

Abstract
Previous studies comparing early-onset Alzheimer’s disease (EOAD) and late-onset AD (LOAD) have been limited by cross-sectional design and a focus on isolated clinical variables. This study aims to explore differentials in clinical features between EOAD and LOAD and to examine longitudinally trends in cognitive function. Data from 3,747 subjects with AD from C-Path Online Data Repository was used to compare demographics, body mass index (BMI), mean arterial pressure (MAP), biochemistry and cognitive assessments, including mini-mental state examination (MMSE) and Alzheimer’s Disease Assessment Scale-cognitive subscale (ADAS-Cog), between EOAD and LOAD. The baseline differences were examined by binominal proportion test and t-test. The trends of cognitive functions, evaluating by MMSE and ADAS-Cog, were examined by the mixed model, controlling for the effect of repeated measures of the same person. No significant difference was found in BMI and MAP. C-reactive protein, creatinine and blood urea nitrogen (BUN) (p<0.05) were significantly higher in LOAD. The APOE ε4 alleles was more likely to be found among LOAD compared to APOE ε2 or APOE ε3. EOAD had significantly lower MMSE at baseline and this difference significantly increased over time. Despite an insignificant differential in ADAS-Cog between EOAD and LOAD at baseline, the differential was enlarged gradually and became more significant with time. Our findings suggest that elevated inflammatory markers, impaired renal function and APOE ε4 alleles are overrepresented in LOAD, possibly indicating that different factors determine the development of EOAD and its more rapid cognitive deterioration.
Keywords: Early-onset dementia, late-onset dementia, Alzheimer’s disease, cognitive assessments, EOAD prognosis
Introduction
The increasing health burden of Alzheimer’s disease (AD), both social and economic, is an international problem [1], particularly in low and middle income countries [2]. In America, there were more than 5 million people living with AD in 2012, and their health expenditure was estimated to exceed $ 200 billion [3]. Despite AD mostly occurs in the elderly, a small proportion of AD occurred before the age of 65, which is called early onset AD (EOAD), and is considered to have a more aggressive course and shorter relative survival time [4]. A more variable presentation including posterior cortical atrophy, frontal variants and linguistic presentations are also recognised - even though the amnesic presentation is observed, it is less common, a conundrum that might lead to incorrect diagnosis [5]. Furthermore, the pathology in EOAD may be more severe with prominent synaptic fallout and neuronal loss [6]. EOAD is also characterized by more severe perfusion and metabolic defects [7].
Despite evidence suggesting differences between EOAD and LOAD, previous investigations are inadequate as they examined isolated clinical features or cognitive functions. The small sample sizes in these investigations limit the value of those differences between EOAD and LOAD. Furthermore, much of the evidence relies on cross-sectional design, particularly the cognitive functions, which is not conclusive. As the disease burden of AD is increasing with aging society, it is important to understand the aetiology and characteristics of EOAD with sufficient sample size and strong epidemiological design. The findings may shed the light on the detection of EOAD, which is more difficult to diagnose and help improve our understanding of AD in general [5].
This study attempts to improve the understanding of EOAD by analyzing a large, global database to answer the following questions: (i) do patients with EOAD have different clinical characteristics from LOAD? (ii) are they biochemically different? (iii) is their APOE genotyping similar to LOAD? and (iv) do they have a different cognitive trajectory?
A large relatively uniform database was required - the Coalition Against Major Diseases (CAMD) has made the C-Path online data repository available. This contains data on over 3,000 patients given placebo from ten AD clinical trials - this data was analysed to provide insights into the differences between EOAD and LOAD, to better understand the nature of EOAD and its origins.
Materials and methods
Database
The CAMD has made the C-Path Online Data Repository (CODR) available for analysis. The dataset is composed of the placebo arm of 10 clinical trials from 7 major pharmaceutical companies into AD; the trials vary in length from 3 months to 2 years; the data is de-identified and contains information on 3,747 subjects (data was retrieved on 16 Feb 2012). Subjects had given informed written consent for entry into the clinical trials according to good clinical research practice guidelines.
