

Two structures of a small protein with a defined tertiary fold, the isolated Pin1 WW domain, have been determined via racemic crystallization with small-molecule additives. These additives, which are either racemic or achiral, appear to stabilize a dynamic loop region of the structure.
Keywords: Pin1 WW domain, loop stabilization, racemic crystallization
Abstract
The human Pin1 WW domain is a small autonomously folding protein that has been useful as a model system for biophysical studies of β-sheet folding. This domain has resisted previous attempts at crystallization for X-ray diffraction studies, perhaps because of intrinsic conformational flexibility that interferes with the formation of a crystal lattice. Here, the crystal structure of the human Pin1 WW domain has been obtained via racemic crystallization in the presence of small-molecule additives. Both enantiomers of a 36-residue variant of the Pin1 WW domain were synthesized chemically, and the l- and d-polypeptides were combined to afford diffracting crystals. The structural data revealed packing interactions of small carboxylic acids, either achiral citrate or a d,l mixture of malic acid, with a mobile loop region of the WW-domain fold. These interactions with solution additives may explain our success in crystallization of this protein racemate. Molecular-dynamics simulations starting from the structure of the Pin1 WW domain suggest that the crystal structure closely resembles the conformation of this domain in solution. The structural data presented here should provide a basis for further studies of this important model system.
1. Introduction
Small protein domains that spontaneously form tertiary structures in aqueous solution have found widespread use in fundamental studies of protein folding (Kubelka et al., 2004 ▶). Such systems are of particular interest if they lack cysteine or metal-ion-mediated cross-links. Examples include the naturally occurring villin headpiece subdomain (VHP; McKnight et al., 1996 ▶) and WW domains (Macias et al., 1996 ▶, 2002 ▶) as well as de novo designs such as the Trp-cage (Neidigh et al., 2002 ▶). These folding modules display the long-range nonbonded contacts that are the hallmark of protein tertiary structure and are small enough to be tractable for computational studies. Polypeptides of ≤50 residues are readily prepared via solid-phase peptide synthesis (SPPS); therefore, tertiary folding motifs in this size range enable the study of relationships between sequence and conformational stability to extend beyond the proteinogenic amino acids (Lengyel & Horne, 2012 ▶; Arnold et al., 2002 ▶, 2013 ▶; Woll et al., 2006 ▶; David et al., 2008 ▶; Fuller et al., 2009 ▶; Reinert et al., 2012 ▶; Goldberg et al., 2012 ▶; Mortenson et al., 2012 ▶).
Among the small set of known mini-protein tertiary structures, examples are available that display predominantly α-helical, predominantly β-sheet or mixed α+β secondary-structural elements (Kubelka et al., 2004 ▶). X-ray crystallographic characterization of these small tertiary structures in isolation is uncommon, particularly for systems that contain predominantly β-sheet secondary structure. For example, although WW domains occur as substructures within many large proteins, only one crystal structure has been reported of an isolated WW domain (Meiyappan et al., 2007 ▶). Several NMR-based structures of isolated WW domains are known (Macias et al., 2000 ▶; Kowalski et al., 2002 ▶; Pires et al., 2005 ▶; Aragón et al., 2012 ▶). X-ray crystallography is an important complement to NMR, as the higher precision with which the atomic positions are determined is valuable as a starting point for computational studies and potentially for mechanistic descriptions of these folding modules.
WW domains contain approximately 35 residues and are found within a variety of proteins (Macias et al., 1996 ▶). The WW designation is based on the two invariant tryptophans that occur in this small autonomously folding domain. The WW-domain tertiary structure is dominated by a three-stranded antiparallel β-sheet. One widely studied WW domain occurs at the N-terminus of the human Pin1 protein (Fig. 1 ▶). This WW domain binds to phosphopeptide segments that contain the consensus sequence (pS/pT)P. The prolyl isomerase domain of Pin1 catalyzes cis–trans isomerization of the proline residue in this consensus sequence (Ranganathan et al., 1997 ▶). This isomerization may play a role in intracellular signal transduction because the cis and trans isomers are dephosphorylated at different rates by phosphatases (Zhang et al., 2012 ▶). Pin1 regulates numerous intracellular processes including the function of RNA polymerase II and the progression through mitosis (Lu & Zhou, 2007 ▶). Several oncogenes fall under the Pin1 cascade. For example, this enzyme has recently been shown to down-regulate the tumor suppressor RUNX3 in breast-cancer cells (Tsang et al., 2013 ▶).
Figure 1.

Structure and unit-cell packing of racemic Pin1 WW domain cocrystallized with dl-malate. Left, the l chain shows the overall three-stranded fold and the invariant tryptophans of the WW domain (also underlined in the sequence). Right, the centrosymmetric packing of l-polypeptides (green) and d-polypeptides (orange).
