sciences. 2008;65(24):3950–3960. doi: 10.1007/s00018-008-8592-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu L, Hausladen A, Zeng M, Que L, Heitman J, Stamler JS. A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature. 2001;410(6827):490–494. doi: 10.1038/35068596. [DOI] [PubMed] [Google Scholar]
- 60.Holmgren A. Thioredoxin. Annual Review of Biochemistry. 1985;54:237–271. doi: 10.1146/annurev.bi.54.070185.001321. [DOI] [PubMed] [Google Scholar]
- 61.Holmgren A. Thioredoxin and glutaredoxin systems. The Journal of Biological Chemistry. 1989;264(24):13963–13966. [PubMed] [Google Scholar]
- 62.Nikitovic D, Holmgren A. S-nitrosoglutathione is cleaved by the thioredoxin system with liberation of glutathione and redox regulating nitric oxide. The Journal of Biological Chemistry. 1996;271(32):19180–19185. doi: 10.1074/jbc.271.32.19180. [DOI] [PubMed] [Google Scholar]
- 63.Benhar M, Forrester MT, Stamler JS. Protein denitrosylation: enzymatic mechanisms and cellular functions. Nature Reviews Molecular Cell Biology. 2009;10(10):721–732. doi: 10.1038/nrm2764. [DOI] [PubMed] [Google Scholar]
- 64.Sliskovic I, Raturi A, Mutus B. Characterization of the S-denitrosation activity of protein disulfide isomerase. The Journal of Biological Chemistry. 2005;280(10):8733–8741. doi: 10.1074/jbc.M408080200. [DOI] [PubMed] [Google Scholar]
- 65.Jourd’heuil D, Laroux FS, Miles AM, Wink DA, Grisham MB. Effect of superoxide dismutase on the stability of S-nitrosothiols. Archives of Biochemistry and Biophysics. 1999;361(2):323–330. doi: 10.1006/abbi.1998.1010. [DOI] [PubMed] [Google Scholar]
- 66.Singh RJ, Hogg N, Joseph J, Kalyanaraman B. Mechanism of nitric oxide release from S-nitrosothiols. The Journal of Biological Chemistry. 1996;271(31):18596–18603. doi: 10.1074/jbc.271.31.18596. [DOI] [PubMed] [Google Scholar]
- 67.Hess DT, Matsumoto A, Kim S-O, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nature Reviews Molecular Cell Biology. 2005;6(2):150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 68.Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nature Reviews Neuroscience. 2010;11(10):682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kalia LV, Kalia SK, Salter MW. NMDA receptors in clinical neurology: excitatory times ahead. The Lancet Neurology. 2008;7(8):742–755. doi: 10.1016/S1474-4422(08)70165-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dawson VL, Dawson TM, London ED, Bredt DS, Snyder SH. Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proceedings of the National Academy of Sciences of the United States of America. 1991;88(14):6368–6371. doi: 10.1073/pnas.88.14.6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dawson VL, Dawson TM. Nitric oxide neurotoxicity. Journal of Chemical Neuroanatomy. 1996;10(3-4):179–190. doi: 10.1016/0891-0618(96)00148-2. [DOI] [PubMed] [Google Scholar]
- 72.Supnet C, Bezprozvanny I. The dysregulation of intracellular calcium in Alzheimer disease. Cell Calcium. 2010;47(2):183–189. doi: 10.1016/j.ceca.2009.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gu Z, Kaul M, Yan B, et al. S-nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science. 2002;297(5584):1186–1190. doi: 10.1126/science.1073634. [DOI] [PubMed] [Google Scholar]
- 74.Stamler JS, Hausladen A. Oxidative modifications in nitrosative stress. Nature Structural Biology. 1998;5(4):247–249. doi: 10.1038/nsb0498-247. [DOI] [PubMed] [Google Scholar]
- 75.Uehara T, Nakamura T, Yao D, et al. S-Nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature. 2006;441(7092):513–517. doi: 10.1038/nature04782. [DOI] [PubMed] [Google Scholar]
- 76.Papadia S, Soriano FX, Léveillé F, et al. Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nature Neuroscience. 2008;11(4):476–487. doi: 10.1038/nn2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cho D-H, Nakamura T, Fang J, et al. β-amyloid-related mitochondrial fission and neuronal injury. Science. 2009;324(5923):102–105. doi: 10.1126/science.1171091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lipton SA, Choi Y-B, Takahashi H, et al. Cysteine regulation of protein function—as exemplified by NMDA-receptor modulation. Trends in Neurosciences. 2002;25(9):474–480. doi: 10.1016/s0166-2236(02)02245-2. [DOI] [PubMed] [Google Scholar]
- 79.Stamler JS, Toone EJ, Lipton SA, Sucher NJ. (S)NO signals: translocation, regulation, and a consensus motif. Neuron. 1997;18(5):691–696. doi: 10.1016/s0896-6273(00)80310-4. [DOI] [PubMed] [Google Scholar]
- 80.Johnson MA, Macdonald TL, Mannick JB, Conaway MR, Gaston B. Accelerated S-nitrosothiol breakdown by amyotrophic lateral sclerosis mutant copper, zinc-superoxide dismutase. The Journal of Biological Chemistry. 2001;276(43):39872–39878. doi: 10.1074/jbc.M102781200. [DOI] [PubMed] [Google Scholar]
