

Abstract
Genome-wide association studies show strong evidence of association with endometriosis for markers on chromosome 1p36 spanning the potential candidate genes WNT4, CDC42 and LINC00339. WNT4 is involved in development of the uterus, and the expression of CDC42 and LINC00339 are altered in women with endometriosis. We conducted fine mapping to examine the role of coding variants in WNT4 and CDC42 and determine the key SNPs with strongest evidence of association in this region. We identified rare coding variants in WNT4 and CDC42 present only in endometriosis cases. The frequencies were low and cannot account for the common signal associated with increased risk of endometriosis. Genotypes for five common SNPs in the region of chromosome 1p36 show stronger association signals when compared with rs7521902 reported in published genome scans. Of these, three SNPs rs12404660, rs3820282, and rs55938609 were located in DNA sequences with potential functional roles including overlap with transcription factor binding sites for FOXA1, FOXA2, ESR1, and ESR2. Functional studies will be required to identify the gene or genes implicated in endometriosis risk.
Keywords: Endometriosis, WNT4, CDC42, chromosome 1p36, rare variants, common variants
Introduction
Large gene mapping studies in endometriosis have consistently identified strong association with disease risk for markers in the region close to wingless-type MMTV integration site family, member 4 (WNT4) on chromosome 1 [1-4]. The single nucleotide marker with the strongest signal rs7521902 identified in the original studies [1,2] is located approximately 20 kb upstream from the transcription start site for WNT4. This is an interesting region as rs7521902 is also implicated in risk for ovarian cancer [5], bone mineral density and risk of fracture [6]. Subsequent studies in endometriosis cases and controls re-analysed data for a higher density of SNPs across this region with genotypes estimated by imputation [3,4]. Results provide strong evidence for replication of association in the region of WNT4 with the highest signals located within the WNT4 genomic sequence. However, the region of strong association extends across adjacent genes and spans the region including the long intergenic non-protein coding RNA LINC00339, cell division cycle 42 (CDC42) and WNT4 [3,4].
WNT4 is a strong candidate for functional changes increasing risk for endometriosis and ovarian cancer. Expression of WNT4 is critical for development of the female reproductive tract [7,8]. WNT4 and other WNT family members are expressed in human endometrium during both the proliferative and secretory phases [9] and expression is up-regulated by oestrogen in an oestrogen receptor independent manner [10]. However, adjacent genes within the DNA block showing association include CDC42 and LINC00339, both implicated in endometriosis. CDC42 is a small GTPase of the Rho-subfamily, which regulates signalling pathways that control diverse cellular functions including cell morphology, migration, endocytosis and cell cycle progression. The gene is expressed in the endometrium and is reported to be differentially expressed in endometriosis [11]. LINC00339 (also known as HSPC157) is differentially expressed in endometriosis lesions versus autologous uterine endometrium [12]. Both CDC42 and LINC00339 must also be considered as candidates for the action of genetic variants on endometriosis risk.
Effects of individual SNPs on disease risk are small and determining the specific genes and pathways affecting disease risk is the best approach to understand mechanisms leading to endometriosis. The aims of this study were to genotype SNPs in the exons of WNT4 and CDC42 to determine whether the association signal could be explained by coding variants, and to genotype common non-coding variants previously implicated from imputed data to confirm results for the best SNPs located in the region of WNT4. We also conducted in silico analyses to evaluate possible functional roles of our top 50 non-coding variants across the region.
Materials and methods
The coding regions of WNT4 were screened for variants in 100 unrelated individuals chosen from 100 case-dense families drawn from the QIMR Berghofer Medical Research Institute dataset [13]. They include 15 families with at least four affected sisters, 71 families with three affected sisters and 14 families with at least two sisters and two other relatives diagnosed with endometriosis. Cases for genotyping were one sample per family chosen as the case with the most severe stage of disease in each family.
Coding variants were genotyped in samples from 958 endometriosis cases with a family history drawn from our Australian study of endometriosis [13,14]. All cases had surgically confirmed endometriosis and disease severity including four-stages (I-IV) was assessed using the revised American Fertility Society (rAFS) classification system [15]. The control group included 959 unrelated controls drawn from women recruited for a study of twins who self-reported that they had never been diagnosed with endometriosis [14]. After removing individuals in case families with non-Caucasian ancestry revealed by our GWA studies [16], 930 cases and 959 controls were included in our analyses.
Ethical approvals
Study protocols were reviewed and approved by the QIMR Human Research Ethics Committee. Participation was voluntary and each participant gave written informed consent.
High Resolution Melt (HRM) assay and sequencing
We screened for coding variants in the exons of WNT4 in DNA from 100 endometriosis cases using a High Resolution Melting (HRM) technique [17]. WNT4 PCR primers were designed to amplify the 5’ and 3’UTRs, non-coding and coding exons, including at least 50 bp of intronic sequence either side of each exon to cover intron-exon boundaries using the Primer 3.0 program [18]. PCR products were screened on a Rotor-Gene 6000 Real-time Rotary Analyser (Corbett Research, QIAGEN, Hilden, Germany). Melting curves were analysed using the Rotor-Gene 6000 analysis software v 1.7 (Corbett Research). Samples showing patterns different from wild-type were sequenced using BigDye 3.0 terminator chemistry (Applied Biosystems).
We also searched for coding variants in both WNT4 and CDC42 using public databases with results from extensive exome sequencing including dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/), 1000 Genomes (http://browser.1000genomes.org/), the NHLBI GO Exome Sequencing Project (ESP, http://evs.gs.washington.edu/EVS/), and the exome chip genotyping array design (http://genome.sph.umich.edu/wiki/Exome_Chip_Design).
Genotyping
Twenty seven coding variants in WNT4, five coding and four potential functional variants (located in the promoter area and splice sites) in CDC42, and six key imputed SNPs identified from the GWA studies [3] were genotyped using Sequenom MassARRAY technology in the 930 cases and 959 controls. Genotyping assays were designed using standard procedures and SNPs were typed using iPLEX™ chemistry on a MALDI-TOF Mass Spectrometer (Sequenom Inc., San Diego, CA, USA) [14].
Data analysis
PLINK software was used to analyse SNP data for quality control measures and to test for association between SNPs and endometriosis risk. Call rates were all greater than 95% and all SNPs were in Hardy-Weinberg equilibrium (P > 0.05). Association tests for endometriosis risk were performed in PLINK using the standard chi-square (--assoc) test for common variants and Fisher’s exact (--fisher) test for low frequency variants [19].
To assess the combined contribution of multiple rare variants towards endometriosis risk, multiple logistic regression was performed. Briefly, genotypes for each SNP were re-coded to allele dosages using PLINK’s --recodeA option (i.e., additive recoding of the number of minor alleles). Multiple logistic regression analysis using the glm function in the R statistical package [20] compared the model with multiple SNPs to the model with zero SNPs to provide a multiple degree of freedom test for association.
Bioinformatics analysis
We performed bioinformatic analyses on a 150 Kb region of chromosome 1 across the CDC42 and WNT4 genes. Linkage disequilibrium (LD) between SNPs and the presence of haplotype blocks was determined using the Haploview program [21]. Potential effects of non-synonymous (coding) variants were predicted using the Sorting Intolerant from Tolerant (SIFT) program [22,23] Polyphen [24], the PANTHER PSEC classification system [25] and the prediction of pathological mutations program (PMut) [26]. SNPs in the 3’UTR were screened for their potential to change microRNA binding sites using the miRBase [27,28], miRDB [29], and MicroInspector databases [30].