Kelly and coworkers have made extensive use of the isolated Pin1 WW domain to explore the effects of both side-chain modification (Jäger et al., 2009 ▶) and backbone modification (Fu et al., 2006 ▶) on the kinetics and thermodynamics of tertiary-structure formation. This work has shown that the Pin1 WW domain is generally tolerant of single-amino-acid substitutions. Alanine substitution is accommodated at all but four of the positions within the Pin1 WW domain sequence; two of the intolerant positions are the conserved tryptophan residues (Jäger et al., 2009 ▶). Cooperative folding and unfolding transitions make the Pin1 WW domain a highly useful scaffold for fundamental studies of protein folding.
In spite of its extensive use as a model system, the Pin1 WW domain has resisted conventional crystallization efforts that could provide structural data to complement solution NMR and other biophysical experiments. Several modified versions of this WW domain have been crystallized in the context of the full Pin1 protein (Jäger et al., 2009 ▶); however, neither the native sequence nor any known variant has been crystallized in isolation. The isolated WW domain is an attractive target for crystallization relative to the full-length Pin1 protein because the length of the latter makes the incorporation of non-proteinogenic residues difficult or impossible, while chemical synthesis of the WW domain alone offers ready access to variants containing novel backbone or side-chain substitutions. Here, we describe the use of racemic crystallization to obtain two X-ray structures of the native Pin1 WW domain.
Racemic protein crystallization was first reported by Zawadzke and Berg, who used chemical synthesis to prepare both enantiomers of rubredoxin (Zawadzke & Berg, 1993 ▶). One motivation for this effort was the recognition that centrosymmetric arrangements of l- and d-polypeptides in the crystal lattice can provide benefits in terms of phase estimation and structural solution based on X-ray diffraction data. A number of subsequent studies have focused on racemic crystallization of polypeptides that are recalcitrant to conventional crystallization approaches, because racemates have a higher statistical likelihood of crystallization than do single enantiomers. This expectation is based on the availability of many space groups in which racemic mixtures may crystallize that are not available to enantiomerically pure molecules (Wukovitz & Yeates, 1995 ▶). The use of racemic or quasi-racemic crystallization for the determination of protein structures is growing (Patterson et al., 1999 ▶; Hung et al., 1999 ▶; Pentelute et al., 2008 ▶, 2010 ▶; Banigan et al., 2010 ▶; Mandal, Pentelute et al., 2012 ▶; Mandal, Uppalapati et al., 2012 ▶; Mortenson et al., 2012 ▶; Yeates & Kent, 2012 ▶), and sophisticated phasing methods unique to these samples are being developed (Sawaya et al., 2012 ▶).
2. Materials and methods
2.1. Chemical synthesis and purification of enantiomeric Pin1 WW domain
l- and d-enantiomers of a previously reported variant of the human Pin1 WW domain (H2N-GSKLPPGWEKRMSRSSGRVYYFNHITNASQWERPSG-COOH) (Kowalski et al., 2002 ▶) were generated via microwave-assisted SPPS using Fmoc-protected amino acids and NovaPEG Wang resin (NovaBioChem). Details are given in the Supplementary Material1. Peptides were cleaved from the resin for 3 h under Reagent K conditions [82.5%(v/v) trifluoroacetic acid (TFA), 5%(v/v) water, 5%(v/v) thioanisole, 5%(v/v) ethanedithiol, 5%(w/v) phenol; Guy & Fields, 1997 ▶], after which time the cleavage cocktails were drained through fritted syringes. The resin was washed several times with additional TFA. Combined TFA fractions were reduced to approximately 50% volume under streams of N2 gas and were then added to cold diethyl ether to precipitate the crude peptide. The crude peptide products were collected via centrifugation and decanting of the ether layers and were then washed with additional volumes of cold ether and centrifuged in the same manner. Crude peptide mixtures were dried and then dissolved in a 1:1 mixture of H2O:acetonitrile for reverse-phase HPLC purification. Peptides were purified on a Shimadzu SCL-10A liquid-chromatography system fitted with a C18-functionalized column (Supelco). The binary solvent system used in the purifications was H2O:TFA [100:0.1(v:v)] as solvent A and acetonitrile:TFA [100:0.1(v:v)] as solvent B. Aliquots of crude peptides were purified over a 20 min gradient of 17–27% solvent B at a total flow rate of 15 ml min−1. Fractions containing the product peptides were pooled and lyophilized to dryness, and their purity was assessed by analytical HPLC. Peptide identity was confirmed by MALDI–TOF mass-spectrometric analysis. All peptides generated for this study were of >95% purity as determined by integration of peak areas from analytical HPLC chromatograms (Supplementary Material).
