

Summary
Small molecule modulators of protein activity are increasingly being utilized as tools to examine the functional roles of proteins. Operating at the post-translational level, these molecules provide enhanced temporal and spatial control and mitigate the potential for compensatory responses in comparison with classical genetic approaches. Proteolysis targeting chimeric molecules, or PROTACs, are small molecules that inhibit the function of their target proteins by targeting them for degradation by the ubiquitin proteasome system (UPS). This chapter summarizes strategies for PROTAC preparation and characterization.
Keywords: Chemical Genetics, PROTAC, Ubiquitin, Proteasome, Small molecule, E3 ubiquitin ligase, Protein degradation
1. Introduction
Traditional genetic techniques have been widely used to elucidate protein function. While these techniques, such as gene knockouts and small interfering RNA (siRNA), inhibit protein expression at the DNA or RNA levels, they are constrained by compensatory mechanisms resulting from protein redundancy or lethality due to eliminating proteins necessary for viability. Unlike these traditional approaches, chemical genetic techniques operate at the post-translational level, allowing efficient temporal and spatial control. However, there are few chemical genetic techniques that can be generalized to a variety of proteins.
Proteolysis targeting chimeric molecules, or PROTACs, provide an example of a post-translational chemical genetic technique capable of more generally targeting the proteome. These chimeric small molecules are designed to induce the degradation of their target proteins via the ubiquitin proteasome system (UPS), thereby eliminating pre-existing proteins. The UPS is the major intracellular pathway for protein degradation in which a series of enzymes known as E1s (ubiquitin activating enzymes), E2s (ubiquitin conjugating enzymes) and E3s (ubiquitin ligases) carry out covalent linkage of the 9kDa, 76 amino acid protein ubiquitin to a target protein. Subsequent enzymatic reactions result in the formation of a polyubiquitin chain, which targets the protein for degradation by the 26S proteasome. Specificity for a particular target protein is associated with the E3 ligase (1), which facilitates the final step of ubiquitin attachment to the target protein.
To promote ubiquitination and proteasomal degradation of a specific target protein via the UPS, PROTACs are structurally comprised of two recognition motifs separated by a linker. One recognition motif is a small molecule ligand for the target protein of interest, and the other recognizes a specific E3 ligase. Interaction of these motifs with their binding partners results in polyubiquitination of the target protein, which is subsequently subjected to proteasomal degradation (Figure 1). The same E3 ligase recognition motif can be attached to a variety of small molecule ligands, thereby generalizing this approach to a broad spectrum of proteins. By inducing proteolytic degradation of their target proteins, PROTACs represent useful tools for both the elucidation of protein function as well as therapeutic applications (2, 3). For example, PROTAC-induced degradation of a particular protein followed by an investigation of the associated phenotypic changes can provide valuable information about the function of that protein.
Fig.1.
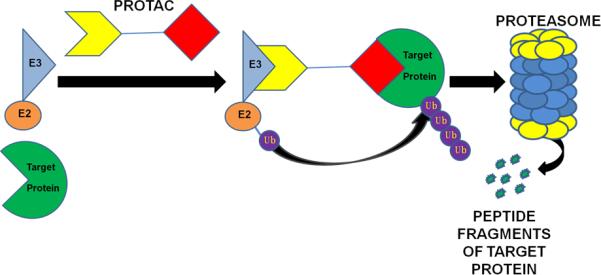
PROTAC-induced degradation of a target protein via the UPS. PROTAC is a heterochimeric molecule composed of an E3 ubiquitin ligase recognition motif on one end and a small-molecule ligand for a target protein on the other end. PROTAC recruits a targeted protein to an E3 ligase complex for ubiquitination and degradation by the proteasome.
While the first generation PROTACs were successfully developed using an E3 ubiquitin ligase-recognizing motif derived from IκBα, they were not cell-permeable (4). The poor cell permeability of the first generation PROTACs was significantly improved by adopting a HIF-1α peptide fragment as an E3 ubiquitin ligase recognition motif in the design of the second generation PROTACs (5-7). So far, androgen receptor (AR) (8) and estrogen receptor (ER) targeting PROTACs (9-11) have been developed, demonstrating the potential applications of these molecules in the treatment of prostate and breast cancers (12, 13) (2, 8). In addition, PROTACs targeting methionine aminopeptidase-2 (MetAP-2) (4), the aryl hydrocarbon receptor (14, 15), and cellular retinoic acid binding proteins (CRABPs) (16) have also been developed. This chapter summarizes the synthetic strategy of PROTAC molecules and includes detailed procedures regarding PROTAC characterization. These procedures involve cell culture, western blotting, and immunoprecipitation techniques and can be utilized to confirm that the PROTAC induces target protein degradation via the UPS.