High throughput functional assays have been conducted by the international ENCODE project to identify functional regions of the genome outside of gene coding regions. The ENCODE data includes areas of open chromatin identified using DNaseI hypersensitivity (HS), Formaldehyde-Assisted Isolation of Regulatory Elements (FAIRE), and Chromatin immunoprecipitation (ChIP) experiments and the locations of functional regulatory elements including promoters, enhancers, silencers, insulators, locus control regions and novel elements [31-33]. Several programs are available to search the ENCODE data. We used HaploReg version 1 [34] to search for SNPs with functional annotations in high LD (r2 > 0.8) to our most strongly associated SNPs and the RegulomeDB program [35] to rank potential functional roles for SNPs based on their location across the entire 150 Kb region. The scoring system for RegulomeDB ranges from 1-6 with the strongest evidence for functional roles as scores 1 (a-f). A score of 2 includes evidence of transcription factor (TF) binding and DNase footprint signals. Scores of 1 require additional evidence of effects of the SNP on specific gene expression.
Results
Search for rare and novel variants in WNT4
Five coding exons (exons 1-5), the 5’ and 3’UTRs, and the intron/exon boundaries for WNT4 were screened by HRM assay. Four variants were detected amongst 100 endometriosis cases using HRM including two known variants and two novel variants (Table 1). One SNP rs34228276 is a synonymous A to G change coding for the amino acid proline at position 277 and was seen at a similar frequency in controls (Table 2). The other one, rs115547783 is in intron 2. A novel non-synonymous T to C change in exon 2 was predicted to change an amino acid from tyrosine to histidine at position 80 (p.Tyr80His). This was detected in one endometriosis patient in heterozygous form. The second novel variant was an insertion of a T base in the non-coding region of the 3’UTR (six base pairs downstream of the termination codon) and found in another patient in heterozygous form (Table 1). All members of both families for whom DNA samples were available, including the proband individuals, were then screened via HRM and/or sequencing for the presence of the relevant variants. The exon 2 variant was observed in two affected sisters and the father of the proband, but an affected aunt did not carry the variant (Figure 1A). The 3’UTR insertion/deletion (in-del) T in the second family was present in all available affected sisters and their father (Figure 1B). These variants were not detected in other cases or in any control samples by Sequenom genotyping (Table 2).
Table 1.
Variants identified in WNT4 during re-sequencing of 100 endometriosis cases including the minor allele frequencies (MAF) observed in public databases and in the cases
| SNP name | Location (Hg19) | Role | Nucleotide variant | Amino acid change | MAF | MAF in 100 cases sequenced | Functional changes predicted |
|---|---|---|---|---|---|---|---|
| rs115547783 | 22456050 | Intronic | c.236 + 59C/G | 0.008# | 0.015 | ||
| Novel* | 22456184 | Exonic | c.161C/T | p.Tyr80His | Not_obs | 0.005 | No |
| rs34228276 | 22446768 | Exonic | c.243A/G | p.Pro277Pro | 0.025# | 0.015 | |
| Novel | 22446536 | 3’UTR | c.473 T/-in-del | Not_obs | 0.005 | micRNA binding sites |
Minor allele frequency (MAF) estimate from phase 1 data from the 1000 Genome Project.
This variant was novel at the time of sequencing, but subsequently reported by Exome.
Sequencing Project (ESP) (release ESP6500 data_20 June 2012). Not_obs: Polymorphisms were not observed.
Table 2.
Results for tests of association for WNT4 and CDC42 variants genotyped in 930 endometriosis cases and 959 controls
| Variants | Nucleotide variant | Role | MAF in cases | MAF in controls | P value | OR | Functional prediction* |
|---|---|---|---|---|---|---|---|
| WNT4 variants | |||||||
| In-del novel variant | T/-in-del | 3’UTR | 0.001 | 0 | 0.242 | NA | micRNA binding sites |
| Exon 2 novel variant | c.161C/T | Non-synonymous | 0.001 | 0 | 0.489 | NA | No damage prediction |
| rs12067696 | A/G | Synonymous | 0.001 | 0 | 0.492 | NA | Splicing (ESE & abolish domain) |
| Conservation: +++ | |||||||
| rs34228276 | A/G | Synonymous | 0.018 | 0.023 | 0.302 | 0.78 (0.50-1.23) | Conservation: +++ |
| rs115547783 | C/G | Intronic | 0.016 | 0.015 | 0.894 | 1.07 (0.64-1.81) | NA |
| CDC42 variants | |||||||
| rs191653816 | C/T | promoter | 0.001 | 0 | 0.492 | NA | TF binding sites |
| rs16860621 | A/G | promoter | 0.125 | 0.130 | 0.708 | 0.96 (0.79-1.17) | TF binding sites |
| rs17837976 | C/T | Intronic, splice site | 0.002 | 0.005 | 0.180 | 0.41 (0.13-1.32) | Conservation: +++ |
Functional prediction using: miRBase and MicroInspector program for micRNA binding site prediction, SIFT and Polyphen program for amino acid change prediction, HaploReg program for TF binding site prediction and ‘snp funtion prediction’ program.
Figure 1.
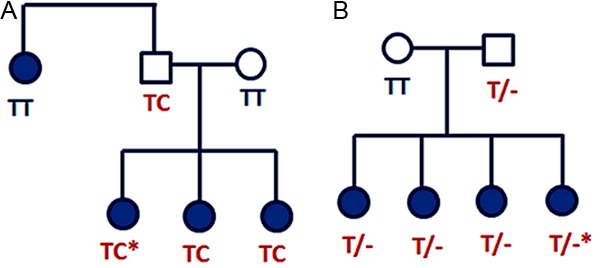
Pedigrees of endometriosis cases carrying novel variants in the WNT4 gene: (A) Pedigree of the endometriosis family with a novel non-synonymous variant in exon 2 (p.Tyr80His) and (B) Pedigree of an endometriosis family with a novel insertion/deletion variant in 3’UTR (in-del T). The proband individual screened by HRM is marked with an asterisk.
Both novel variants were examined for potential functional effects in silico. There was no predicted effect of the p.Tyr80His substitution in exon 2 (corresponding to the variant rs115547783) using the Polyphen and SIFT programs and the PANTHER PSEC classification system. The novel in-del variant located in the 3’UTR is predicted to be in a binding site of microRNA-4767-5p (miRBase) and was predicted to change a binding site from hsa-miR-3151 to hsa-miR-4507 by MicroInspector (Table 2).
Genotyping known coding variants in WNT4 and CDC42
We genotyped 36 coding variants in WNT4 and CDC42 documented in public databases (Table 3), in addition to the novel variants identified by HRM. Genotyping of these variants in our set of 930 endometriosis cases and 959 controls revealed only 3 of 27 WNT4 variants and 3 of 9 variants in CDC42 were polymorphic in our sample (Tables 2 and 3, Figures 2 and 3). Two variants, rs12067696 in WNT4 and rs191653816 in CDC42, were found only in cases and not in controls, although the differences in allele frequencies were not significant (P = 0.492). Frequencies for these variants were < 0.001 and they were not observed in the 100 cases screened by HRM. The WNT4 synonymous variant rs12067696 is conserved through mammalian and vertebrate species and predicted to be an exonic splicing enhancer (ESE) by the SNP Function Prediction (FuncPred) program. The SNP rs191653816 in the CDC42 promoter was located in the binding sites of 22 TFs by HaploReg (Table 2).