- 81.Schonhoff CM, Matsuoka M, Tummala H, et al. S-nitrosothiol depletion in amyotrophic lateral sclerosis. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(7):2404–2409. doi: 10.1073/pnas.0507243103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Marino SM, Gladyshev VN. Structural analysis of cysteine S-nitrosylation: a modified Aacid-based motif and the emerging role of trans-nitrosylation. Journal of Molecular Biology. 2010;395(4):844–859. doi: 10.1016/j.jmb.2009.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bartberger MD, Mannion JD, Powell SC, Stamler JS, Houk KN, Toone EJ. S-N dissociation energies of S-nitrosothiols: on the origins of nitrosothiol decomposition rates. Journal of the American Chemical Society. 2001;123(36):8868–8869. doi: 10.1021/ja0109390. [DOI] [PubMed] [Google Scholar]
- 84.Stamler JS, Toone EJ. The decomposition of thionitrites. Current Opinion in Chemical Biology. 2002;6(6):779–785. doi: 10.1016/s1367-5931(02)00383-6. [DOI] [PubMed] [Google Scholar]
- 85.Seth D, Stamler JS. The SNO-proteome: causation and classifications. Current Opinion in Chemical Biology. 2011;15(1):129–136. doi: 10.1016/j.cbpa.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chung KKK, Dawson TM, Dawson VL. Nitric oxide, S-nitrosylation and neurodegeneration. Cellular and Molecular Biology. 2005;51(3):247–254. [PubMed] [Google Scholar]
- 87.Nakamura T, Cieplak P, Cho D-H, Godzik A, Lipton SA. S-Nitrosylation of Drp1 links excessive mitochondrial fission to neuronal injury in neurodegeneration. Mitochondrion. 2010;10(5):573–578. doi: 10.1016/j.mito.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tsang AHK, Lee Y-IL, Ko HS, et al. S-nitrosylation of XIAP compromises neuronal survival in Parkinson’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(12):4900–4905. doi: 10.1073/pnas.0810595106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Walker AK, Farg MA, Bye CR, McLean CA, Horne MK, Atkin JD. Protein disulphide isomerase protects against protein aggregation and is S-nitrosylated in amyotrophic lateral sclerosis. Brain. 2010;133(1):105–116. doi: 10.1093/brain/awp267. [DOI] [PubMed] [Google Scholar]
- 90.Yao D, Gu Z, Nakamura T, et al. Nitrosative stress linked to sporadic Parkinson’s disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(29):10810–10814. doi: 10.1073/pnas.0404161101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Choi Y-B, Tenneti L, Le DA, et al. Molecular basis of NMDA receptor-coupled ion channel modulation by S-nitrosylation. Nature Neuroscience. 2000;3(1):15–21. doi: 10.1038/71090. [DOI] [PubMed] [Google Scholar]
- 92.Townsend DM. S-glutathionylation: indicator of cell stress and regulator of the unfolded protein response. Molecular Interventions. 2008;7(6):313–324. doi: 10.1124/mi.7.6.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hansen RE, Roth D, Winther JR. Quantifying the global cellular thiol-disulfide status. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(2):422–427. doi: 10.1073/pnas.0812149106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jacob C, Knight I, Winyard PG. Aspects of the biological redox chemistry of cysteine: from simple redox responses to sophisticated signalling pathways. Biological Chemistry. 2006;387(10-11):1385–1397. doi: 10.1515/BC.2006.174. [DOI] [PubMed] [Google Scholar]
- 95.Sabens Liedhegner EA, Gao X-H, Mieyal JJ. Mechanisms of altered redox regulation in neurodegenerative diseases-focus on S-glutathionylation. Antioxidants and Redox Signaling. 2012;16(6):543–566. doi: 10.1089/ars.2011.4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ziegler DM. Role of reversible oxidation-reduction of enzyme thiols-disulfides in metabolic regulation. Annual Review of Biochemistry. 1985;54:305–329. doi: 10.1146/annurev.bi.54.070185.001513. [DOI] [PubMed] [Google Scholar]
- 97.Gallogly MM, Mieyal JJ. Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Current Opinion in Pharmacology. 2007;7(4):381–391. doi: 10.1016/j.coph.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 98.Mieyal JJ, Gallogly MM, Qanungo S, Sabens EA, Shelton MD. Molecular mechanisms and clinical implications of reversible protein S-glutathionylation. Antioxidants and Redox Signaling. 2008;10(11):1941–1988. doi: 10.1089/ars.2008.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Brasil AA, Belati A, Mannarino SC, Panek AD, Eleutherio ECA, Pereira MD. The involvement of GSH in the activation of human Sod1 linked to FALS in chronologically aged yeast cells. FEMS Yeast Research. 2013;13(5):433–440. doi: 10.1111/1567-1364.12045. [DOI] [PubMed] [Google Scholar]
- 100.Carletti B, Passarelli C, Sparaco M, et al. Effect of protein glutathionylation on neuronal cytoskeleton: a potential link to neurodegeneration. Neuroscience. 2011;192:285–294. doi: 10.1016/j.neuroscience.2011.05.060. [DOI] [PubMed] [Google Scholar]