2.2. Crystallization conditions
Conditions for racemic Pin1 WW domain crystals suitable for X-ray data collection were identified using a sparse-matrix screen (Index, Hampton Research). Crystals were grown at room temperature (295 K) via vapor diffusion using the hanging-drop method (McPherson, 1982 ▶) by combining 1 µl racemic peptide stock (2.5 mg ml−1 of each enantiomeric peptide in pure water) with 1 µl precipitant solution on silanized glass slides (Hampton Research) and equilibrating against reservoir volumes of 500 µl precipitant solution in Linbro-style plates. Single crystals grew overnight from precipitant solutions containing either 2.1 M dl-malic acid pH 7.0 or 1.8 M triammonium citrate pH 7.0. Attempts at cryoprotection using various organic additives were unsuccessful; consequently, diffraction data were collected at room temperature.
2.3. Data collection, processing and refinement
For racemic Pin1 WW domain cocrystallized with dl-malic acid, diffraction data were collected at 298 K from crystals mounted in quartz capillaries using an AXS Microstar X-ray generator and a SMART6000 CCD detector (Bruker). Indexing, integration and scaling of the data to 2.25 Å resolution were carried out using the HKL-2000 package (Otwinowski & Minor, 1997 ▶). A similar procedure was used for data collection and processing from crystals of racemic Pin1 WW domain cocrystallized with triammonium citrate, with a resolution limit of 3.05 Å (Table 1 ▶).
Table 1. Data-collection statistics.
Values in parentheses are for the outer shell.
| Pin1 WW with malate (4gwt) | Pin1 WW with citrate (4gwv) | |
|---|---|---|
| Temperature (K) | 298 | 298 |
| Source | Bruker AXS | Bruker AXS |
| Detector | SMART6000 | SMART6000 |
| Wavelength () | 1.5418 | 1.5418 |
| Resolution range | 33.322.25 (2.292.25) | 34.253.05 (3.163.05) |
| Unique reflections | 4031 (190) | 1615 (154) |
| Completeness (%) | 100.0 (100.0) | 98.2 (96.4) |
| R merge † | 0.069 (0.290) | 0.129 (0.356) |
| I/(I) | 23.6 (6.1) | 7.2 (3.6) |
| Multiplicity | 13.3 (9.1) | 4.0 (4.0) |
| Wilson B factor (2) | 41.6 | 75.3 |
| Space group | I41/a | I41/a |
| Unit-cell parameters (, ) | a = b = 66.0, c = 38.6, = = = 90 | a = b = 64.6, c = 40.4, = = = 90 |
| Water content (%) | 52 | 52 |
The structure of Pin1 WW domain cocrystallized with dl-malic acid was initially solved in space group P−1 via molecular replacement in Phaser (McCoy et al., 2007 ▶) using a three-stranded β-sheet fragment obtained from the structure of the human FE65 WW domain (PDB entry 2idh; Meiyappan et al., 2007 ▶) as an initial model. The partial solution of four copies of this model per asymmetric unit was extended through a combined density-modification and chain-tracing protocol in SHELXE (Sheldrick, 2010 ▶), and manual rebuilding of the resulting model was carried out in Coot (Emsley & Cowtan, 2004 ▶). The space group of the structure was then assigned as I41/a, reducing the contents of the asymmetric unit to a single polypeptide chain. Refinement of this model was carried out in REFMAC5 (Murshudov et al., 2011 ▶; Table 2 ▶).
Table 2. Refinement statistics.
Values in parentheses are for the outer shell.
| Pin1 WW with malate (4gwt) | Pin1 WW with citrate (4gwv) | |
|---|---|---|
| Resolution range () | 33.232.25 | 34.253.05 |
| R work | 0.238 (0.348) | 0.205 (0.323) |
| R free | 0.260 (0.334) | 0.269 (0.325) |
| No. of reflections, working set | 3685 | 1375 |
| No. of reflections, test set | 172 | 151 |
| No. of non-H atoms | ||
| Protein | 276 | 276 |
| Solvent | 11 | 2 |
| Ligand | 9 | 13 |
| R.m.s.d., bond lengths () | 0.010 | 0.011 |
| R.m.s.d., bond angles () | 1.5 | 1.4 |
| Coordinate e.s.u. () | 0.086 | 0.189 |
| Average B factor (2) | ||
| Protein | 39.6 | 36.9 |
| Solvent | 51.8 | 36.1 |
| Ligand | 42.7 | 62.1 |
| Ramachandran plot | ||
| Favored regions (%) | 100 | 96.8 |
| Additionally allowed regions (%) | 0 | 3.2 |
| Outliers (%) | 0 | 0 |
The data obtained from racemic Pin1 WW domain cocrystallized with triammonium citrate were merged and scaled in space group I41/a from the outset (Table 1 ▶). The refined model obtained from the dl-malic acid-containing crystals was used as a molecular-replacement model for the citrate-containing structure. Refinement of this structure was then carried out in REFMAC5 at 3.05 Å resolution (Table 2 ▶). In order to present the two structures in equivalent positions within their respective unit cells, data were reindexed ex post facto by the h, k, l → k, h, −l transformation and the atomic coordinates were adjusted by the x, y, z → y, x, −z operation.