The CAMD Data Working Group, in association with the Clinical Data Interchange Students Consortium (CDISC), has developed standards for collecting, sorting and interchanging AD clinical trial data. The companies reworked the clinical trial data into a new format and agreed to share the information to aid research into AD. Ephibian, an information technology solutions company, have transformed the data into a workable database.
The database was interrogated by demographics, medical history, subject characteristics, body mass index (BMI), mean blood pressure (MAP), apolipoprotein E genotypes and alleles (APOE), and laboratory measures (biochemistry). Data for cerebrospinal fluid analysis and neuroimaging were not available at the time of this analysis. Cognitive functions of individuals were assessed by Mini mental state examination (MMSE) and Alzheimer’s Disease Assessment Scale - Cognitive (ADAS-Cog) at 9 of 10 visits (data at the third visit was not available). The completeness of data in each trial varied as it is not compulsory to report all information in the C-Path online data repository.
Statistical analysis
The chi-square test was used to compare the distributions of demographics by EOAD and LOAD. The unpaired t-test was used to examine differences in clinical features including BMI, MAP, biochemistries (e.g. C-reactive protein and creatinine). Allele and genotype frequencies were analysed using the binominal proportion test. The mixed model was used to examine the trend of MMSE and ADAS-Cog data over time, adjusting for patients from the same clinical trial. All statistical analyses were conducted by SAS 9.2.
Results
Of 3,747 AD patients, EOAD accounted for 16.4%. Table 1 shows the distributions of demographics by EOAD and LOAD. The proportion of males was similar in both groups, which accounted for approximately 45% of the total ADs. The majority of ADs were Whites (over 90%), and the proportion of EOAD were from other races (native American Indian, Alaskan, Hawaiian and others) was higher than that of LOAD. A significantly higher proportion of LOAD patients reported to have a first degree relative with AD.
Table 1.
The demographics of the study population: late-onset and early onset of Alzheimer’s disease
| All (n=3747) | LOAD (n=3133) | EOAD (n=614) | |||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|||||||||
| N | % | N | % | N | % | Chi-square | p-value | ||
| Gender | Male | 1672 | 44.6 | 1385 | 44.2 | 287 | 46.7 | χ²=1.36 | 0.25 |
| Race | While | 3467 | 92.5 | 2913 | 93.0 | 554 | 90.2 | χ²=14.8 | 0.002 |
| Asian | 173 | 4.6 | 143 | 4.6 | 30 | 4.9 | |||
| Black | 45 | 1.2 | 36 | 1.2 | 9 | 1.5 | |||
| Others* | 62 | 1.7 | 41 | 1.3 | 21 | 3.4 | |||
| Parental/Sibling AD | Yes | 1147 | 30.6 | 973 | 31.1 | 174 | 28.3 | χ²=1.79 | 0.18 |
| First degree relative AD | Yes | 2510 | 67.0 | 2157 | 68.9 | 353 | 57.5 | χ²=29.94 | <0.0001 |
Including native American Indians, Alaskans and Hawaiians.
The BMI and MAP were not different between the EOAD versus LOAD groups at baseline (Table 2). The laboratory measures of C-reactive protein (CRP), creatinine and blood urea nitrogen (BUN) were significantly higher in the LOAD population (Table 2). The differences in the blood glucose and lipid studies, B12 and red blood cell folate, thyroid function and other measures were not significant (Table 2).
Table 2.