Table 3.
Known coding variants in WNT4 and CDC42 identified from dbSNP, 1000 Genomes (1000G), Exome Sequencing Project (ESP), Exome Chips manifest (EC), and COSMIC project
| Gene | Variants | Location (Hg19) | Source | Role | MAF | Nucleotide variant | Amino acid change |
|---|---|---|---|---|---|---|---|
| WNT4 | rs112942159 | 22446645 | dbSNP, 1000G | Synonymous | Not_obs | T/C | p.318Ala/Ala |
| WNT4 | rs112452625 | 22446690 | dbSNP, 1000G | Synonymous | 0.003# | A/G | p.303Ile/Ile |
| WNT4 | rs145169034 | 22446782 | dbSNP, 1000G | Synonymous | 0.001# | G/A | p.273Leu/Leu |
| WNT4 | rs201963772 | 22446798 | ESP5400 release | Non-synonymous | Not_obs | T/A | p.267Asp/Glu |
| WNT4 | rs140080433 | 22446860 | dbSNP, 1000G | Non-synonymous | Not_obs | A/G | p.247Arg/Cys |
| WNT4 | rs41441349 | 22446902 | dbSNP, 1000G | Non-synonymous | 0.002# | A/G | p.233Ala/Thr |
| WNT4 | rs140262773 | 22446925 | dbSNP, 1000G | Non-synonymous | Not_obs | A/G | p.225Pro/Leu |
| WNT4 | rs193047338 | 22446929 | dbSNP, 1000G | Non-synonymous | 0.001# | T/C | p.224Val/Met |
| WNT4 | rs121908650 | 22446952 | dbSNP, 1000G | Non-synonymous | Not_obs | G/A | p.216Glu/Gly |
| WNT4 | rs138491414 | 22447774 | dbSNP, 1000G | Non-synonymous | Not_obs | G/C | p.173Arg/Pro |
| WNT4 | rs12067696 | 22447821 | dbSNP, 1000G | Synonymous | 0.017# | A/G | p.157Asp/Asp |
| WNT4 | rs139165736 | 22447940 | dbSNP, 1000G | Non-synonymous | 0.002# | C/T | p.148Gln/Arg |
| WNT4 | rs121908651 | 22448042 | dbSNP, 1000G | Non-synonymous | Not_obs | T/C | p.114Ala/Val |
| WNT4 | rs139045509 | 22448044 | dbSNP, 1000G | Synonymous | Not_obs | A/G | p.113Tyr/Tyr |
| WNT4 | rs16826648 | 22456146 | dbSNP, 1000G | Synonymous | 0.010# | A/G | p.92Leu/Leu |
| WNT4 | rs121908652 | 22456175 | dbSNP, 1000G | Non-synonymous | Not_obs | T/C | p.83Arg/Trp |
| WNT4 | rs34611251 | 22456205 | dbSNP, 1000G | Frame shift | Not_obs | G/- | p.73Leu/Trp |
| WNT4 | rs144407094 | 22456326 | dbSNP, 1000G | Synonymous | 0.001# | T/C | p.32Ser/Ser |
| WNT4 | rs121908653 | 22469381 | dbSNP, 1000G | Non-synonymous | Not_obs | C/T | p.12Leu/Pro |
| WNT4 | unknown | 22446572 | ESP | Non-synonymous | 0.0001§ | A/G | p.343Trp/Arg |
| WNT4 | unknown | 22446857 | ESP | Non-synonymous | 0.0002§ | A/G | p.248Cys/Arg |
| WNT4 | unknown | 22446859 | ESP | Non-synonymous | 0.0001§ | T/C | p.247His/Arg |
| WNT4 | unknown | 22447712 | ESP | Non-synonymous | 0.0001§ | T/C | p.194Ser/Gly |
| WNT4 | unknown | 22456127 | ESP | Non-synonymous | 0.0001§ | T/C | p.99Ser/Gly |
| WNT4 | unknown | 22456174 | ESP | Non-synonymous | 0.0001§ | T/C | p.83Gln/Arg |
| WNT4 | unknown | 22446566 | EC | Non-synonymous | 0.0007§ | G/C | |
| WNT4 | unknown | 22446856 | EC | Non-synonymous | 0.0003§ | T/C | |
| CDC42 | unknown | 22412982 | ESP | Non-synonymous | 0.0001§ | C/G | p.77Leu/Val |
| CDC42 | COSM46468 | 22405060 | 1000G, COSMIC | Non-synonymous | Not_obs | T/C | p.30Ser/Leu |
| CDC42 | rs142108830 | 22413282 | ESP, dbSNP | Non-synonymous | Not_obs | G/A | p.137Ile/Val |
| CDC42 | COSM260019 | 22417991 | COSMIC | Non-synonymous | Not_obs | A/G | p.557Arg/His |
| CDC42 | COSM229859 | 22405006 | COSMIC | Non-synonymous | Not_obs | T/G | p.12Gly/Val |
| CDC42 | rs16860621 | 22379360 | dbSNP, 1000G | promoter | 0.146# | A/G | |
| CDC42 | rs16860623 | 22379375 | dbSNP, 1000G | promoter | Not_obs | T/G | |
| CDC42 | rs191653816 | 22379314 | dbSNP, 1000G | promoter | 0.001# | T/C | |
| CDC42 | rs17837976 | 22408212 | dbSNP, 1000G | Intron, splice site | 0.014# | C/T |
MAF estimate from Exome Sequencing Project.
MAF estimate from phase 1 data from 1000 Genomes.
COSMIC, Catalogue of Somatic Mutations in Cancer.
Figure 2.
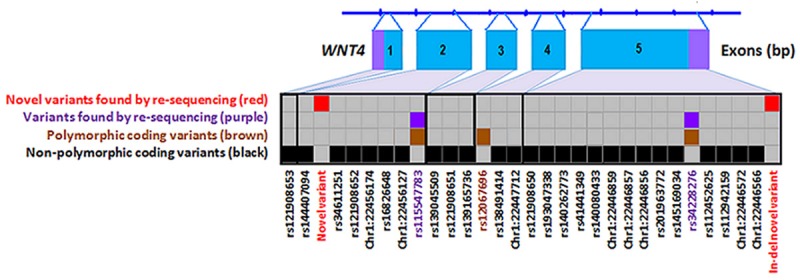
Rare coding variants in WNT4 identified by High Resolution Melt analysis or from public databases and genotyped in 930 cases and 959 controls.
Figure 3.
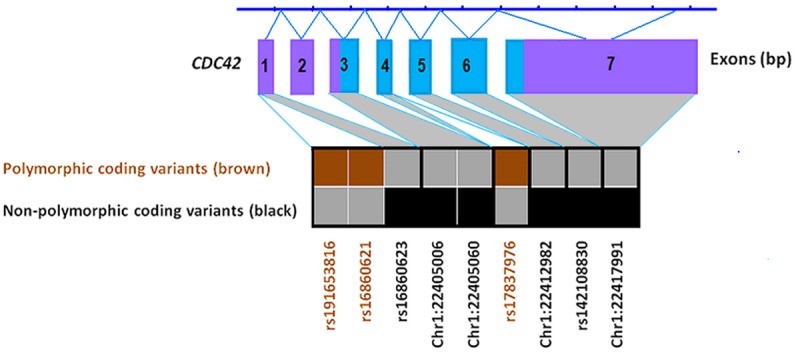
Rare coding variants in CDC42 identified by High Resolution Melt analysis or from public databases and genotyped in 930 cases and 959 controls.