The full resolution ranges of the two data sets were used throughout the model refinements. Isotropic temperature factors were used in both cases and were not restrained. Solvent positions in the two models were initially determined automatically in REFMAC5 and were updated manually in Coot. In both models, two amino acids at the N-terminus and one at the C-terminus were omitted owing to disorder.
Following model refinement, we found that the average B factor of the citrate-containing structure (36.9 Å2) had converged at a value of roughly half of the Wilson B factor (75.3 Å2) determined from the corresponding reflection data. In order to investigate this apparent discrepancy, a set of calculated amplitudes was generated from the refined citrate-containing model using the program SFALL in CCP4 (Winn et al., 2011 ▶); the program SFCHECK (Vaguine et al., 1999 ▶) was then used to obtain a Wilson plot from these simulated data. The value of the Wilson B factor determined from the model-derived data was 75.3 Å2,which is in very good agreement with the value obtained from the Wilson plot. Although we cannot fully explain the origin of the difference, it is partially accounted for if the three omitted amino acids and unmodeled waters in this structure are highly disordered, leading to an artificially lowered average B factor for the refined atoms.
2.4. Molecular-dynamics simulation
CHARMM27 force-field parameters and the TIP3P water model were used in the simulation, which was carried out in the GROMACS 4.6 software package (MacKerell et al., 1998 ▶; Mackerell et al., 2004 ▶; Hess et al., 2008 ▶). The l-polypeptide used in the simulation was taken from the structure of the racemic Pin1 WW domain cocrystallized with dl-malic acid. The polypeptide termini were treated as neutral. Coot (Emsley & Cowtan, 2004 ▶) was used to build in the side chain of Lys6, which was disordered in the X-ray structure. The peptide was solvated in a periodic rhombic dodecahedral box with a 10 Å solute-box cutoff. To neutralize the +4 net charge of the peptide, four Cl− counterions were added to the box. The system underwent a steepest-descent minimization for 50 000 steps with a 10 J mol−1 step size. Next, a 100 ps position-restrained NVT equilibration with a 2 fs step size was carried out on the system. The temperature was set and maintained at 300 K with a Berendsen thermostat (Berendsen et al., 1984 ▶). A subsequent NPT equilibration was carried out on the system using similar parameters. A Parrinello–Rahman barostat was used to maintain the reference pressure at 100 kPa during NPT equilibration (Bussi et al., 2007 ▶). For the 100 ns simulation the position restraint was removed and the simulation was allowed to run for 100 ns with a 2 fs step size. The temperature and pressure coupling schemes used during the equilibration runs were also used during the 100 ns simulation.
For equilibration and production MD runs a leap-frog integrator was used, and the particle mesh Ewald method was applied with a 10 Å cutoff to treat long-range Coulombic interactions (Essmann et al., 1995 ▶). The LINCS algorithm was used to constrain covalent bonds (Hess et al., 1997 ▶). The 100 ns production run was carried out on the UW-Madison Department of Chemistry Phoenix Computing Cluster.
3. Results and discussion
3.1. The structure of Pin1 WW domain
We have solved and refined two crystal structures of the isolated Pin1 WW domain obtained by cocrystallization of the enantiomers of this polypeptide. In the two crystal forms, l and d Pin1 WW domain polypeptides are packed in virtually identical centrosymmetric arrangements corresponding to space group I41/a (Fig. 1 ▶). The structure that contains malate was refined at 2.25 Å resolution to R and R free values of 0.238 and 0.260, respectively, while the structure that contains citrate was refined at 3.05 Å resolution to R and R free values of 0.205 and 0.270, respectively. Based on the expectation that centrosymmetric reflection intensities should be more broadly distributed than non-centrosymmetric data, the R factors that we report for the structures of racemic Pin1 WW domain correspond to R and R free values of 0.159 and 0.175, repectively, for the malate-containing structure, and of 0.136 and 0.182, respectively, for the citrate-containing structure for non-centrosymmetric structures with similar coordinate error (Wilson, 1949 ▶; Luzzati, 1952 ▶). The two structures have similar unit-cell parameters (Table 1 ▶), and the conformations of the Pin1 WW domain found within them are closely related (r.m.s.d. of 0.3 Å over all Cα atoms).