- 101.di Domenico F, Cenini G, Sultana R, et al. Glutathionylation of the pro-apoptotic protein p53 in alzheimer’s disease brain: implications for AD pathogenesis. Neurochemical Research. 2009;34(4):727–733. doi: 10.1007/s11064-009-9924-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhu Y, Carvey PM, Ling Z. Age-related changes in glutathione and glutathione-related enzymes in rat brain. Brain Research. 2006;1090(1):35–44. doi: 10.1016/j.brainres.2006.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sian J, Dexter DT, Lees AJ, et al. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Annals of Neurology. 1994;36(3):348–355. doi: 10.1002/ana.410360305. [DOI] [PubMed] [Google Scholar]
- 104.Choo YS, Mao Z, Johnson GV, Lesort M. Increased glutathione levels in cortical and striatal mitochondria of the R6/2 Huntington’s disease mouse model. Neuroscience Letters. 2005;386(1):63–68. doi: 10.1016/j.neulet.2005.05.065. [DOI] [PubMed] [Google Scholar]
- 105.Fox JH, Barber DS, Singh B, et al. Cystamine increases L-cysteine levels in Huntington’s disease transgenic mouse brain and in a PC12 model of polyglutamine aggregation. Journal of Neurochemistry. 2004;91(2):413–422. doi: 10.1111/j.1471-4159.2004.02726.x. [DOI] [PubMed] [Google Scholar]
- 106.Wilcox KC, Zhou L, Jordon JK, et al. Modifications of superoxide dismutase (SOD1) in human erythrocytes: a possible role in amyotrophic lateral sclerosis. The Journal of Biological Chemistry. 2009;284(20):13940–13947. doi: 10.1074/jbc.M809687200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hwang C, Sinskey AJ, Lodish HF. Oxidized redox state of glutathione in the endoplasmic reticulum. Science. 1992;257(5076):1496–1502. doi: 10.1126/science.1523409. [DOI] [PubMed] [Google Scholar]
- 108.Atkin JD, Farg MA, Walker AK, McLean C, Tomas D, Horne MK. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiology of Disease. 2008;30(3):400–407. doi: 10.1016/j.nbd.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 109.Colla E, Coune P, Liu Y, et al. Endoplasmic reticulum stress is important for the manifestations of α-synucleinopathy in vivo. Journal of Neuroscience. 2012;32(10):3306–3320. doi: 10.1523/JNEUROSCI.5367-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Doyle KM, Kennedy D, Gorman AM, Gupta S, Healy SJM, Samali A. Unfolded proteins and endoplasmic reticulum stress in neurodegenerative disorders. Journal of Cellular and Molecular Medicine. 2011;15(10):2025–2039. doi: 10.1111/j.1582-4934.2011.01374.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lashuel HA, Hirling H. Rescuing defective vesicular trafficking protects against alpha-synuclein toxicity in cellular and animal models of Parkinson’s disease. ACS Chemical Biology. 2006;1(7):420–424. doi: 10.1021/cb600331e. [DOI] [PubMed] [Google Scholar]
- 112.Viana RJS, Nunes AF, Rodrigues CMP. Endoplasmic reticulum enrollment in Alzheimer’s disease. Molecular Neurobiology . 2012;46(2):522–534. doi: 10.1007/s12035-012-8301-x. [DOI] [PubMed] [Google Scholar]
- 113.Vidal R, Caballero B, Couve A, Hetz C. Converging pathways in the occurrence of endoplasmic reticulum (ER) stress in Huntington’s disease. Current Molecular Medicine. 2011;11(1):1–12. doi: 10.2174/156652411794474419. [DOI] [PubMed] [Google Scholar]
- 114.Walker AK, Atkin JD. Stress signaling from the endoplasmic reticulum: a central player in the pathogenesis of amyotrophic lateral sclerosis. IUBMB Life. 2011;63(9):754–763. doi: 10.1002/iub.520. [DOI] [PubMed] [Google Scholar]
- 115.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nature Reviews Molecular Cell Biology. 2007;8(7):519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 116.Schröder M. Endoplasmic reticulum stress responses. Cellular and Molecular Life Sciences. 2007;65(6):862–894. doi: 10.1007/s00018-007-7383-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yoshida H. ER stress and diseases. FEBS Journal. 2007;274(3):630–658. doi: 10.1111/j.1742-4658.2007.05639.x. [DOI] [PubMed] [Google Scholar]
- 118.Hitomi J, Katayama T, Eguchi Y, et al. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Aβ-induced cell death. Journal of Cell Biology. 2004;165(3):347–356. doi: 10.1083/jcb.200310015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Urano F, Wang X, Bertolotti A, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287(5453):664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 120.Ellgaard L, Ruddock LW. The human protein disulphide isomerase family: substrate interactions and functional properties. EMBO Reports. 2005;6(1):28–32. doi: 10.1038/sj.embor.7400311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kozlov G, Määttänen P, Thomas DY, Gehring K. A structural overview of the PDI family of proteins. FEBS Journal. 2010;277(19):3924–3936. doi: 10.1111/j.1742-4658.2010.07793.x. [DOI] [PubMed] [Google Scholar]
- 122.Ferrari DM, Söling H-D. The protein disulphide-isomerase family: unravelling a string of folds. Biochemical Journal. 1999;339(1):1–10. [PMC free article] [PubMed] [Google Scholar]
- 123.Andreu CI, Woehlbier U, Torres M, Hetz C. Protein disulfide isomerases in neurodegeneration: from disease mechanisms to biomedical applications. FEBS Letters. 2012;586(18):2826–2834. doi: 10.1016/j.febslet.2012.07.023. [DOI] [PubMed] [Google Scholar]