There was no evidence that individual coding variants contributed to endometriosis risk, but several rare variants were detected only in cases. We therefore analysed data for the six variants listed in Table 2 using multiple logistic regression and found no significant evidence for combined effects on association with endometriosis risk (x 6 2 = 6.973; P = 0.323).
Common variants in WNT4
We previously reported significant association across the CDC42-WNT4 region with the strongest signal from genotypes estimated by imputation. To confirm imputation results we genotyped the five top imputed SNPs together with rs7521902, the top SNP from the original GWA studies, in our 930 cases and 959 controls. Our genotyping results showed high concordance between genotyped and imputed data (98-100%) and confirmed stronger evidence for association with SNPs located in intron 1 of WNT4 (Table 4) than for rs7521902 located ~20 kb upstream of WNT4.
Table 4.
Results for association tests for imputed common variants in WNT4 genotyped in 930 endometriosis cases and 959 controls
| Variants | Location (Hg19) | Role | N_variant | MAF | MAF in cases | MAF in controls | P value | OR |
|---|---|---|---|---|---|---|---|---|
| rs61768001 | 22465820 | Intron 1-2 | C/T | 0.234# | 0.202 | 0.156 | 2.002 x10-4 | 1.38 (1.16-1.63) |
| rs12037376 | 22462111 | Intron 1-2 | A/G | 0.224# | 0.201 | 0.156 | 2.735 x10-4 | 1.37 (1.15-1.61) |
| rs56318008 | 22470407 | Promoter | T/C | 0.220# | 0.190 | 0.147 | 3.860 x10-4 | 1.36 (1.15-1.62) |
| rs55938609 | 22470451 | Promoter | C/G | 0.230# | 0.190 | 0.148 | 4.730 x10-4 | 1.36 (1.14-1.61) |
| rs7412010 | 22436446 | 3’ near gene | C/G | 0.186* | 0.203 | 0.160 | 7.696 x10-4 | 1.33 (1.13-1.57) |
| rs7521902 | 22490724 | Genomic | A/C | 0.235* | 0.267 | 0.241 | 0.067 | 1.15 (0.99-1.33) |
MAF estimate from HapMap CEU population (HapMap data release 28; phase 1, 2 & 3; August 10).
MAF estimate from phase 1 data from 1000 Genomes.
The five SNPs are in high LD and together the risk alleles form a single risk haplotype (P = 7 x10-4) with a similar estimate for disease risk as the individual SNPs.
Functional annotation for WNT4 common variants
Functional annotation of our top SNPs revealed limited evidence for a functional role for the sentinel SNP rs61768001 (defined as the SNP with best association signal in a multi-SNP analysis) or for the original genotyped SNP rs7521902. Analysis of 50 SNPs (with strongest evidence of association from the GWAS data) across the 150 Kb region using RegulomeDB identified seven variants in strong LD (r2 > 0.8) with rs61768001 and two SNPs in high LD with rs7521902 with evidence of predicted functional roles (Table 5, Figures 3 and 4I). The SNP with the best score (2b) was rs12404660 (r2 = 0.84 with rs61768001; Table 5, Figure 4II and 4III). SNP rs12404660 is located in an area of histone protein H3K4me1 chromatin modification associated with transcription enhancer sequences, open chromatin, altered regulatory motifs for the transcription factor YY1 and a binding site for the transcription factor CTCF, all of which are suggestive of regulatory potential. Two additional SNPs may also have functional roles as predicted by the HaploReg, and JASPAR core programs. SNP rs3820282, located in intron 1 of WNT4, (r2 = 0.94 to rs61768001), is predicted to lie in a conserved region within regulatory motifs bound by the transcription factors oestrogen receptor 1 (ESR1) and oestrogen receptor 2 (ESR2). No TF binding sites were predicted at the original G allele of this variant. However, the change to the minor A allele introduced potential regulatory sites for ESR1 and ESR2 identified by HaploReg and JASPAR (Figure 4III, Table 5). The SNP rs55938609, in high LD with rs61768001 (r2 = 0.98, Figure 5), is located in a region of histone protein H3K4Me2 modification, associated with both promoters and enhancers (Figure 4III), and potential binding sites for the transcription factors forkhead box A1 (FOXA1) and A2 (FOXA2) (Figure 4III, Table 5).
Table 5.
Functional regulation annotation for common SNPs in high LD (r2 ≥ 0.8) with rs61768001 and rs7521902 using HaploReg and RegulomeDB
| Variants | Location (hg19) | SNP score* | Cons | DNAseI HS peak (N) | TFs binding | Motifs changed | Gene | Genomic position |
|---|---|---|---|---|---|---|---|---|
| Common SNPs in high LD with rs61768001 | ||||||||
| rs12038474 | 22403357 | - | SEF-1 | CDC42 | intronic | |||
| rs7412010 | 22436446 | - | HES1 | 7.4 kb 3’ of WNT4 | intergenic | |||
| rs12404660 | 22458794 | 2b | 45 | CTCF | YY1# | WNT4 | intronic | |
| rs12037376 | 22462111 | 5 | 6 | SEF-1, | WNT4 | intronic | ||
| rs61768001 | 22465820 | 5 | 9 | Gabpa# | WNT4 | intronic | ||
| rs3820282 | 22468215 | 5 | +++ | 33 | Esr2, Esr1 | WNT4 | intronic | |
| rs56318008 | 22470407 | 5 | 34 | Gfi1b | 887 bp 5’ of WNT4 | promoter | ||
| rs55938609 | 22470451 | 4 | 31 | FOXA1, FOXA2 | 931 bp 5’ of WNT4 | promoter | ||
| Common SNPs in high LD with rs7521902 | ||||||||
| rs72478520 | 22489567 | 4 | 11 | IKZF1# | 20 kb 5’ of WNT4 | intergenic | ||
| rs7521902 | 22490724 | 5 | 1 | 20 kb 5’ of WNT4 | intergenic | |||
| rs3920498 | 22492887 | 5 | 5 | Smad3, TP53# | 23 kb 5’ of WNT4 | Intergenic | ||
Scores from RegulomeDB.
Prediction for SNP with score = 2b: TF binding + any motif + DNase Footprint + DNase peak. Prediction for SNP with score = 4: TF binding and DNase peak. Prediction for SNP with score = 5: TF binding or DNase peak.
Identified by various methods in a number of cell types (http://regulome.stanford.edu/).
N is number of cell types in which DNase peaks marked.
Figure 4.