BMI, mean arterial pressure and biochemical differences between EOAD and LOAD
| LOAD | EOAD | T-test p-value | |||||
|---|---|---|---|---|---|---|---|
|
|
|||||||
| n | Mean | STD | n | Mean | STD | ||
| BMI | 1211 | 25.3 | 3.97 | 237 | 25.5 | 4.32 | 0.58 |
| MAP* | 1217 | 95.0 | 9.50 | 237 | 95.2 | 9.81 | 0.68 |
| C-reactive Protein (mg/L) | 169 | 2.21 | 2.218 | 32 | 1.71 | 0.929 | 0.03 |
| Creatinine (umol/L) | 299 | 0.0835 | 0.0200 | 61 | 0.0765 | 0.0151 | 0.003 |
| Blood Urea Nitrogen (mmol/L) | 294 | 6.26 | 1.656 | 61 | 5.56 | 1.761 | 0.003 |
| Glucose (mmol/L) | 180 | 5.2 | 0.86 | 35 | 5.1 | 0.81 | 0.58 |
| Cholesterol (mmol/L) | 175 | 5.8 | 0.86 | 34 | 6.0 | 0.96 | 0.12 |
| Triglyceride (mmol/L) | 174 | 1.6 | 0.81 | 34 | 1.4 | 0.68 | 0.26 |
| HDL (mg/dL) | 162 | 62.5 | 18.44 | 32 | 66.6 | 17.5 | 0.25 |
| LDL (mg/dL) | 162 | 137.1 | 25.6 | 32 | 143.3 | 28.7 | 0.22 |
| VLDL (mmol/L) | 161 | 0.61 | 0.409 | 31 | 0.54 | 0.335 | 0.39 |
| Apolipoprotein E (g/L) | 155 | 0.04 | 0.012 | 32 | 0.04 | 0.013 | 0.89 |
| Apolipoprotein B (g/L) | 160 | 1.10 | 0.215 | 32 | 1.12 | 0.218 | 0.57 |
| Vitamin B12 (pmol/L) | 277 | 374.7 | 236.00 | 57 | 389.3 | 224.10 | 0.67 |
| TSH (mu/L) | 292 | 1.78 | 1.132 | 55 | 1.94 | 2.919 | 0.70 |
| Total Thyroxine (nmol/L) | 116 | 102.6 | 18.76 | 22 | 101.7 | 15.54 | 0.84 |
| Free Thyroxine (nmol/L) | 161 | 13.9 | 2.36 | 32 | 14.5 | 2.67 | 0.20 |
MAP: mean arterial pressure=(2×diastolic pressure) + systolic pressure]/3.
The distribution of APOE allele1*allele2 by EOAD and LOAD patients is presented in Table 3A and 3B. Most ADs were found to have a combination of APOE3*APOE3 or APOE3*APOE4, which accounted for 69% of total EOAD and 75% of total LOAD. The proportion of APOE2 seems to be more frequently found among EOADs (EOAD 18% vs. LOAD 10%). Among LOAD patients, the proportion of APOE4 was significantly higher compared with APOE2 or APOE3. This tendency was not found among EOADs.
Table 3A.
The distributions of APOE Allele1*Allele2 by LOAD vs. EOAD
| All | LOAD | EOAD | ||||
|---|---|---|---|---|---|---|
|
|
||||||
| Allele1×Allele2 | N | % | N | % | N | % |
| 2×2 | 2 | 0.12 | 2 | 0.15 | 0 | 0.00 |
| 2×3 | 79 | 4.93 | 59 | 4.35 | 20 | 8.10 |
| 2×4 | 121 | 7.55 | 94 | 6.94 | 27 | 10.93 |
| 3×3 | 537 | 33.52 | 451 | 33.28 | 86 | 34.82 |
| 3×4 | 662 | 41.32 | 577 | 42.58 | 85 | 34.41 |
| 4×4 | 201 | 12.55 | 172 | 12.69 | 29 | 11.74 |
| All | 1602 | 100.00 | 1355 | 100.00 | 247 | 100.00 |
Table 3B.
Binomial proportion test
| LOAD | EOAD | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
|
||||||||||
| Allele1×Allele2 | N | % | 95% CI | p-value | N | % | 95% CI | p-value | ||
| 2×3 | 59 | 38.56 | 0.31 | 0.47 | 0.005 | 20 | 42.55 | 0.28 | 0.58 | 0.3 |
| 2×4 | 94 | 61.44 | 27 | 57.45 | ||||||
| 3×3 | 451 | 43.87 | 0.41 | 0.47 | <0.0001 | 86 | 50.29 | 0.43 | 0.58 | 0.94 |
| 3×4 | 577 | 56.13 | 85 | 49.71 | ||||||
The results of mixed models of the cognitive assessments are presented in Table 4. Table 5 shows cognitive assessments at each visit for LOAD vs EOAD. Compared to LOAD, EOAD had significantly lower MMSE at baseline (EOAD 19.3 vs. LOAD 20.9, p<0.05) and this difference was gradually enlarged over time with a significant time effect (p<0.0001). The significant interaction between time and MMSE (p<0.05) indicated that EOAD has lower MMSE status compared to LOAD and this difference increases with time. In contrast, the difference in ADAS-Cog was not significant at baseline (EOAD 33.6 vs. LOAD 29.9, p=0.28). Nevertheless, the difference was significant at the conclusion of the studies, with a significant interaction between ADAS-Cog and time (p=0.04). This finding was consistent with the MMSE data and reveals that compared to LOAD, EOAD has greater cognitive deterioration with time. The trends of MMSE and ADAS-Cog over time are illustrated in Figure 1.