- 124.Benham AM. The protein disulfide isomerase family: key players in health and disease. Antioxidants and Redox Signaling. 2012;16(8):781–789. doi: 10.1089/ars.2011.4439. [DOI] [PubMed] [Google Scholar]
- 125.Galligan JJ, Petersen DR. The human protein disulfide isomerase gene family. Human Genomics. 2012;6, article 6 doi: 10.1186/1479-7364-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sevier CS, Kaiser CA. Conservation and diversity of the cellular disulfide bond formation pathways. Antioxidants and Redox Signaling. 2006;8(5-6):797–811. doi: 10.1089/ars.2006.8.797. [DOI] [PubMed] [Google Scholar]
- 127.Hawkins HC, Blackburn EC, Freedman RB. Comparison of the activities of protein disulphide-isomerase and thioredoxin in catalysing disulphide isomerization in a protein substrate. Biochemical Journal. 1991;275(2):349–353. doi: 10.1042/bj2750349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Turano C, Coppari S, Altieri F, Ferraro A. Proteins of the PDI family: unpredicted non-ER locations and functions. Journal of Cellular Physiology. 2002;193(2):154–163. doi: 10.1002/jcp.10172. [DOI] [PubMed] [Google Scholar]
- 129.Higo T, Hattori M, Nakamura T, Natsume T, Michikawa T, Mikoshiba K. Subtype-specific and ER lumenal environment-dependent regulation of inositol 1,4,5-trisphosphate receptor type 1 by ERp44. Cell. 2005;120(1):85–98. doi: 10.1016/j.cell.2004.11.048. [DOI] [PubMed] [Google Scholar]
- 130.Li Y, Camacho P. Ca2+-dependent redox modulation of SERCA 2b by ERp57. Journal of Cell Biology. 2004;164(1):35–46. doi: 10.1083/jcb.200307010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Jansens A, van Duijn E, Braakman I. Coordinated nonvectorial folding in a newly synthesized multidomain protein. Science. 2002;298(5602):2401–2403. doi: 10.1126/science.1078376. [DOI] [PubMed] [Google Scholar]
- 132.Schwaller M, Wilkinson B, Gilbert HF. Reduction-reoxidation cycles contribute to catalysis of disulfide isomerization by protein-disulfide isomerase. The Journal of Biological Chemistry. 2003;278(9):7154–7159. doi: 10.1074/jbc.M211036200. [DOI] [PubMed] [Google Scholar]
- 133.Walker KW, Gilbert HF. Scanning and escape during protein-disulfide isomerase-assisted protein folding. The Journal of Biological Chemistry. 1997;272(14):8845–8848. doi: 10.1074/jbc.272.14.8845. [DOI] [PubMed] [Google Scholar]
- 134.Lyles MM, Gilbert HF. Catalysis of the oxidative folding of ribonuclease A by protein disulfide isomerase: dependence of the rate on the composition of the redox buffer. Biochemistry. 1991;30(3):613–619. doi: 10.1021/bi00217a004. [DOI] [PubMed] [Google Scholar]
- 135.Gething M-J, Sambrook J. Protein folding in the cell. Nature. 1992;355(6355):33–45. doi: 10.1038/355033a0. [DOI] [PubMed] [Google Scholar]
- 136.Ellis RJ, Hemmingsen SM. Molecular chaperones: proteins essential for the biogenesis of some macromolecular structures. Trends in Biochemical Sciences. 1989;14(8):339–342. doi: 10.1016/0968-0004(89)90168-0. [DOI] [PubMed] [Google Scholar]
- 137.Puig A, Gilbert HF. Protein disulfide isomerase exhibits chaperone and anti-chaperone activity in the oxidative refolding of lysozyme. The Journal of Biological Chemistry. 1994;269(10):7764–7771. [PubMed] [Google Scholar]
- 138.Quan H, Fan G, Wang C-C. Independence of the chaperone activity of protein disulfide isomerase from its thioredoxin-like active site. The Journal of Biological Chemistry. 1995;270(29):17078–17080. doi: 10.1074/jbc.270.29.17078. [DOI] [PubMed] [Google Scholar]
- 139.Dai Y, Wang C-C. A mutant truncated protein disulfide isomerase with no chaperone activity. The Journal of Biological Chemistry. 1997;272(44):27572–27576. doi: 10.1074/jbc.272.44.27572. [DOI] [PubMed] [Google Scholar]
- 140.Tian G, Xiang S, Noiva R, Lennarz WJ, Schindelin H. The crystal structure of yeast protein disulfide isomerase suggests cooperativity between its active sites. Cell. 2006;124(1):61–73. doi: 10.1016/j.cell.2005.10.044. [DOI] [PubMed] [Google Scholar]
- 141.Klappa P, Ruddock LW, Darby NJ, Freedman RB. The b′ domain provides the principal peptide-binding site of protein disulfide isomerase but all domains contribute to binding of misfolded proteins. EMBO Journal. 1998;17(4):927–935. doi: 10.1093/emboj/17.4.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kozlov G, Maattanen P, Schrag JD, et al. Crystal structure of the bb′ domains of the protein disulfide isomerase ERp57. Structure. 2006;14(8):1331–1339. doi: 10.1016/j.str.2006.06.019. [DOI] [PubMed] [Google Scholar]
- 143.Kozlov G, Määttänen P, Schrag JD, et al. Structure of the noncatalytic domains and global fold of the protein disulfide isomerase ERp72. Structure. 2009;17(5):651–659. doi: 10.1016/j.str.2009.02.016. [DOI] [PubMed] [Google Scholar]