2. Materials
2.1. PROTAC Synthesis
For a list of materials used for PROTAC synthesis, refer to ref. (2). Depending on the selected ligand and E3 ligase recognition motif, different sets of chemicals will be required.
Of note, the following equipment should be accessible for chemical characterization and purification: Nuclear magnetic resonance (NMR), Mass spectrometry (MS), High performance liquid chromatography (HPLC).
2.2. Confirmation of PROTAC-Induced Target Protein Degradation
MCF7 cells (ATCC)
Cell culture medium: Phenol red-free RPMI 1640 medium (Invitrogen) supplemented with 10% (v/v) fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin.
Cell treatment medium: Phenol red-free RPMI 1640 medium (Invitrogen) supplemented with 5% (v/v) charcoal/dextran treated fetal bovine serum (Hyclone), 100 U/mL penicillin, and 100 μg/mL streptomycin.
24-well tissue culture plates.
DMSO (Sigma-Aldrich).
- PROTAC stock solution: Calculate the volume of DMSO required to dilute the desired mass of the PROTAC to obtain the desired final concentration of the PROTAC stock solution using the following formula:
For example, to create a 10 mM PROTAC stock solution starting with 5 mg of a PROTAC with a molecular weight of 1,172g/mol, first calculate:
Resuspend 5 mg of the PROTAC in 0.427 mL of DMSO to obtain a 10 mM PROTAC stock solution. Negative PROTAC control (hydroxyproline→norleucine): Prepare via the procedure summarized in section 3.1., but substitute norleucine for hydroxyproline.
Hank's Balanced Salt Solution HBSS (Gibco)
1× Trypsin-EDTA: Prepare from 10× Trypsin-EDTA (Gibco) by adding 5 mL of 10× Trypsin-EDTA to 45 mL of HBSS and mixing.
Non-denaturing lysis buffer: 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 15 μL Nonidet P40 (NP40), 1% Triton X-100, add deionized water to 1 mL. Combine in a 1.5 mL microcentrifuge tube and store at −20°C. Immediately prior to use, add 1% protease inhibitor cocktail (Sigma-Aldrich).
Protein Assay Dye Reagent Concentrate (Bio-Rad).
Bovine serum albumin standard (Bio-Rad).
Laemmli Sample Buffer 2× Concentrate (Sigma-Aldrich).
10-12% SDS-PAGE gel.
Prestained SDS-PAGE Standards (Bio-Rad).
Immuno-Blot PVDF Membrane (Bio-Rad).
10× TBS: 6 g of Tris-HCl, 45 g NaCL, add 800 mL of de-ionized water and dissolve. Adjust the pH to 7.4. Add de-ionized water to a final volume of 1L.
0.05% (v/v) TBST: Dilute 10×X TBS to 1× by adding 50 mL of 10× TBS to 450mL of deionized water for a final volume of 500mL. Add 250μL of Tween-20 (Sigma).
5% (w/v) blocking grade nonfat dry milk (Bio-Rad) in 0.05% (v/v) TBST.
Anti-ERα antibody (Santa Cruz, cat. no. sc543).
Anti-β-actin antibody (Novus Biologicals, cat. no. NB600-501).
Anti-rabbit IgG horseradish peroxidase (Amersham).
Anti-mouse IgG horseradish peroxidase (Amersham).
ECL Western Blotting Substrate (Pierce).
Kodak BioMax XAR Film (Sigma-Aldrich).
2.3. Confirmation of Target Protein Ubiquitination
Refer to the materials listed in section 2.2. Additionally, the following are required:
Epoxomicin (Calbiochem).
Protein G-agarose (Roche).
Anti-ubiquitin antibody (Santa Cruz, cat. no. sc-8017).
Non-specific control antibodies: anti-rabbit and anti-mouse IgG (Santa Cruz).
2.4. Confirmation of Specific E3 Ligase-Dependent Target Protein Degradation
Refer to the materials for section 2.2., with the exception of numbers 1-3. Additional materials:
786-O cells
786-O/VHL cells
RPMI-1640 Medium (ATCC) supplemented with 10% (v/v) fetal bovine serum
2.5. Confirmation of Proteasome-Dependent Target Protein Degradation
Refer to the materials listed in section 2.2. Additionally, Epoxomicin (Calbiochem) is required.