Table 4.
The effect of EOAD (vs. LOAD ) and time on cognitive assessments by the mixed model (repeated measure with correlation type=UN)
| p-value | |
|---|---|
| MMSE | |
| EOAD vs. LOAD | <0.05 |
| Time | <0.0001 |
| EOAD*Time | <0.05 |
| ADAS-Cog | |
| EOAD vs. LOAD | 0.28 |
| Time | <0.0001 |
| EOAD*Time | 0.04 |
Table 5.
Cognitive assessments at each visit by LOAD vs. EOAD: 9 visits at approximately 2-monthly intervals over 2 years
| MMSE | LOAD | EOAD | ||||
|
|
||||||
| n | Mean | STD | n | Mean | STD | |
|
| ||||||
| 1st Visit | 56 | 20.9 | 3.42 | 7 | 19.3 | 3.55 |
| 2nd Visit | 56 | 20.9 | 4.02 | 7 | 19.1 | 3.76 |
| 4th Visit | 52 | 21.6 | 4.22 | 6 | 19.8 | 3.43 |
| 5th Visit | 49 | 20.4 | 4.43 | 6 | 17.2 | 5.08 |
| 6th Visit | 45 | 20.2 | 4.95 | 6 | 18.0 | 4.47 |
| 7th Visit | 44 | 20.6 | 4.83 | 5 | 14.4 | 3.78 |
| 8th Visit | 40 | 20.1 | 5.87 | 4 | 14.8 | 7.41 |
| 9th Visit | 46 | 19.4 | 5.67 | 4 | 14.0 | 3.92 |
| 10th Visit | 39 | 19.1 | 5.85 | 4 | 12.0 | 4.69 |
|
| ||||||
| ADAS-Cog | n | Mean | STD | n | Mean | STD |
|
| ||||||
| 1st Visit | 56 | 29.9 | 11.03 | 7 | 33.6 | 8.58 |
| 2nd Visit | 56 | 31.2 | 10.87 | 7 | 31.0 | 10.35 |
| 4th Visit | 52 | 31.2 | 10.18 | 6 | 32.1 | 11.39 |
| 5th Visit | 49 | 30.3 | 11.59 | 6 | 35.0 | 11.63 |
| 6th Visit | 45 | 33.0 | 13.04 | 6 | 36.5 | 11.11 |
| 7th Visit | 44 | 33.5 | 12.48 | 5 | 42.1 | 11.81 |
| 8th Visit | 41 | 34.2 | 13.92 | 4 | 50.8 | 10.63 |
| 9th Visit | 46 | 35.2 | 14.18 | 4 | 49.6 | 9.41 |
| 10th Visit | 37 | 33.1 | 14.06 | 4 | 51.4 | 13.78 |
Figure 1.
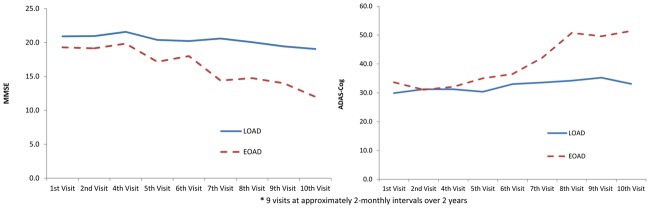
The trend of MMSE and ADAS-Cog over time by LOAD versus EOAD.
Discussion
This study has examined the differences between EOAD and LOAD using some clinical features, biochemistry data, APOE types and cognitive assessment for a large cohort of ADs mainly from North America and Europe. Although a proportion of the cohort had incomplete data, probably due to different design and protocol of each trial, this study found that EOAD had different neurobiological features compared to older AD patients.