- 144.Rutkevich LA, Williams DB. Participation of lectin chaperones and thiol oxidoreductases in protein folding within the endoplasmic reticulum. Current Opinion in Cell Biology. 2011;23(2):157–166. doi: 10.1016/j.ceb.2010.10.011. [DOI] [PubMed] [Google Scholar]
- 145.Tian G, Kober F-X, Lewandrowski U, Sickmann A, Lennarz WJ, Schindelin H. The catalytic activity of protein-disulfide isomerase requires a conformationally flexible molecule. The Journal of Biological Chemistry. 2008;283(48):33630–33640. doi: 10.1074/jbc.M806026200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Conn KJ, Gao W, McKee A, et al. Identification of the protein disulfide isomerase family member PDIp in experimental Parkinson’s disease and Lewy body pathology. Brain Research. 2004;1022(1-2):164–172. doi: 10.1016/j.brainres.2004.07.026. [DOI] [PubMed] [Google Scholar]
- 147.Klappa P, Stromer T, Zimmermann R, Ruddock LW, Freedman RB. A pancreas-specific glycosylated protein disulphide-isomerase binds to misfolded proteins and peptides with an interaction inhibited by oestrogens. European Journal of Biochemistry. 1998;254(1):63–69. doi: 10.1046/j.1432-1327.1998.2540063.x. [DOI] [PubMed] [Google Scholar]
- 148.Volkmer J, Guth S, Nastainczyk W, et al. Pancreas specific protein disulfide isomerase, PDIp, is in transient contact with secretory proteins during late stages of translocation. FEBS Letters. 1997;406(3):291–295. doi: 10.1016/s0014-5793(97)00288-3. [DOI] [PubMed] [Google Scholar]
- 149.Alanen HI, Salo KEH, Pekkala M, Siekkinen HM, Pirneskoski A, Ruddock LW. Defining the domain boundaries of the human protein disulfide isomerases. Antioxidants and Redox Signaling. 2003;5(4):367–374. doi: 10.1089/152308603768295096. [DOI] [PubMed] [Google Scholar]
- 150.Raykhel I, Alanen H, Salo K, et al. A molecular specificity code for the three mammalian KDEL receptors. Journal of Cell Biology. 2007;179(6):1193–1204. doi: 10.1083/jcb.200705180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Klappa P, Freedman RB, Zimmermann R. Protein disulphide isomerase and a lumenal cyclophilin-type peptidyl prolyl cis-trans isomerase are in transient contact with secretory proteins during late stages of translocation. European Journal of Biochemistry. 1995;232(3):755–764. [PubMed] [Google Scholar]
- 152.Freedman RB, Hirst TR, Tuite MF. Protein disulphide isomerase: building bridges in protein folding. Trends in Biochemical Sciences. 1994;19(8):331–336. doi: 10.1016/0968-0004(94)90072-8. [DOI] [PubMed] [Google Scholar]
- 153.Hirano N, Shibasaki F, Sakai B, et al. Molecular cloning of the human glucose-regulated protein ERp57/GRP58, a thiol-dependent reductase. Identification of its secretory form and inducible expression by the oncogenic transformation. European Journal of Biochemistry. 1995;234(1):336–342. doi: 10.1111/j.1432-1033.1995.336_c.x. [DOI] [PubMed] [Google Scholar]
- 154.Jessop CE, Chakravarthi S, Garbi N, Hämmerling GJ, Lovell S, Bulleid NJ. ERp57 is essential for efficient folding of glycoproteins sharing common structural domains. EMBO Journal. 2007;26(1):28–40. doi: 10.1038/sj.emboj.7601505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Bourdi M, Demady D, Martin JL, et al. cDNA cloning and baculovirus expression of the human liver endoplasmic reticulum P58: characterization as a protein disulfide isomerase isoform, but not as a protease or a carnitine acyltransferase. Archives of Biochemistry and Biophysics. 1995;323(2):397–403. doi: 10.1006/abbi.1995.0060. [DOI] [PubMed] [Google Scholar]
- 156.Frickel E-M, Frei P, Bouvier M, et al. ERp57 is a multifunctional thiol-disulfide oxidoreductase. The Journal of Biological Chemistry. 2004;279(18):18277–18287. doi: 10.1074/jbc.M314089200. [DOI] [PubMed] [Google Scholar]
- 157.Dingwall C, Laskey RA. Nuclear import: a tale of two sites. Current Biology. 1998;8(25):R922–R924. doi: 10.1016/s0960-9822(98)00010-4. [DOI] [PubMed] [Google Scholar]
- 158.Turano C, Gaucci E, Grillo C, Chichiarelli S. ERp57/GRP58: a protein with multiple functions. Cellular and Molecular Biology Letters. 2011;16(4):539–563. doi: 10.2478/s11658-011-0022-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Van PN, Rupp K, Lampen A, Soling H-D. CaBP2 is a rat homolog of ERp72 with proteindisulfide isomerase activity. European Journal of Biochemistry. 1993;213(2):789–795. doi: 10.1111/j.1432-1033.1993.tb17821.x. [DOI] [PubMed] [Google Scholar]
- 160.Spee P, Subjeck J, Neefjes J. Identification of novel peptide binding proteins in the endoplasmic reticulum: ERp72, calnexin, and grp170. Biochemistry. 1999;38(32):10559–10566. doi: 10.1021/bi990321r. [DOI] [PubMed] [Google Scholar]
- 161.Gulerez IE, Kozlov G, Rosenauer A, Gehring K. Structure of the third catalytic domain of the protein disulfide isomerase ERp46. Acta Crystallographica Section F. 2012;68(4):378–381. doi: 10.1107/S1744309112005866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Knoblach B, Keller BO, Groenendyk J, et al. ERp19 and ERp46, new members of the thioredoxin family of endoplasmic reticulum proteins. Molecular & Cellular Proteomics. 2003;2(10):1104–1119. doi: 10.1074/mcp.M300053-MCP200. [DOI] [PubMed] [Google Scholar]