Crystals of racemic Pin1 WW grew from two of the 96 precipitant conditions tested (Index from Hampton Research). One of these conditions contained a racemic small molecule, dl-malic acid, while the other contained a structurally related achiral small molecule, citric acid. Citric acid can be considered to be a derivative of malic acid that contains an extra CH2CO2H unit (Fig. 2 ▶). Interestingly, succinic acid, an achiral molecule similar to malic acid but lacking the hydroxyl substituent (Fig. 2 ▶), did not promote the growth of racemic Pin1 WW domain crystals. In both structures reported here, l- and d-versions of the Pin1 WW domain are related by inversion symmetry elements; hence, a small-molecule additive must be either achiral or racemic in order to cocrystallize with the racemic polypeptide, according to the lattice symmetry.
Figure 2.
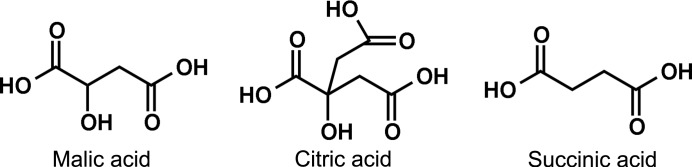
Structures of the organic additives malic acid, citric acid and succinic acid. The first two molecules facilitated crystallization of racemic Pin1 WW domain, whereas succinic acid did not.
In previous structures, loop I has been the most variable portion of the WW-domain sequence (Peng et al., 2007 ▶). This loop is involved in substrate binding, and the sequence diversity in this segment presumably underlies the binding preferences of different WW domains. NMR analysis indicates that the loop I region of the Pin1 WW domain (S16-R-S-S-G-R21), which forms a type 4:6 β-hairpin, is dynamic in solution (Kowalski et al., 2002 ▶). Additionally, bioinformatics analysis of type 4:6 β-hairpin supersecondary structures in the protein structure database suggests that many different specific backbone conformations are consistent with this motif (Sibanda et al., 1989 ▶). It seems likely that the intrinsic flexibility of loop I could act as a significant entropic barrier to crystallization of the isolated Pin1 WW domain.
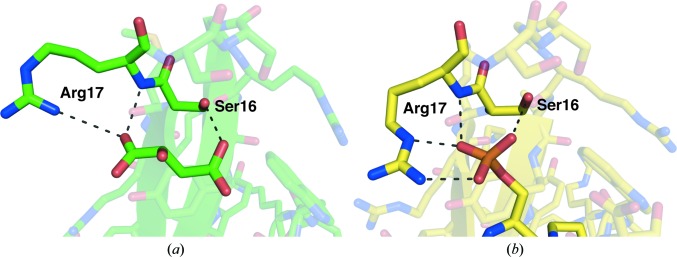
In the two structures of racemic Pin1 WW domain reported here, we observe extensive hydrogen bonding between residues in loop I and either malate or citrate; these additives also make contacts with other regions of symmetry-related polypeptides (Fig. 3 ▶). The inter-peptide contacts mediated by citrate and malate take place between polypeptides of the same chirality: in the malate structure, l–l contacts are mediated by d-malate and d–d contacts are mediated by l-malate. Interestingly, this arrangement requires that the racemic small molecule be ‘resolved’ into its enantiomers to satisfy mirror-image versions of the same packing contact within the centrosymmetric lattice. We speculate that the interactions involving malate or citrate serve to lock loop I in specific conformations that favor crystal growth. Consistent with this hypothesis, we observe that the values of the crystallographic B factor determined for loop I residues do not deviate significantly from the structure averages and that these residues are well resolved in the electron-density maps. The pattern of intramolecular hydrogen bonds between backbone amide groups that is characteristic of a 4:6 β-hairpin is not disrupted by the interactions with citrate or malate (Fig. 4 ▶).
Figure 3.

Views of crystal-packing interactions involving loop I residues and malate (a) or citrate (b). Hydrogen bonds are indicated by dashed lines. Residues from symmetry-related polypeptides are denoted by primes.
Figure 4.

Fold of the Pin1 WW domain as crystallized with dl-malate and refined at 2.25 Å resolution. Main-chain hydrogen bonds are indicated by dashed lines. Amino-acid residue numbering (red for carbonyl O atoms and blue for main-chain N atoms) highlights the 4:6 β-hairpin structure of loop I.
In the structures of the racemic Pin1 WW domain, the small molecules malate and citrate form intermolecular hydrogen bonds to both backbone and side-chain groups in loop I. Interestingly, the binding of these additives to the loop is closely analogous to the interaction of the Pin1 WW domain with its native substrates. Comparison to an X-ray structure of Pin1 complexed with a substrate peptide (PDB entry 1f8a; Verdecia et al., 2000 ▶) indicates that the hydrogen bonds formed between malate and residues in loop I are closely related to the contacts made with phosphate (Fig. 5 ▶). Additionally, a carboxylate anion of either malate or citrate may form an ionic interaction with Arg17 in a manner similar to phosphate. There is a 0.44 Å r.m.s.d. between this Pin1–substrate complex and the racemic structures that we report over Cα atoms 6–36 with the same loop I conformation. This comparison indicates that the structures of racemic Pin1 WW domain cocrystallized with malate or citrate reflect the properties of this domain in its native chiral form.