This study did not find significant difference in BMI and MAP between EOAD and LOAD. While previous studies suggested that lower BMI [8] and higher blood pressure [9] were risk factors for developing a dementia or AD among the older population; the role of BMI and blood pressure among EOAD requires further investigation, as some studies suggest there might not be a relationship [10].
Significantly increased levels of CRP, creatinine and BUN were found among LOAD versus EOAD. The CRP is an acute phase reactant produced by the liver in response to injury, infection or inflammation. Inflammatory mechanisms are believed to have a role in cognitive decline and AD in the elderly [11,12]. Our observations demonstrate an increase in CRP in LOAD in comparison to the younger onset patients, suggesting inflammatory mechanisms might be more important in older than younger people with AD. However, older patients have an increased risk for infection in general, including urine and periodontal disease [13]. It is possible that the increase in CRP might be a consequence of increased infection burden in the elderly and not implicated in the pathophysiology of AD [14]. Furthermore, some studies suggest the CRP might decrease in AD, after midlife elevations [15]. It cannot be discounted that in LOAD patients that, in comparison to EOAD, inflammatory mechanisms might be more important.
Both creatinine and BUN are indicators of renal health. Renal impairment is associated with vascular ageing and vascular complications as a result of accelerated atherosclerosis [16]. Patients with chronic renal disease are at increased risk of stroke [17]. The significantly impaired renal function in the LOAD group in our study might just be a consequence of age; however, kidney dysfunction might promote cerebrovascular pathology and AD in the elderly. Furthermore, patients with end-stage renal disease receiving peritoneal dialysis have excessive white hyperintensities as a result of small vessel ischaemic disease of the brain, which might trigger proinflammatory and endothelial reactions resulting in AD [18]; such a mechanistic pathway might not be relevant in younger patients.
Genetic factors are important in determining a person’s risk of developing AD: rare mutations in APP and PSN 1 and 2 cause autosomal dominant familial AD of early onset; polymorphisms in genes like APOE represent a major population risk factor for the development of both EOAD and LOAD [10,19] - polymorphism in other genes confer less of a risk (eg, PICALM, CR1, SORLI). Our findings suggest that APOE4 allele is a more important risk factor for LOAD - a variable which interacts with cerebrovascular risk factors, proinflammatory mediators, renal dysfunction in older but not younger populations; other environmental and genetic factors might be conferring the earlier onset of dementia and more rapid progression in younger adults. Interestingly, APOE ε4 carriers have a load effect in comparison to non ε4 carriers on brain structure, functional activation, brain glucose metabolism and other variables [20]. However, these factors might not be operational in younger patients. We did observe that APOE ε2 alleles and genotypes might be overrepresented in younger onset patients - a finding which supports earlier observations that APOE ε2 allele is associated with increased risk and reduced survival in EOAD [21] and does not seem to be protective of its development as suggested by Corder et al. [22] and Panza et al [23]. The absence of the APOE ε4 allele in EOAD might determine atypical presentations of EOAD including posterior cortical atrophy syndrome, linguistic presentations and frontal lobe syndromes; the presence of APOE allele possibly determining mesial temporal involvement and the typical episodic memory impairment of AD [24].
Despite the relatively small sample size, EOAD patients showed a more rapid rate of cognitive decline using both the ADAS-Cog and MMSE in comparison to LOAD. This study allowed access to multiple follow-up data and to implement a sophisticated statistical method to examine the effect of time on cognitive function in EOAD and LOAD. Our observations are supported by others with larger sample sizes [4,25,26]. Our findings might relate to observations showing that there is more cortical atrophy, hypoperfusion and hypometabolism in EOAD [27,28]. Furthermore, there might be more advanced neuritic plaque and neurofibrillary burden in EOAD [6,29,30] and neurochemical differences with greater deficits in acetylcholine and adrenaline [31,32].