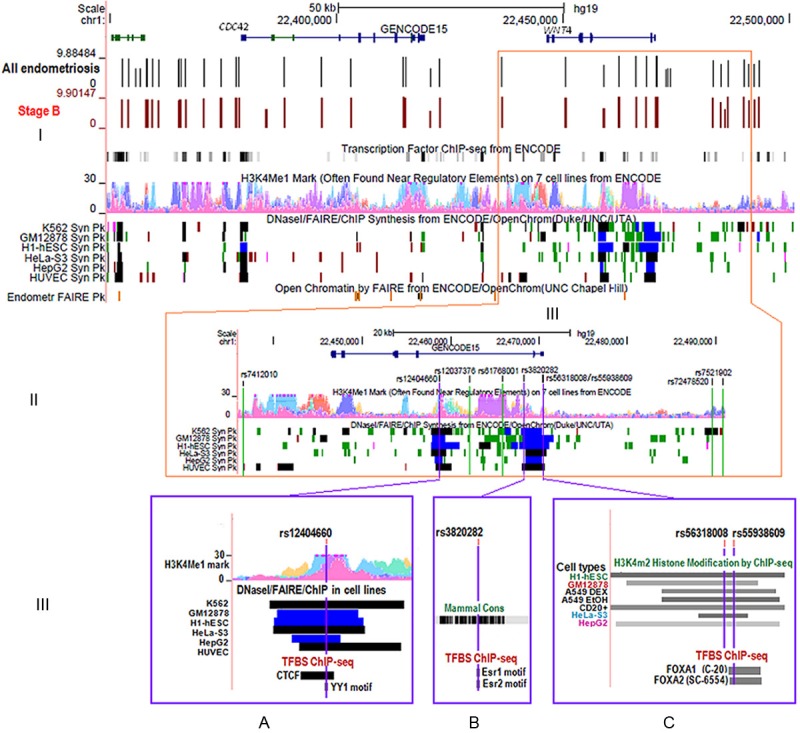
ENCODE annotation of WNT4 GWA and imputed variants in endometriosis. (I) Overview of the region of chromosome 1 including the CDC42 and WNT4 genes. Genotyped markers are shown by vertical lines with results (log-10 P values) for all endometriosis cases in black and stage B endometriosis cases in brown. Peaks for histone modification marks included in the ENCODE data [39] are shown for H3K4Me1 on 7 cell types (Tier 1 and Tier 2). The ENCODE data also shows areas of open chromatin as revealed by DNaseI, FAIRE and ChIP experiments in Tier 1 and Tier 2 cell types indicated by solid blocks (Regions showing peaks defined by both DNase I and FAIRE assays are in black, highly significant regions with combinations of peaks from these assays are in blue. Less significant regions identified by only DNase I HS as peaks are in green). The peak signals of open chromatin by FAIRE for normal endometrial tissue are shown in the Endometrium FAIRE Pk track. (II) Regions of interest around nine imputed SNPs. Three SNPs with the strongest association signals are located in regions of open chromatin as shown by DNaseI signals in the ENCODE data. (III) A zoomed-in image for the three top SNPs; (A) rs12404660, (B) rs3820282, and (C) rs55938609 (purple vertical lines) and functional annotation by ENCODE. Regulatory protein bound regions and histone modifications indicated by ChIP-seq of H3K4m2 in a subset of Tier 1 and 2 cell types (in greyscale light (lowest) to dark (highest)). The level of conservation across the region is shown for mammals. (A) SNP rs12404660 lies in histone modification area for H3K4Me1, open chromatin location by DNaseI, FAIRE, and ChIP assays, transcription factors CTCF by ChIP and sequence motifs of transcription factor YY1 by footprinting assays (B) rs3820282 is located within sequence motifs for transcription factors ESR1 and ESR2 and (C) rs55938609 lies within sequence bound by the transcription factors FOXA1 and FOXA2.
Figure 5.
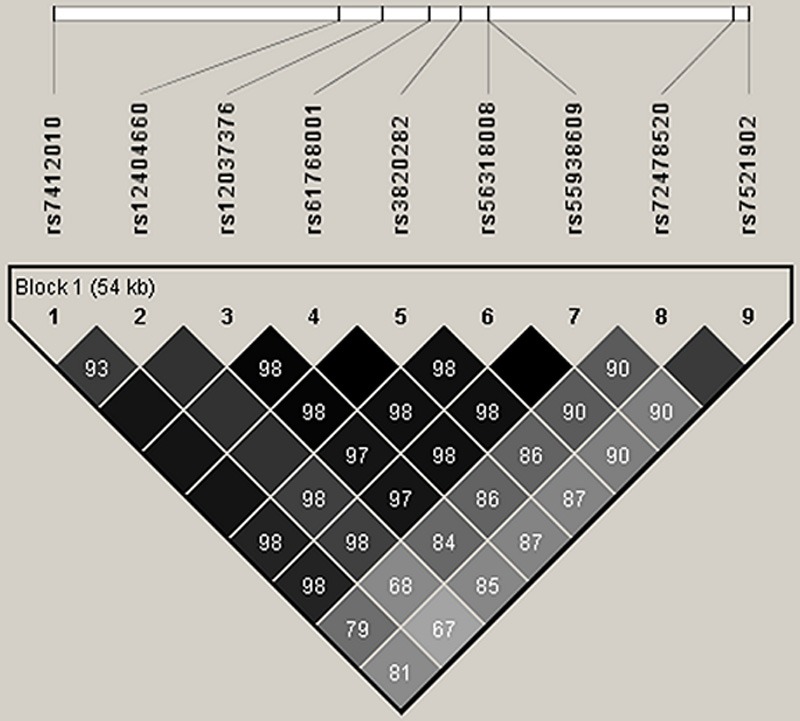
Linkage disequilibrium (r2) between genotyped and imputed common variants across the region of WNT4 and CDC42.
Discussion
GWAS for endometriosis report strong associations for disease risk with markers at chromosome 1p36, a region which spans potential candidate genes WNT4, CDC42 and LINC00339 [1-4]. To further investigate the WNT4 locus we first sequenced the WNT4 coding regions and identified two novel variants. One variant was a non-synonymous variant coding for an amino acid substitution p.Tyr80His with no predicted deleterious effect from in silico analysis. The second variant was a 3’UTR insertion/deletion predicted to be in a micro-RNA binding site for miR-4767-5p and to change a binding site for miR-3151, associated with melanoma [36] and childhood acute lymphoblastic leukemia [37]. Both novel variants were seen only in the families in which they were first detected.
During the project, data on coding variants from sequencing ~12,000 individuals were released from the Exome Sequencing Project (ESP6500 data, Jun 2012) reducing the need for further sequencing. For example the exome data included our novel p.Tyr80His variant (rs115547783) with a minor allele frequency (MAF) of 0.00012 in the European American population. Genotyping all coding variants in WNT4 and CDC42 reported in public databases identified only 6 variants that were polymorphic in our sample. Two of these variants were rare, had predicted functional effects and were found only in cases. Tested either individually or together, the six rare variants did not provide evidence for association with disease risk in our case-control sample. Our results demonstrate no evidence that rare exonic or promoter region variants in WNT4 or CDC42 were associated with disease risk.
Our case-control sample for this study has only 80% power to detect a genotype relative risk (GRR) of 1.4 at an allele frequency of 0.25 [14] and very low power to identify association with rare SNPs with low MAF unless the GRR was much greater (e.g. a GRR of at least 1.7 at an allele frequency of 0.05 is required for 80% power [14]). We chose familial cases for initial genotyping because the frequencies of alleles contributing to disease are generally enriched in these designs [38]. We have a further 4000 cases available for genotyping to increase power, but concluded that while effects of some rare variants contributing to disease risk in carriers cannot be excluded, the evidence did not justify further genotyping. All coding variants were rare and the association signal in this region cannot be explained by coding variants in either WNT4 or CDC42.