Figure 5.
Comparison of hydrogen bonds (dashed lines) between loop I residues and malate from the structure of racemic Pin1 WW domain (a) with hydrogen bonds formed between the same residues and a phosphate group from a substrate phosphopeptide (b) (PDB entry 1f8a; Verdecia et al., 2000 ▶).
3.2. Simulation supports the racemic structure
The NMR structure of the isolated Pin1 WW domain reported by Kowalski et al. (2002 ▶) displays a high degree of conformational variability in the loop I region and at both termini. We wished to assess whether our structures of racemic Pin1 WW domain, which are subject to packing interactions between the loop I region and small-molecule additives, reflect a conformation that would be favorable for an isolated Pin1 WW domain in solution. Molecular-dynamics (MD) simulations were carried out to address this question, starting from the Pin1 WW domain structure cocrystallized with dl-malic acid.
The simulated Pin1 WW domain tertiary structure was stable in aqueous solution over the course of 100 ns. This result suggests that the conformation of the WW domain that we observe in the racemic crystal form is representative of the range of conformations that this polypeptide can adopt in solution. Although the loop I region of the WW domain is well resolved in the racemic crystal structure, we observed that this region of the structure was conformationally dynamic over the course of the simulation (Fig. 6 ▶). This finding is consistent with the results of Kowalski et al. (2002 ▶) and suggests that the small-molecule additives rigidify loop I in the racemic crystal.
Figure 6.

Comparison of the root-mean-square fluctuation (RMSF) for backbone atoms (N, Cα and C) from MD simulation and crystallographic B factors determined for the same residues in the structure of racemic Pin1 WW domain cocrystallized with dl-malate. The largest fluctuations predicted by simulation in aqueous solution correspond to loop I residues (17–20), whereas the values of the B factors determined for these residues do not differ significantly from the structure average.
4. Conclusions
Our efforts to crystallize racemic Pin1 WW domain suggest that small-molecule additives can play an intimate role in protein crystallization by mimicking a natural binding partner. In the case of the chiral additive malate, we were fortunate to have used the racemic form because the two enantiomers take part in mirror-image versions of the same interactions in the centrosymmetric crystal. A related achiral additive, citrate, also facilitated the growth of diffraction-quality crystals from the racemic protein.
Molecular-dynamics simulations and comparison with a previous NMR-based structure suggest that the Pin1 WW domain structure that we have determined in crystallo is a good model for the conformation of this small protein module in solution. The generally good agreement among our crystal structures, crystal structures of the WW domain within full-length Pin1 and the previously reported NMR structure of the Pin1 WW domain suggests that racemic crystallization can provide insight into the intrinsic folding behavior of an isolated polypeptide chain.
The racemic approach provides no guarantee of success in terms of the growth of diffraction-quality crystals; indeed, our efforts to crystallize racemic mixtures of other polypeptides have often met with failure. However, given the prediction that racemic mixtures of polypeptides should crystallize more readily than either enantiomer alone (Wukovitz & Yeates, 1995 ▶), racemic crystallization offers a useful alternative when conventional crystallization approaches fail, as in the case of the isolated Pin1 WW domain. The two structures reported here suggest that loop I stabilization is an important prerequisite for crystal growth and that the crystallization of racemic Pin1 WW domain in the presence of either dl-malate or citrate locks this loop in a defined conformation. Interestingly, we find that the interactions between small-molecule additives and the loop in our crystals are analogous to interactions between this loop and native substrate peptides. For other proteins that contain flexible substrate-binding loops, it may be generally productive to include small-molecule additives that can mimic substrate interactions among the factors evaluated in the screening of conditions for crystallization.
Supplementary Material
PDB reference: Pin1 WW domain with malate, 4gwt
PDB reference: with citrate, 4gwv
Methodological details of peptide synthesis.. DOI: 10.1107/S090744491302444X/bw5418sup1.pdf
Acknowledgments
This work was supported by the Wisconsin Alumni Research Foundation and the NIH (GM061238). DEM was supported in part by a Molecular Biophysics Training Grant (NIH T32 GM008293) and DFK in part by a Biotechnology Training Grant (NIH T32 GM008349). Use of the UW-Madison Chemistry Phoenix Cluster was supported by National Science Foundation Grant CHE-0840494.
Footnotes
Supplementary material has been deposited in the IUCr electronic archive (Reference: BW5418). Services for accessing this material are described at the back of the journal.