The findings of this study should be interpreted with caution. First, most data was not complete for all ADs in the data repository. Nevertheless, the C-Path online data repository (CODR) only provides a platform for sharing information which facilitates AD research: for example, data collection for the MMSE may only be collected in one but not other trials. CODR may need to use more restricted guidelines to include and exclude participating studies/trials. In spite of this limitation, the sample size on most measures is considerably larger than other AD studies and the findings are also consistent with others. Another limitation of this study is the difficulty in teasing out an age effect. For instance, the CRP was found to be higher among the older population; nonetheless, it is unlikely to reveal if an increased CRP among LOAD is due to age alone or relates to true biological differences between EOAD and LOAD. Selecting an age-matched, non AD cohort would help clarify this issue.
Conclusion
In conclusion, our investigations suggest that CRP, creatinine and BUN were higher among LOAD and the proportion of LOAD with APOE ε4 was greater. The proportion of EOAD with APOE ε2 allele was greater than that of LOAD, suggesting a possible genetic difference between EOAD and LOAD. Further investigations correlating our findings with CSF analysis of Aβ and tau, in combination with neuroimaging including MRI and amyloid scans, will help to elucidate these differences in future studies, when this data becomes available on the C-Path online repository. Recent studies suggest that EOAD is not related to amyloid burden using Pittsburgh compound B scans [33]; therefore, amyloid mechanisms possibly involving Aβ oligomer function, and not deposition, might be more important in EOAD [34]. This study also support observations that EOAD has a more aggressive cognitive deterioration than LOAD. This finding implies that patients with EOAD require earlier diagnosis and intervention.
Disclosure of conflict of interest
The authors of this paper have no conflict of interest to declare.
References
- 1.Xu W, Ferrari C, Wang HX. In: Epidemiology of Alzheimer’s Disease. Inga Z, editor. InTech: Understanding Alzheimer’s Disease; 2013. [Google Scholar]
- 2.Prince M, Bryce R, Albanese E, Wimo A, Ribeiro W, Ferri CP. The global prevalence of dementia: A systematic review and metaanalysis. Alzheimers Dement. 2013;9:63–75. e62. doi: 10.1016/j.jalz.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 3.Alzheimer’s Association. 2012 Alzheimer’s disease facts and figures. Alzheimers Dement. 2012;8:131–168. doi: 10.1016/j.jalz.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 4.Seltzer B, Sherwin I. A comparison of clinical features in early- and late-onset primary degenerative dementia. One entity or two? Arch Neurol. 1983;40:143–146. doi: 10.1001/archneur.1983.04050030037006. [DOI] [PubMed] [Google Scholar]
- 5.Balasa M, Gelpi E, Antonell A, Rey MJ, Sánchez-Valle R, Molinuevo JL, Lladó A Neurol-ogical Tissue Bank/University of Barcelona/Hospital Clínic NTB/UB/HC Collaborative Group. Clinical features and APOE genotype of pathologically proven early-onset Alzheimer disease. Neurology. 2011;76:1720–1725. doi: 10.1212/WNL.0b013e31821a44dd. [DOI] [PubMed] [Google Scholar]
- 6.Nochlin D, van Belle G, Bird TD, Sumi SM. Comparison of the severity of neuropathologic changes in familial and sporadic Alzheimer’s disease. Alzheimer Dis Assoc Disord. 1993;7:212–222. [PubMed] [Google Scholar]