- 163.Hayano T, Kikuchi M. Molecular cloning of the cDNA encoding a novel protein disulfide isomerase-related protein (PDIR) FEBS Letters. 1995;372(2-3):210–214. doi: 10.1016/0014-5793(95)00996-m. [DOI] [PubMed] [Google Scholar]
- 164.Cunnea PM, Miranda-Vizuete A, Bertoli G, et al. ERdj5, an endoplasmic reticulum (ER)-resident protein containing DnaJ and thioredoxin domains, is expressed in secretory cells or following ER stress. The Journal of Biological Chemistry. 2003;278(2):1059–1066. doi: 10.1074/jbc.M206995200. [DOI] [PubMed] [Google Scholar]
- 165.Soldà T, Garbi N, Hämmerling GJ, Molinari M. Consequences of ERp57 deletion on oxidative folding of obligate and facultative clients of the calnexin cycle. The Journal of Biological Chemistry. 2006;281(10):6219–6226. doi: 10.1074/jbc.M513595200. [DOI] [PubMed] [Google Scholar]
- 166.Cabibbo A, Pagani M, Fabbri M, et al. ERO1-L, a human protein that favors disulfide bond formation in the endoplasmic reticulum. The Journal of Biological Chemistry. 2000;275(7):4827–4833. doi: 10.1074/jbc.275.7.4827. [DOI] [PubMed] [Google Scholar]
- 167.Frand AR, Kaiser CA. Ero1p oxidizes protein disulfide isomerase in a pathway for disulfide bond formation in the endoplasmic reticulum. Molecular Cell. 1999;4(4):469–477. doi: 10.1016/s1097-2765(00)80198-7. [DOI] [PubMed] [Google Scholar]
- 168.Lundstrom J, Holmgren A. Protein disulfide-isomerase is a substrate for thioredoxin reductase and has thioredoxin-like activity. The Journal of Biological Chemistry. 1990;265(16):9114–9120. [PubMed] [Google Scholar]
- 169.Holaska JM, Black BE, Love DC, Hanover JA, Leszyk J, Paschal BM. Calreticulin is a receptor for nuclear export. Journal of Cell Biology. 2001;152(1):127–140. doi: 10.1083/jcb.152.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Johnson S, Michalak M, Opas M, Eggleton P. The ins and outs of calreticulin: from the ER lumen to the extracellular space. Trends in Cell Biology. 2001;11(3):122–129. doi: 10.1016/s0962-8924(01)01926-2. [DOI] [PubMed] [Google Scholar]
- 171.Roderick HL, Campbell AK, Llewellyn DH. Nuclear localisation of calreticulin in vivo is enhanced by its interaction with glucocorticoid receptors. FEBS Letters. 1997;405(2):181–185. doi: 10.1016/s0014-5793(97)00183-x. [DOI] [PubMed] [Google Scholar]
- 172.Burns K, Duggan B, Atkinson EA, et al. Modulation of gene expression by calreticulin binding to the glucocorticoid receptor. Nature. 1994;367(6462):476–480. doi: 10.1038/367476a0. [DOI] [PubMed] [Google Scholar]
- 173.Dedhar S. Novel functions for calreticulin: interaction with integrins and modulation of gene expression? Trends in Biochemical Sciences. 1994;19(7):269–271. doi: 10.1016/0968-0004(94)90001-9. [DOI] [PubMed] [Google Scholar]
- 174.Fraser SA, Michalak M, Welch WH, Hudig D. Calreticulin, a component of the endoplasmic reticulum and of cytotoxic lymphocyte granules, regulates perforin-mediated lysis in the hemolytic model system. Biochemistry and Cell Biology. 1998;76(5):881–887. doi: 10.1139/bcb-76-5-881. [DOI] [PubMed] [Google Scholar]
- 175.Michalak M, Corbett EF, Mesaeli N, Nakamura K, Opas M. Calreticulin: one protein, one gene, many functions. Biochemical Journal. 1999;344(2):281–292. [PMC free article] [PubMed] [Google Scholar]
- 176.Ni M, Zhang Y, Lee AS. Beyond the endoplasmic reticulum: atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochemical Journal. 2011;434(2):181–188. doi: 10.1042/BJ20101569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wroblewski VJ, Masnyk M, Khambatta SS, Becker GW. Mechanisms involved in degradation of human insulin by cytosolic fractions of human, monkey, and rat liver. Diabetes. 1992;41(4):539–547. doi: 10.2337/diab.41.4.539. [DOI] [PubMed] [Google Scholar]
- 178.Tabata Y, Takano K, Ito T, et al. Vaticanol B, a resveratrol tetramer, regulates endoplasmic reticulum stress and inflammation. American Journal of Physiology: Cell Physiology. 2007;293(1):C411–C418. doi: 10.1152/ajpcell.00095.2007. [DOI] [PubMed] [Google Scholar]
- 179.Yang YS, Harel NY, Strittmatter SM. Reticulon-4A (Nogo-A) redistributes protein disulfide isomerase to protect mice from SOD1-dependent amyotrophic lateral sclerosis. Journal of Neuroscience. 2009;29(44):13850–13859. doi: 10.1523/JNEUROSCI.2312-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Bernardoni P, Fazi B, Costanzi A, et al. Reticulon1-C modulates protein disulphide isomerase function. Cell Death & Disease. 2013;4, article e581 doi: 10.1038/cddis.2013.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Ramachandran N, Root P, Jiang X-M, Hogg PJ, Mutus B. Mechanism of transfer of NO from extracellular S-nitrosothiols into the cytosol by cell-surface protein disulfide isomerase. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(17):9539–9544. doi: 10.1073/pnas.171180998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Zai A, Rudd MA, Scribner AW, Loscalzo J. Cell-surface protein disulfide isomerase catalyzes transnitrosation and regulates intracellular transfer of nitric oxide. Journal of Clinical Investigation. 1999;103(3):393–399. doi: 10.1172/JCI4890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Shah CM, Bell SE, Locke IC, Chowdrey HS, Gordge MP. Interactions between cell surface protein disulphide isomerase and S-nitrosoglutathione during nitric oxide delivery. Nitric Oxide. 2007;16(1):135–142. doi: 10.1016/j.niox.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 184.Watts JC, Huo H, Bai Y, et al. Interactome analyses identify ties of PrP and its mammalian paralogs to oligomannosidic N-glycans and endoplasmic reticulum-derived chaperones. PLoS Pathogens. 2009;5(10) doi: 10.1371/journal.ppat.1000608.e1000608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Altieri F, Maras B, Eufemi M, Ferraro A, Turano C. Purification of a 57kDa nuclear matrix protein associated with thiol:protein-disulfide oxidoreductase and phospholipase C activities. Biochemical and Biophysical Research Communications. 1993;194(3):992–1000. doi: 10.1006/bbrc.1993.1919. [DOI] [PubMed] [Google Scholar]
- 186.Gerner C, Holzmann K, Meissner M, Gotzmann J, Grimm R, Sauermann G. Reassembling proteins and chaperones in human nuclear matrix protein fractions. Journal of Cellular Biochemistry. 1999;74(2):145–151. [PubMed] [Google Scholar]
- 187.Rigobello MP, Donella-Deana A, Cesaro L, Bindoli A. Isolation, purification, and characterization of a rat liver mitochondrial protein disulfide isomerase. Free Radical Biology and Medicine. 2000;28(2):266–272. doi: 10.1016/s0891-5849(99)00237-3. [DOI] [PubMed] [Google Scholar]
- 188.Müller JM, Milenkovic D, Guiard B, Pfanner N, Chacinska A. Precursor oxidation by Mia40 and Erv1 promotes vectorial transport of proteins into the mitochondrial intermembrane space. Molecular Biology of the Cell. 2008;19(1):226–236. doi: 10.1091/mbc.E07-08-0814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Wrobel L, Trojanowska A, Sztolsztener ME, Chacinska A. Mitochondrial protein import: Mia40 facilitates Tim22 translocation into the inner membrane of mitochondria. Molecular Biology of the Cell. 2013;24(5):543–554. doi: 10.1091/mbc.E12-09-0649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Hoffstrom BG, Kaplan A, Letso R, et al. Inhibitors of protein disulfide isomerase suppress apoptosis induced by misfolded proteins. Nature Chemical Biology. 2010;6(12):900–906. doi: 10.1038/nchembio.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Tanaka S, Uehara T, Nomura Y. Up-regulation of protein-disulfide isomerase in response to hypoxia/brain ischemia and its protective effect against apoptotic cell death. The Journal of Biological Chemistry. 2000;275(14):10388–10393. doi: 10.1074/jbc.275.14.10388. [DOI] [PubMed] [Google Scholar]
- 192.Lee JH, Won SM, Suh J, et al. Induction of the unfolded protein response and cell death pathway in Alzheimer’s disease, but not in aged Tg2576 mice. Experimental and Molecular Medicine. 2010;42(5):386–394. doi: 10.3858/emm.2010.42.5.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Erickson RR, Dunning LM, Olson DA, et al. In cerebrospinal fluid ER chaperones ERp57 and calreticulin bind β-amyloid. Biochemical and Biophysical Research Communications. 2005;332(1):50–57. doi: 10.1016/j.bbrc.2005.04.090. [DOI] [PubMed] [Google Scholar]
- 194.Honjo Y, Ito H, Horibe T, et al. Derlin-1-immunopositive inclusions in patients with Alzheimer’s disease. Neuroreport. 2012;23(10):611–615. doi: 10.1097/WNR.0b013e3283552a75. [DOI] [PubMed] [Google Scholar]
- 195.Honjo Y, Ito H, Horibe T, Takahashi R, Kawakami K. Protein disulfide isomerase-immunopositive inclusions in patients with Alzheimer disease. Brain Research. 2010;1349:90–96. doi: 10.1016/j.brainres.2010.06.016. [DOI] [PubMed] [Google Scholar]
- 196.Kim-Han JS, O’Malley KL. Cell stress induced by the parkinsonian mimetic, 6-hydroxydopamine, is concurrent with oxidation of the chaperone, ERp57, and aggresome formation. Antioxidants and Redox Signaling. 2007;9(12):2255–2264. doi: 10.1089/ars.2007.1791. [DOI] [PubMed] [Google Scholar]
- 197.Cheng H, Wang L, Wang C-C. Domain a of protein disulfide isomerase plays key role in inhibiting α-synuclein fibril formation. Cell Stress and Chaperones. 2010;15(4):415–421. doi: 10.1007/s12192-009-0157-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Yoo BC, Krapfenbauer K, Cairns N, Belay G, Bajo M, Lubec G. Overexpressed protein disulfide isomerase in brains of patients with sporadic Creutzfeldt-Jakob disease. Neuroscience Letters. 2002;334(3):196–200. doi: 10.1016/s0304-3940(02)01071-6. [DOI] [PubMed] [Google Scholar]