References
- Aragón, E., Goerner, N., Xi, Q., Gomes, T., Gao, S., Massagué, J. & Macias, M. J. (2012). Structure, 20, 1726–1736. [DOI] [PMC free article] [PubMed]
- Arnold, U., Hinderaker, M. P., Nilsson, B. L., Huck, B. R., Gellman, S. H. & Raines, R. T. (2002). J. Am. Chem. Soc. 124, 8522–8523. [DOI] [PubMed]
- Arnold, U., Huck, B. R., Gellman, S. H. & Raines, R. T. (2013). Protein Sci. 22, 274–279. [DOI] [PMC free article] [PubMed]
- Banigan, J. R., Mandal, K., Sawaya, M. R., Thammavongsa, V., Hendrickx, A. P. A., Schneewind, O., Yeates, T. O. & Kent, S. B. H. (2010). Protein Sci. 19, 1840–1849. [DOI] [PMC free article] [PubMed]
- Berendsen, H. J. C., Postma, J. P. M., van Gunsteren, W. F., DiNola, A. & Haak, J. R. (1984). J. Chem. Phys. 81, 3684.
- Bussi, G., Donadio, D. & Parrinello, M. (2007). J. Chem. Phys. 126, 014101. [DOI] [PubMed]
- David, R., Günther, R., Baumann, L., Lühmann, T., Seebach, D., Hofmann, H.-J. & Beck-Sickinger, A. G. (2008). J. Am. Chem. Soc. 130, 15311–15317. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Essmann, U., Perera, L., Berkowitz, M. L., Darden, T., Lee, H. & Pedersen, L. G. (1995). J. Chem. Phys. 103, 8577.
- Fu, Y., Gao, J., Bieschke, J., Dendle, M. A. & Kelly, J. W. (2006). J. Am. Chem. Soc. 128, 15948–15949. [DOI] [PMC free article] [PubMed]
- Fuller, A. A., Du, D., Liu, F., Davoren, J. E., Bhabha, G., Kroon, G., Case, D. A., Dyson, H. J., Powers, E. T., Wipf, P., Gruebele, M. & Kelly, J. W. (2009). Proc. Natl Acad. Sci. USA, 106, 11067–11072. [DOI] [PMC free article] [PubMed]
- Goldberg, J. M., Speight, L. C., Fegley, M. W. & Petersson, E. J. (2012). J. Am. Chem. Soc. 134, 6088–6091. [DOI] [PMC free article] [PubMed]
- Guy, C. A. & Fields, G. B. (1997). Methods Enzymol. 289, 67–83. [DOI] [PubMed]
- Hess, B., Bekker, H., Berendsen, H. J. C. & Fraaije, J. G. E. M. (1997). J. Comput. Chem. 18, 1463–1472.
- Hess, B., Kutzner, C., van der Spoel, D. & Lindahl, E. (2008). J. Chem. Theory Comput. 4, 435–447. [DOI] [PubMed]
- Hung, L.-W., Kohmura, M., Ariyoshi, Y. & Kim, S.-H. (1999). J. Mol. Biol. 285, 311–321. [DOI] [PubMed]
- Jäger, M., Dendle, M. & Kelly, J. W. (2009). Protein Sci. 18, 1806–1813. [DOI] [PMC free article] [PubMed]
- Kowalski, J. A., Liu, K. & Kelly, J. W. (2002). Biopolymers, 63, 111–121. [DOI] [PubMed]
- Kubelka, J., Hofrichter, J. & Eaton, W. A. (2004). Curr. Opin. Struct. Biol. 14, 76–88. [DOI] [PubMed]
- Lengyel, G. A. & Horne, W. S. (2012). J. Am. Chem. Soc. 134, 15906–15913. [DOI] [PubMed]
- Lu, K. P. & Zhou, X. Z. (2007). Nature Rev. Mol. Cell Biol. 8, 904–916. [DOI] [PubMed]
- Luzzati, V. (1952). Acta Cryst. 5, 802–810.
- Macias, M. J., Gervais, V., Civera, C. & Oschkinat, H. (2000). Nature Struct. Biol. 7, 375–379. [DOI] [PubMed]
- Macias, M. J., Hyvönen, M., Baraldi, E., Schultz, J., Sudol, M., Saraste, M. & Oschkinat, H. (1996). Nature (London), 382, 646–649. [DOI] [PubMed]
- Macias, M. J., Wiesner, S. & Sudol, M. (2002). FEBS Lett. 513, 30–37. [DOI] [PubMed]
- MacKerell, A. D. Jr et al. (1998). J. Phys. Chem. B, 102, 3586–3616. [DOI] [PubMed]
- Mackerell, A. D. Jr, Feig, M. & Brooks, C. L. III (2004). J. Comput. Chem. 25, 1400–1415. [DOI] [PubMed]
- Mandal, K., Pentelute, B. L., Bang, D., Gates, Z. P., Torbeev, V. Y. & Kent, S. B. H. (2012). Angew. Chem. Int. Ed. 51, 1481–1486. [DOI] [PubMed]
- Mandal, K., Uppalapati, M., Ault-Riché, D., Kenney, J., Lowitz, J., Sidhu, S. S. & Kent, S. B. H. (2012). Proc. Natl Acad. Sci. USA, 109, 14779–14784. [DOI] [PMC free article] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- McKnight, C. J., Doering, D. S., Matsudaira, P. T. & Kim, P. S. (1996). J. Mol. Biol. 260, 126–134. [DOI] [PubMed]
- McPherson, A. (1982). Preparation and Analysis of Protein Crystals. Chichester: John Wiley & Sons.