- 7.Yasuno F, Imamura T, Hirono N, Ishii K, Sasaki M, Ikejiri Y, Hashimoto M, Shimomura T, Yamashita H, Mori E. Age at onset and regional cerebral glucoseh metabolism in Alzheimer’s disease. Dement Geriatr Cogn Disord. 1998;9:63–67. doi: 10.1159/000017024. [DOI] [PubMed] [Google Scholar]
- 8.Buchman AS, Wilson RS, Bienias JL, Shah RC, Evans DA, Bennett DA. Change in body mass index and risk of incident Alzheimer disease. Neurology. 2005;65:892–897. doi: 10.1212/01.wnl.0000176061.33817.90. [DOI] [PubMed] [Google Scholar]
- 9.Skoog I, Nilsson L, Persson G, Lernfelt B, Landahl S, Palmertz B, Andreasson LA, Odén A, Svanborg A. 15-year longitudinal study of blood pressure and dementia. Lancet. 1996;347:1141–1145. doi: 10.1016/s0140-6736(96)90608-x. [DOI] [PubMed] [Google Scholar]
- 10.Atkins ER, Bulsara MK, Panegyres PK. Cerebrovascular risk factors in early-onset dementia. J Neurol Neurosurg Psychiatry. 2012;83:666–667. doi: 10.1136/jnnp.2009.202846. [DOI] [PubMed] [Google Scholar]
- 11.Dik MG, Jonker C, Hack CE, Smit JH, Comijs HC, Eikelenboom P. Serum inflammatory proteins and cognitive decline in older persons. Neurology. 2005;64:1371–1377. doi: 10.1212/01.WNL.0000158281.08946.68. [DOI] [PubMed] [Google Scholar]
- 12.Engelhart MJ, Geerlings MI, Meijer J, Kiliaan A, Ruitenberg A, van Swieten JC, Stijnen T, Hofman A, Witteman JC, Breteler MM. Inflammatory proteins in plasma and the risk of dementia: the rotterdam study. Arch Neurol. 2004;61:668–672. doi: 10.1001/archneur.61.5.668. [DOI] [PubMed] [Google Scholar]
- 13.Kamer AR, Craig RG, Dasanayake AP, Brys M, Glodzik-Sobanska L, de Leon MJ. Inflammation and Alzheimer’s disease: possible role of periodontal diseases. Alzheimers Dement. 2008;4:242–250. doi: 10.1016/j.jalz.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 14.Nordestgaard BG, Zacho J. Lipids, atherosclerosis and CVD risk: is CRP an innocent bystander? Nutr Metab Cardiovasc Dis. 2009;19:521–524. doi: 10.1016/j.numecd.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 15.O’Bryant SE, Waring SC, Hobson V, Hall JR, Moore CB, Bottiglieri T, Massman P, Diaz-Arrastia R. Decreased C-reactive protein levels in Alzheimer disease. J Geriatr Psychiatry Neurol. 2010;23:49–53. doi: 10.1177/0891988709351832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kutyrina IM, Rudenko TE, Shvetsov M, Kushnir VV. [Risk factors of vascular complications in patients at a predialysis stage of chronic renal failure] . Ter Arkh. 2006;78:45–50. [PubMed] [Google Scholar]
- 17.Fernandez-Andrade C. [Renal markers and predictors, and renal and cardiovascular risk factors] . Nefrologia. 2002;22(Suppl 1):2–29. [PubMed] [Google Scholar]
- 18.Kim CD, Lee HJ, Kim DJ, Kim BS, Shin SK, Do JY, Jang MH, Park SH, Kim YS, Kim YL. High prevalence of leukoaraiosis in cerebral magnetic resonance images of patients on peritoneal dialysis. Am J Kidney Dis. 2007;50:98–107. doi: 10.1053/j.ajkd.2007.03.019. [DOI] [PubMed] [Google Scholar]
- 19.Atkins ER, Panegyres PK. The clinical utility of gene testing for Alzheimer’s disease. Neurol Int. 2011;3:e1. doi: 10.4081/ni.2011.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bendlin BB, Ries ML, Canu E, Sodhi A, Lazar M, Alexander AL, Carlsson CM, Sager MA, Asthana S, Johnson SC. White matter is altered with parental family history of Alzheimer’s disease. Alzheimers Dement. 2010;6:394–403. doi: 10.1016/j.jalz.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Duijn CM, de Knijff P, Wehnert A, De Voecht J, Bronzova JB, Havekes LM, Hofman A, Van Broeckhoven C. The apolipoprotein E epsilon 2 allele is associated with an increased risk of early-onset Alzheimer’s disease and a reduced survival. Ann Neurol. 1995;37:605–610. doi: 10.1002/ana.410370510. [DOI] [PubMed] [Google Scholar]