- 199.Wang SB, Shi Q, Xu Y, et al. Protein disulfide isomerase regulates endoplasmic reticulum stress and the apoptotic process during prion infection and PrP mutant-induced cytotoxicity. PLoS ONE. 2012;7(6, article e38221) doi: 10.1371/journal.pone.0038221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Farg MA, Soo KY, Walker AK, et al. Mutant FUS induces endoplasmic reticulum stress in amyotrophic lateral sclerosis and interacts with protein disulfide-isomerase. Neurobiology of Aging. 2012;33(12):2855–2868. doi: 10.1016/j.neurobiolaging.2012.02.009. [DOI] [PubMed] [Google Scholar]
- 201.Honjo Y, Kaneko S, Ito H, et al. Protein disulfide isomerase-immunopositive inclusions in patients with amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis. 2011;12(6):444–450. doi: 10.3109/17482968.2011.594055. [DOI] [PubMed] [Google Scholar]
- 202.Kwok CT, Morris AG, Frampton J, Smith B, Shaw CE, de Belleroche J. Association studies indicate that protein disulfide isomerase is a risk factor in amyotrophic lateral sclerosis. Free Radical Biology and Medicine. 2013;58:81–86. doi: 10.1016/j.freeradbiomed.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 203.Atkin JD, Farg MA, Turner BJ, et al. Induction of the unfolded protein response in familial amyotrophic lateral sclerosis and association of protein-disulfide isomerase with superoxide dismutase 1. The Journal of Biological Chemistry. 2006;281(40):30152–30165. doi: 10.1074/jbc.M603393200. [DOI] [PubMed] [Google Scholar]
- 204.Ilieva EV, Ayala V, Jové M, et al. Oxidative and endoplasmic reticulum stress interplay in sporadic amyotrophic lateral sclerosis. Brain. 2007;130(12):3111–3123. doi: 10.1093/brain/awm190. [DOI] [PubMed] [Google Scholar]
- 205.Niwa J-I, Yamada S-I, Ishigaki S, et al. Disulfide bond mediates aggregation, toxicity, and ubiquitylation of familial amyotrophic lateral sclerosis-linked mutant SOD1. The Journal of Biological Chemistry. 2007;282(38):28087–28095. doi: 10.1074/jbc.M704465200. [DOI] [PubMed] [Google Scholar]
- 206.Jaronen M, Vehviläinen P, Malm T, et al. Protein disulfide isomerase in ALS mouse glia links protein misfolding with NADPH oxidase-catalyzed superoxide production. Human Molecular Genetics. 2012;22(4):646–655. doi: 10.1093/hmg/dds472. [DOI] [PubMed] [Google Scholar]
- 207.Uehara T. Accumulation of misfolded protein through nitrosative stress linked to neurodegenerative disorders. Antioxidants and Redox Signaling. 2007;9(5):597–601. doi: 10.1089/ars.2006.1517. [DOI] [PubMed] [Google Scholar]
- 208.Xu L, Eu JP, Meissner G, Stamler JS. Activation of the cardiac calcium release channel (Ryanodoine receptor) by poly-S-nitrosylation. Science. 1998;279(5348):234–237. doi: 10.1126/science.279.5348.234. [DOI] [PubMed] [Google Scholar]
- 209.Ozawa K, Tsumoto H, Wei W, et al. Proteomic analysis of the role of S-nitrosoglutathione reductase in lipopolysaccharide-challenged mice. PROTEOMICS. 2012;12(12):2024–2035. doi: 10.1002/pmic.201100666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Townsend DM, Manevich Y, Lin H, et al. Nitrosative stress-induced S-glutathionylation of protein disulfide isomerase leads to activation of the unfolded protein response. Cancer Research. 2009;69(19):7626–7634. doi: 10.1158/0008-5472.CAN-09-0493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Uys JD, Xiong Y, Townsend DM. Nitrosative stress-induced S-glutathionylation of protein disulfide isomerase. Methods in Enzymology. 2011;490(C):321–332. doi: 10.1016/B978-0-12-385114-7.00018-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Xiong Y, Manevich Y, Tew KD, Townsend DM. S-glutathionylation of protein disulfide isomerase regulates estrogen receptor α stability and function. International Journal of Cell Biology. 2012;2012:9 pages. doi: 10.1155/2012/273549.273549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Schultz-Norton JR, McDonald WH, Yates JR, Nardulli AM. Protein disulfide isomerase serves as a molecular chaperone to maintain estrogen receptor α structure and function. Molecular Endocrinology. 2006;20(9):1982–1995. doi: 10.1210/me.2006-0006. [DOI] [PubMed] [Google Scholar]
- 214.Tager M, Kroning H, Thiel U, Ansorge S. Membrane-bound proteindisulfide isomerase (PDI) is involved in regulation of surface expression of thiols and drug sensitivity of B-CLL cells. Experimental Hematology. 1997;25(7):601–607. [PubMed] [Google Scholar]
- 215.Swiatkowska M, Szymański J, Padula G, Cierniewski CS. Interaction and functional association of protein disulfide isomerase with αvβ3 integrin on endothelial cells. FEBS Journal. 2008;275(8):1813–1823. doi: 10.1111/j.1742-4658.2008.06339.x. [DOI] [PubMed] [Google Scholar]
- 216.Park B, Lee S, Kim E, et al. Redox regulation facilitates optimal peptide selection by MHC class I during antigen processing. Cell. 2006;127(2):369–382. doi: 10.1016/j.cell.2006.08.041. [DOI] [PubMed] [Google Scholar]
- 217.Goplen D, Wang J, Enger PØ, et al. Protein disulfide isomerase expression is related to the invasive properties of malignant glioma. Cancer Research. 2006;66(20):9895–9902. doi: 10.1158/0008-5472.CAN-05-4589. [DOI] [PubMed] [Google Scholar]