Analysis with imputed SNPs previously reported genome-wide significant association for a number of common SNPs close to WNT4 and CDC42 [3]. Genotyping key common SNPs demonstrated strong concordance (>98%) between observed and imputed genotypes and confirmed previous suggestions that the strongest association signals lie in the non-coding regions around WNT4. Haplotype analyses of our common SNPs spanning the region show a single common risk haplotype and this result suggests the observed association signal at this locus is not being driven by a rare causal variant in either the coding or non-coding regions. The association signal is most likely due to functional effects of one or more of the common non-coding SNPs in this haplotype block.
Up to 88% of variants associated with common diseases are found within introns and intergenic regions [39] and it is likely that causal variants function by altering gene regulation. To annotate potential functional roles of our top variants, we screened 50 SNPs with the lowest P values from imputed data [3]. Three variants, rs12404660, rs3820282, and rs55938609, all in high LD with our top SNP rs61768001, had the best evidence for potential functional effects in the ENCODE data as identified by the HaploReg and RegulomeDB programs [35]. All three variants are located in validated and significant regions of open chromatin with combinations of DNaseI HS and FAIRE peaks in multiple cell types (Figure 4III) [39]. The three variants were also located in binding sites for TFs or altered regulatory motifs. SNP rs12404660 is located at a binding site for the transcription factor CTCF, and the regulatory motif for YY1, TFs which function as enhancer blockers, transcriptional repressors and activators, and initiators of transcription [40-43].
SNP rs55938609 is located at a predicted TF binding site for FOXA1 in T-47D cells and FOXA2 in HepG2 cells in the ENCODE data. FOXA1 mediates oestrogen receptor (ER) function and is a critical factor for interactions between ER and chromatin [44]. FOXA1 and H3K4me2 interact at enhancers to increase gene transcription [45]. The minor A allele at rs3820282 may alter regulatory motifs for oestrogen receptor (ESR1) identified in various cell types by footprinting experiments [35] and ESR2 [34]. ESR1 and ESR2 are involved in differentiation and proliferation of cells in target tissues including ovaries, uterus, and mammary gland [46-48]. Regulatory elements at these sites occur in multiple cell types, but functional studies of potential regulatory sites must be carried out in relevant target tissues to determine any role in endometriosis risk.
In conclusion, we identified coding variants in WNT4 and CDC42 present only in women with endometriosis, but these variants were rare and there was no evidence for association with increased disease risk. Genotyping key common non-coding variants confirmed imputation results showing a strong association signal covering the chromosome region that includes LINC00339, CDC42 and WNT4. The strong association signal is not explained by coding variants in WNT4 or CDC42 and our genetic data cannot determine which of the genes in this region are affected. Functional studies on gene expression and gene regulation in relevant target tissues will be necessary to identify the gene or genes responsible for increased endometriosis risk. SNPs located in sites with potential functional roles in binding of transcription factors FOXA1, FOXA2, ESR1 and ESR2 indicate potential targets to prioritise for future functional experiments to understand the molecular pathophysiology of endometriosis.
Acknowledgements
We thank L. Wallace, B. Haddon, D. Smyth, H. Beeby, and O. Zheng for project and database management and sample processing. We thank Brisbane gynaecologist D.T. O’Connor for confirmation of diagnosis and staging of disease from clinical records of many cases, including 251 in these analyses. We acknowledge with appreciation all the women who participated in the QIMR and OXEGENE studies. We thank Endometriosis Associations for supporting the study recruitment and the many hospital directors and staff, gynaecologists, general practitioners and pathology services in Australia who provided assistance with confirmation of diagnoses. We thank S. Nicolaides and the Queensland Medical Laboratory for pro bono collection and delivery of blood samples and other pathology services for assistance with blood collection. The study was supported by grants from the National Health and Medical Research Council (NHMRC) of Australia (241944, 339462, 389927, 389875, 389891, 389892, 389938, 443036, 442915, 442981, 496610, 496739 552485, 552498, and 1026033), the Cooperative Research Centre for Discovery of Genes for Common Human Diseases (CRC), Cerylid Biosciences (Melbourne) and donations from N. Hawkins and S. Hawkins. D.R.N. was supported by the NHMRC Fellowship (339462 and 613674) and the ARC Future Fellowship (FT0991022) schemes and G.W.M. was supported by the NHMRC Fellowships Scheme (339446, 619667).
Disclosure of conflict of interest
None.
References
- 1.Uno S, Zembutsu H, Hirasawa A, Takahashi A, Kubo M, Akahane T, Aoki D, Kamatani N, Hirata K, Nakamura Y. A genome-wide association study identifies genetic variants in the CDKN2BAS locus associated with endometriosis in Japanese. Nat Genet. 2010;42:707–788. doi: 10.1038/ng.612. [DOI] [PubMed] [Google Scholar]
- 2.Painter JN, Anderson CA, Nyholt DR, Macgregor S, Lin JH, Lee SH, Lambert A, Zhao ZZ, Roseman F, Guo Q, Gordon SD, Wallace L, Henders AK, Visscher PM, Kraft P, Martin NG, Morris AP, Treloar A, Kennedy SH, Missmer SA, Montgomery GW, Zondervan KT. Genome-wide association study identifies a locus at 7p15.2 associated with endometriosis. Nat Genet. 2011;43:51–4. doi: 10.1038/ng.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nyholt DR, Low SK, Anderson CA, Painter JN, Uno S, Morris AP, Macgregor S, Gordon SD, Henders AK, Martin NG, Attia J, Holliday EG, McEvoy M, Scott RJ, Kennedy SH, Treloar SA, Missmer SA, Adachi S, Tanaka K, Nakamura Y, Zondervan KT, Zembutsu H, Montgomery GW. Genome-wide association meta-analysis identifies new endometriosis risk loci. Nat Genet. 2012;44:1355–1359. doi: 10.1038/ng.