- 22.Corder EH, Saunders AM, Risch NJ, Strittmatter WJ, Schmechel DE, Gaskell PC Jr, Rimmler JB, Locke PA, Conneally PM, Schmader KE, et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet. 1994;7:180–184. doi: 10.1038/ng0694-180. [DOI] [PubMed] [Google Scholar]
- 23.Panza F, Solfrizzi V, Torres F, Mastroianni F, Colacicco AM, Basile AM, Capurso C, D’Introno A, Del Parigi A, Capurso A. Apolipoprotein E in Southern Italy: protective effect of epsilon 2 allele in early- and late-onset sporadic Alzheimer’s disease. Neurosci Lett. 2000;292:79–82. doi: 10.1016/s0304-3940(00)01447-6. [DOI] [PubMed] [Google Scholar]
- 24.van der Flier WM, Pijnenburg YA, Fox NC, Scheltens P. Early-onset versus late-onset Alzheimer’s disease: the case of the missing APOE epsilon4 allele. Lancet Neurol. 2011;10:280–288. doi: 10.1016/S1474-4422(10)70306-9. [DOI] [PubMed] [Google Scholar]
- 25.Jacobs D, Sano M, Marder K, Bell K, Bylsma F, Lafleche G, Albert M, Brandt J, Stern Y. Age at onset of Alzheimer’s disease: relation to pattern of cognitive dysfunction and rate of decline. Neurology. 1994;44:1215–1220. doi: 10.1212/wnl.44.7.1215. [DOI] [PubMed] [Google Scholar]
- 26.Smits LL, Pijnenburg YA, Koedam EL, van der Vlies AE, Reuling IE, Koene T, Teunissen CE, Scheltens P, van der Flier WM. Early onset Alzheimer’s disease is associated with a distinct neuropsychological profile. J Alzheimers Dis. 2012;30:101–108. doi: 10.3233/JAD-2012-111934. [DOI] [PubMed] [Google Scholar]
- 27.Frisoni GB, Testa C, Sabattoli F, Beltramello A, Soininen H, Laakso MP. Structural correlates of early and late onset Alzheimer’s disease: voxel based morphometric study. J Neurol Neurosurg Psychiatry. 2005;76:112–114. doi: 10.1136/jnnp.2003.029876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim EJ, Cho SS, Jeong Y, Park KC, Kang SJ, Kang E, Kim SE, Lee KH, Na DL. Glucose metabolism in early onset versus late onset Alzheimer’s disease: an SPM analysis of 120 patients. Brain. 2005;128:1790–1801. doi: 10.1093/brain/awh539. [DOI] [PubMed] [Google Scholar]
- 29.Bigio EH, Hynan LS, Sontag E, Satumtira S, White CL. Synapse loss is greater in presenile than senile onset Alzheimer disease: implications for the cognitive reserve hypothesis. Neuropathol Appl Neurobiol. 2002;28:218–227. doi: 10.1046/j.1365-2990.2002.00385.x. [DOI] [PubMed] [Google Scholar]
- 30.Marshall GA, Fairbanks LA, Tekin S, Vinters HV, Cummings JL. Early-onset Alzheimer’s disease is associated with greater pathologic burden. J Geriatr Psychiatry Neurol. 2007;20:29–33. doi: 10.1177/0891988706297086. [DOI] [PubMed] [Google Scholar]
- 31.Bird TD, Stranahan S, Sumi SM, Raskind M. Alzheimer’s disease: choline acetyltransferase activity in brain tissue from clinical and pathological subgroups. Ann Neurol. 1983;14:284–293. doi: 10.1002/ana.410140306. [DOI] [PubMed] [Google Scholar]
- 32.Rossor MN, Iversen LL, Reynolds GP, Mountjoy CQ, Roth M. Neurochemical characteristics of early and late onset types of Alzheimer’s disease. Br Med J (Clin Res Ed) 1984;288:961–964. doi: 10.1136/bmj.288.6422.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rabinovici GD, Furst AJ, Alkalay A, Racine CA, O’Neil JP, Janabi M, Baker SL, Agarwal N, Bonasera SJ, Mormino EC, Weiner MW, Gorno-Tempini ML, Rosen HJ, Miller BL, Jagust WJ. Increased metabolic vulnerability in early-onset Alzheimer’s disease is not related to amyloid burden. Brain. 2010;133:512–28. doi: 10.1093/brain/awp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Braak H, Del Tredici K. Alzheimer’s disease: pathogenesis and prevention. Alzheimers Dement. 2012;8:227–33. doi: 10.1016/j.jalz.2012.01.011. [DOI] [PubMed] [Google Scholar]