- Meiyappan, M., Birrane, G. & Ladias, J. A. (2007). J. Mol. Biol. 372, 970–980. [DOI] [PMC free article] [PubMed]
- Mortenson, D. E., Satyshur, K. A., Guzei, I. A., Forest, K. T. & Gellman, S. H. (2012). J. Am. Chem. Soc. 134, 2473–2476. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Neidigh, J. W., Fesinmeyer, R. M. & Andersen, N. H. (2002). Nature Struct. Biol. 9, 425–430. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Patterson, W. R., Anderson, D. H., DeGrado, W. F., Cascio, D. & Eisenberg, D. (1999). Protein Sci. 8, 1410–1422. [DOI] [PMC free article] [PubMed]
- Peng, T., Zintsmaster, J. S., Namanja, A. T. & Peng, J. W. (2007). Nature Struct. Mol. Biol. 14, 325–331. [DOI] [PubMed]
- Pentelute, B. L., Gates, Z. P., Tereshko, V., Dashnau, J. L., Vanderkooi, J. M., Kossiakoff, A. A. & Kent, S. B. H. (2008). J. Am. Chem. Soc. 130, 9695–9701. [DOI] [PMC free article] [PubMed]
- Pentelute, B. L., Mandal, K., Gates, Z. P., Sawaya, M. R., Yeates, T. O. & Kent, S. B. H. (2010). Chem. Commun., 46, 8174–8176. [DOI] [PubMed]
- Pires, J. R., Parthier, C., do Aido-Machado, R., Wiedemann, U., Otte, L., Böhm, G., Rudolph, R. & Oschkinat, H. (2005). J. Mol. Biol. 348, 399–408. [DOI] [PubMed]
- Ranganathan, R., Lu, K. P., Hunter, T. & Noel, J. P. (1997). Cell, 89, 875–886. [DOI] [PubMed]
- Reinert, Z. E., Musselman, E. D., Elcock, A. H. & Horne, W. S. (2012). Chembiochem, 13, 1107–1111. [DOI] [PubMed]
- Sawaya, M. R., Pentelute, B. L., Kent, S. B. H. & Yeates, T. O. (2012). Acta Cryst. D68, 62–68. [DOI] [PubMed]
- Sheldrick, G. M. (2010). Acta Cryst. D66, 479–485. [DOI] [PMC free article] [PubMed]
- Sibanda, B. L., Blundell, T. L. & Thornton, J. M. (1989). J. Mol. Biol. 206, 759–777. [DOI] [PubMed]
- Tsang, Y.-H. N., Wu, X.-W., Lim, J.-S., Wee Ong, C., Salto-Tellez, M., Ito, K., Ito, Y. & Chen, L.-F. (2013). Oncogene, 32, 1488–1496. [DOI] [PMC free article] [PubMed]
- Vaguine, A. A., Richelle, J. & Wodak, S. J. (1999). Acta Cryst. D55, 191–205. [DOI] [PubMed]
- Verdecia, M. A., Bowman, M. E., Lu, K. P., Hunter, T. & Noel, J. P. (2000). Nature Struct. Biol. 7, 639–643. [DOI] [PubMed]
- Wilson, A. J. C. (1949). Acta Cryst. 2, 318–321.
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Woll, M. G., Hadley, E. B., Mecozzi, S. & Gellman, S. H. (2006). J. Am. Chem. Soc. 128, 15932–15933. [DOI] [PubMed]
- Wukovitz, S. W. & Yeates, T. O. (1995). Nature Struct. Biol. 2, 1062–1067. [DOI] [PubMed]
- Yeates, T. O. & Kent, S. B. H. (2012). Annu. Rev. Biophys. 41, 41–61. [DOI] [PubMed]
- Zawadzke, L. E. & Berg, J. M. (1993). Proteins, 16, 301–305. [DOI] [PubMed]
- Zhang, M., Wang, X. J., Chen, X., Bowman, M. E., Luo, Y., Noel, J. P., Ellington, A. D., Etzkorn, F. A. & Zhang, Y. (2012). ACS Chem. Biol. 7, 1462–1470. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: Pin1 WW domain with malate, 4gwt
PDB reference: with citrate, 4gwv
Methodological details of peptide synthesis.. DOI: 10.1107/S090744491302444X/bw5418sup1.pdf