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Albertsen HM, Chettier R, Farrington P, Ward K. Genome-wide association study link novel loci to endometriosis. PLoS One. 2013;8:e58257. doi: 10.1371/journal.pone.0058257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goode EL, Chenevix-Trench G, Song H, Ramus SJ, Notaridou M, Lawrenson K, Widschwendter M, Vierkant RA, Larson MC, Kjaer SK, Birrer MJ, Berchuck A, Schildkraut J, Tomlinson I, Kiemeney LA, Cook LS, Gronwald J, Garcia-Closas M, Gore ME, Campbell I, Whittemore AS, Sutphen R, Phelan C, Anton-Culver H, Pearce CL, Lambrechts D, Rossing MA, Chang-Claude J, Moysich KB, Goodman MT, Dörk T, Nevanlinna H, Ness RB, Rafnar T, Hogdall C, Hogdall E, Fridley BL, Cunningham JM, Sieh W, McGuire V, Godwin AK, Cramer DW, Hernandez D, Levine D, Lu K, Iversen ES, Palmieri RT, Houlston R, van Altena AM, Aben KK, Massuger LF, Brooks-Wilson A, Kelemen LE, Le ND, Jakubowska A, Lubinski J, Medrek K, Stafford A, Easton DF, Tyrer J, Bolton KL, Harrington P, Eccles D, Chen A, Molina AN, Davila BN, Arango H, Tsai YY, Chen Z, Risch HA, McLaughlin J, Narod SA, Ziogas A, Brewster W, Gentry-Maharaj A, Menon U, Wu AH, Stram DO, Pike MC Wellcome Trust Case-Control Consortium; Beesley J, Webb PM Australian Cancer Study (Ovarian Cancer); Australian Ovarian Cancer Study Group; Ovarian Cancer Association Consortium (OCAC); Chen X, Ekici AB, Thiel FC, Beckmann MW, Yang H, Wentzensen N, Lissowska J, Fasching PA, Despierre E, Amant F, Vergote I, Doherty J, Hein R, Wang-Gohrke S, Lurie G, Carney ME, Thompson PJ, Runnebaum I, Hillemanns P, Dürst M, Antonenkova N, Bogdanova N, Leminen A, Butzow R, Heikkinen T, Stefansson K, Sulem P, Besenbacher S, Sellers TA, Gayther SA, Pharoah PD Ovarian Cancer Association Consortium (OCAC) A genome-wide association study identifies susceptibility loci for ovarian cancer at 2q31 and 8q24. Nat Genet. 2010;42:874–879. doi: 10.1038/ng.668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Estrada K, Styrkarsdottir U, Evangelou E, Hsu YH, Duncan EL, Ntzani EE, Oei L, Albagha OM, Amin N, Kemp JP, Koller DL, Li G, Liu CT, Minster RL, Moayyeri A, Vandenput L, Willner D, Xiao SM, Yerges-Armstrong LM, Zheng HF, Alonso N, Eriksson J, Kammerer CM, Kaptoge SK, Leo PJ, Thorleifsson G, Wilson SG, Wilson JF, Aalto V, Alen M, Aragaki AK, Aspelund T, Center JR, Dailiana Z, Duggan DJ, Garcia M, Garcia-Giralt N, Giroux S, Hallmans G, Hocking LJ, Husted LB, Jameson KA, Khusainova R, Kim GS, Kooperberg C, Koromila T, Kruk M, Laaksonen M, Lacroix AZ, Lee SH, Leung PC, Lewis JR, Masi L, Mencej-Bedrac S, Nguyen TV, Nogues X, Patel MS, Prezelj J, Rose LM, Scollen S, Siggeirsdottir K, Smith AV, Svensson O, Trompet S, Trummer O, van Schoor NM, Woo J, Zhu K, Balcells S, Brandi ML, Buckley BM, Cheng S, Christiansen C, Cooper C, Dedoussis G, Ford I, Frost M, Goltzman D, González-Macías J, Kähönen M, Karlsson M, Khusnutdinova E, Koh JM, Kollia P, Langdahl BL, Leslie WD, Lips P, Ljunggren Ö, Lorenc RS, Marc J, Mellström D, Obermayer-Pietsch B, Olmos JM, Pettersson-Kymmer U, Reid DM, Riancho JA, Ridker PM, Rousseau F, Slagboom PE, Tang NL, Urreizti R, Van Hul W, Viikari J, Zarrabeitia MT, Aulchenko YS, Castano-Betancourt M, Grundberg E, Herrera L, Ingvarsson T, Johannsdottir H, Kwan T, Li R, Luben R, Medina-Gómez C, Palsson ST, Reppe S, Rotter JI, Sigurdsson G, van Meurs JB, Verlaan D, Williams FM, Wood AR, Zhou Y, Gautvik KM, Pastinen T, Raychaudhuri S, Cauley JA, Chasman DI, Clark GR, Cummings SR, Danoy P, Dennison EM, Eastell R, Eisman JA, Gudnason V, Hofman A, Jackson RD, Jones G, Jukema JW, Khaw KT, Lehtimäki T, Liu Y, Lorentzon M, McCloskey E, Mitchell BD, Nandakumar K, Nicholson GC, Oostra BA, Peacock M, Pols HA, Prince RL, Raitakari O, Reid IR, Robbins J, Sambrook PN, Sham PC, Shuldiner AR, Tylavsky FA, van Duijn CM, Wareham NJ, Cupples LA, Econs MJ, Evans DM, Harris TB, Kung AW, Psaty BM, Reeve J, Spector TD, Streeten EA, Zillikens MC, Thorsteinsdottir U, Ohlsson C, Karasik D, Richards JB, Brown MA, Stefansson K, Uitterlinden AG, Ralston SH, Ioannidis JP, Kiel DP, Rivadeneira F. Genome-wide meta-analysis identifies 56 bone mineral density loci and reveals 14 loci associated with risk of fracture. Nat Genet. 2012;44:491–501. doi: 10.1038/ng.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kobayashi A, Behringer RR. Developmental genetics of the female reproductive tract in mammals. Nat Rev Genet. 2003;4:969–980. doi: 10.1038/nrg1225. [DOI] [PubMed] [Google Scholar]
- 8.MacLaughlin DT, Teixeira J, Donahoe PK. Perspective: Reproductive tract development – New discoveries and future directions. Endocrinology. 2001;142:2167–2172. doi: 10.1210/endo.142.6.8262. [DOI] [PubMed] [Google Scholar]
- 9.Bui TD, Zhang L, Rees MCP, Bicknell R, Harris AL. Expression and hormone regulation of Wnt2, 3, 4, 5a, 7a, 7b and 10b in normal human endometrium and endometrial carcinoma. Br J Cancer. 1997;75:1131–1136. doi: 10.1038/bjc.1997.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hou XN, Tan Y, Li ML, Dey SK, Das SK. Canonical Wnt signaling is critical to estrogen-mediated uterine growth. Mol Endocrinol. 2004;18:3035–3049. doi: 10.1210/me.2004-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goteri G, Ciavottini A, Lucarini G, Montik N, Filosa A, Stramazzotti D, Biagini G, Tranquilli AL. Expression of motility-related molecule Cdc42 in endometrial tissue in women with adenomyosis and ovarian endometriomata. Fertil Steril. 2006;86:559–565. doi: 10.1016/j.fertnstert.2006.01.031. [DOI] [PubMed] [Google Scholar]
- 12.Hu WP, Tay SK, Zhao Y. Endometriosis-specific genes identified by real-time reverse transcription-polymerase chain reaction expression profiling of endometriosis versus autologous uterine endometrium. J Clin Endocrinol Metab. 2006;91:228–238. doi: 10.1210/jc.2004-1594. [DOI] [PubMed] [Google Scholar]
- 13.Treloar S, Hadfield R, Montgomery G, Lambert A, Wicks J, Barlow DH, O’Connor DT, Kennedy S International Endogene Study Group. The International Endogene Study: a collection of families for genetic research in endometriosis. Fertil Steril. 2002;78:679–685. doi: 10.1016/s0015-0282(02)03341-1. [DOI] [PubMed] [Google Scholar]
- 14.Zhao ZZ, Nyholt DR, Le L, Martin NG, James MR, Treloar SA, Montgomery GW. KRAS variation and risk of endometriosis. Mol Hum Reprod. 2006;12:671–676. doi: 10.1093/molehr/gal078. [DOI] [PubMed] [Google Scholar]
- 15.Canis M, Donnez JG, Guzick DS, Halme JK, Rock JA, Schenken RS, Vernon MW. Revised American Society for Reproductive Medicine classification of endometriosis: 1996. Fertil Steril. 1997;67:817–821. doi: 10.1016/s0015-0282(97)81391-x. [DOI] [PubMed] [Google Scholar]
- 16.Painter JN, Nyholt DR, Morris A, Zhao ZZ, Henders AK, Lambert A, Wallace L, Martin NG, Kennedy SH, Treloar SA, Zondervan KT, Montgomery GW. High-density fine-mapping of a chromosome 10q26 linkage peak suggests association between endometriosis and variants close to CYP2C19. Fertil Steril. 2011;95:2236–2240. doi: 10.1016/j.fertnstert.2011.03.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luong HTT, Chaplin J, McRae AF, Medland SE, Willemsen G, Nyholt DR, Henders AK, Hoekstra C, Duffy DL, Martin NG, Boomsma DI, Montgomery GW, Painter JN. Variation in BMPR1B, TGFRB1 and BMPR2 and Control of Dizygotic Twinning. Twin Res Hum Genet. 2011;14:408–416. doi: 10.1375/twin.14.5.408. [DOI] [PubMed] [Google Scholar]
- 18.Rozen S, Skaletsky H. Primer3 on the WWW for General Users and for Biologist Programmers. Methods Mol Biol. 1999;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- 19.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, Sham PC. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.R Development Core Team. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2008. ISBN 3-900051-07-0, URL http://www.R-project.org. [Google Scholar]
- 21.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 22.Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ng PC, Henikoff S. Accounting for human polymorphisms predicted to affect protein function. Genome Res. 2002;12:436–446. doi: 10.1101/gr.212802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mi HY, Muruganujan A, Thomas PD. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013;41:D377–D386. doi: 10.1093/nar/gks1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferrer-Costa C, Gelpi JL, Zamakola L, Parraga I, de la Cruz X, Orozco M. PMUT: a web-based tool for the annotation of pathological mutations on proteins. Bioinformatics. 2005;21:3176–3178. doi: 10.1093/bioinformatics/bti486. [DOI] [PubMed] [Google Scholar]
- 27.Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36:D154–D158. doi: 10.1093/nar/gkm952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011;39:D152–D157. doi: 10.1093/nar/gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang XW. miRDB: A microRNA target prediction and functional annotation database with a wiki interface. RNA. 2008;14:1012–1017. doi: 10.1261/rna.965408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rusinov V, Baev V, Minkov IN, Tabler M. MicroInspector: a web tool for detection of miRNA binding sites in an RNA sequence. Nucleic Acids Res. 2005;33:W696–W700. doi: 10.1093/nar/gki364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crawford GE, Davis S, Scacheri PC, Renaud G, Halawi MJ, Erdos MR, Green RM, Paul SW, Tyra G, Collins FS. DNase-chip: a high-resolution method to identify DNase I hypersensitive sites using tiled microarrays. Nat Methods. 2006;3:503–509. doi: 10.1038/NMETH888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giresi PG, Lieb JD. Isolation of active regulatory elements from eukaryotic chromatin using FAIRE (Formaldehyde Assisted Isolation of Regulatory Elements) Methods. 2009;48:233–239. doi: 10.1016/j.ymeth.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Simon JM, Giresi PG, Davis IJ, Lieb JD. Using formaldehyde-assisted isolation of regulatory elements (FAIRE) to isolate active regulatory DNA. Nat Protoc. 2012;7:256–267. doi: 10.1038/nprot.2011.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40:D930–D934. doi: 10.1093/nar/gkr917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, Karczewski KJ, Park J, Hitz BC, Weng S, Cherry JM, Snyder M. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22:1790–1797. doi: 10.1101/gr.137323.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stark MS, Tyagi S, Nancarrow DJ, Boyle GM, Cook AL, Whiteman DC, Parsons PG, Schmidt C, Sturm RA, Hayward NK. Characterization of the Melanoma miRNAome by Deep Sequencing. PLoS One. 2010;5:e9685. doi: 10.1371/journal.pone.0009685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schotte D, Moqadam FA, Lange-Turenhout EAM, Chen C, van Ljcken WFJ, Pieters R, den Boer ML. Discovery of new microRNAs by small RNAome deep sequencing in childhood acute lymphoblastic leukemia. Leukemia. 2011;25:1389–1399. doi: 10.1038/leu.2011.105. [DOI] [PubMed] [Google Scholar]
- 38.Emond MJ, Louie T, Emerson J, Zhao W, Mathias RA, Knowles MR, Wright FA, Rieder MJ, Tabor HK, Nickerson DA, Barnes KC National Heart, Lung, and Blood Institute (NHLBI) GO Exome Sequencing Project. Lung GO, Gibson RL, Bamshad MJ. Exome sequencing of extreme phenotypes identifies DCTN4 as a modifier of chronic Pseudomonas aeruginosa infection in cystic fibrosis. Nat Genet. 2012;44:886–889. doi: 10.1038/ng.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.ENCODE Project Consortium. Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bell AC, West AG, Felsenfeld G. The protein CTCF is required for the enhancer blocking activity of vertebrate insulators. Cell. 1999;98:387–396. doi: 10.1016/s0092-8674(00)81967-4. [DOI] [PubMed] [Google Scholar]
- 41.Ohlsson R, Renkawitz R, Lobanenkov V. CTCF is a uniquely versatile transcription regulator linked to epigenetics and disease. Trends Genet. 2001;17:520–527. doi: 10.1016/s0168-9525(01)02366-6. [DOI] [PubMed] [Google Scholar]
- 42.Vostrov AA, Quitschke WW. The zinc finger protein CTCF binds to the APB beta domain of the amyloid beta-protein precursor promoter - Evidence for a role in transcriptional activation. J Biol Chem. 1997;272:33353–33359. doi: 10.1074/jbc.272.52.33353. [DOI] [PubMed] [Google Scholar]
- 43.Shi Y, Lee JS, Galvin KM. Everything you have ever wanted to know about Yin Yang 1. Biochim Biophys Acta. 1997;1332:F49–F66. doi: 10.1016/s0304-419x(96)00044-3. [DOI] [PubMed] [Google Scholar]
- 44.Hurtado A, Holmes KA, Ross-Innes CS, Schmidt D, Carroll JS. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat Genet. 2011;43:27–33. doi: 10.1038/ng.730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lupien M, Eeckhoute J, Meyer CA, Wang QB, Zhang Y, Li W, Carroll JS, Liu XS, Brown M. FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell. 2008;132:958–970. doi: 10.1016/j.cell.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, Tujague M, Strom A, Treuter E, Warner M, Gustafsson JA. Estrogen receptors: How do they signal and what are their targets. Physiol Rev. 2007;87:905–931. doi: 10.1152/physrev.00026.2006. [DOI] [PubMed] [Google Scholar]
- 47.Lamp M, Peters M, Reinmaa E, Haller-Kikkatalo K, Kaart T, Kadastik U, Karro H, Metspalu A, Salumets A. Polymorphisms in ESR1, ESR2 and HSD17B1 genes are associated with fertility status in endometriosis. Gynecol Endocrinol. 2011;27:425–433. doi: 10.3109/09513590.2010.495434. [DOI] [PubMed] [Google Scholar]
- 48.Osborne CK, Schiff R, Fuqua SAW, Shou J. Estrogen receptor: Current understanding of its activation and modulation. Clin Cancer Res. 2001;7:4338S–4342S. [PubMed] [Google Scholar]