3. Methods
3.1. PROTAC Synthesis
A convergent synthetic approach can be applied to PROTAC synthesis (Fig. 2). The E3 ligase recognition motif is first synthesized using a standard peptide synthetic strategy, followed by the synthesis of a ligand for the target protein with an attached handle (see Notes 1-2). This handle is then utilized to link the two components together, creating the chimeric molecule. The synthetic procedure should be specifically designed for a particular small molecule ligand.
Fig.2.
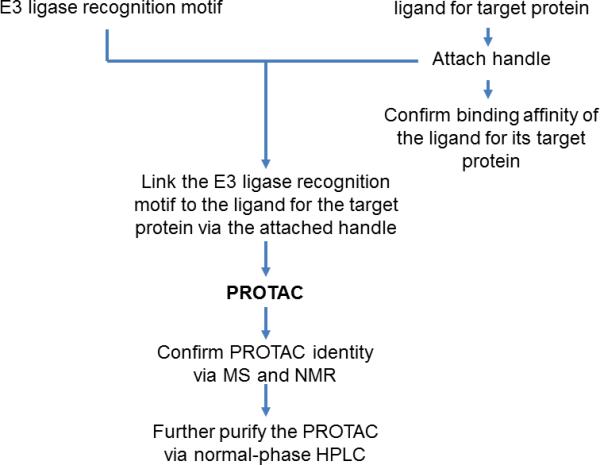
PROTAC synthesis strategy. Chimeric PROTAC molecules are prepared through a convergent approach, first synthesizing two fragments separately and then coupling them to produce a fully assembled PROTAC.
Two E3 ligase recognition motifs have previously been utilized in PROTAC synthesis, an IκBα-derived phosphopeptide motif and a HIF-1α-derived pentapeptide motif. These motifs are recognized by the E3 ligases SCFβ-TRCP (4) and pVHL (5, 7), respectively. The HIF-1α-derived motif is recommended due to its increased cell permeability and bioavailability.
Following synthesis of the PROTAC (see Note 3), its identity must be confirmed via mass spectrometry such as electrospray ionization mass spectrometry (ESI-MS) or MALDI-TOF (matrix-assisted laser desorption ionization-time of flight) MS as well as nuclear magnetic resonance (NMR) spectroscopy. The PROTAC can then be further purified by normal-phase HPLC.
3.2. Confirmation of PROTAC-Induced Target Protein Degradation
PROTAC-induced degradation of the target protein can be confirmed by treatment of the selected cell line with increasing concentrations of the PROTAC followed by analysis of protein abundance via western blotting. The following procedure describes the detection of estrogen receptor (ER) protein levels following treatment with an ER-targeting PROTAC.
Plate the cells (see Note 4) onto a 24-well plate at a density of ~20,000 cells/well (see Note 5) in 500 μL of medium/well.
Incubate the cells at 37°C, 95% humidity, and 5% carbon dioxide for 24 h.
Change the cell culture medium to medium supplemented with 5% charcoal/dextran treated fetal bovine serum and incubate the cells for another 24 h prior to treatment (see Note 6).
Treat the cells with increasing concentrations of the PROTAC and vehicle control in the same medium used in step 3 (see Note 7). Additionally, a mutant PROTAC (hydroxyproline→norleucine) can serve as negative control (see Note 8).
Incubate the cells for the desired period of time (see Note 9).
Wash the cells with ~200 μL of Hank's Balanced Salt Solution and detach the cells by incubating them in ~200 μL of trypsin-EDTA for ~3 min (see Note 10).
Collect the cells from each well in separate centrifuge tubes by resuspending the cells in ~800 μL of medium.
Centrifuge at 20,000 × g and 4°C for 10 min. Aspirate the medium from the pellet and store the pellet at −80°C until lysis.
Place the cell pellets in an ice-filled container (see Note 11) and resuspend them in lysis buffer (see Note 12). Incubate on ice for at least 1 h.
Centrifuge at 20,000 × g at 4°C for 10 min and collect the supernatants.
Determine the protein concentration of each supernatant (see Note 13).
Dilute the samples in an equivalent volume of 2× Laemmli sample buffer. Mix well and denature at 100°C for 10 min. The samples may be stored at 4°C for ~2 weeks or at −20°C for longer time periods.
Load equal protein concentrations (see Note 14) of each sample into the wells of a 10-12% SDS-PAGE gel along with prestained protein standards (see Note 15).
Electrophorese (see Note 16) and transfer proteins onto a PVDF membrane (see Note 17).
Wash the membrane with 1× TBST by agitating on a platform rotator for ~10 min.
Block the membrane in 5% (w/v) nonfat dry milk in TBST for ~1 h at room temperature while gently agitating on a platform rotator, or overnight at 4°C without agitation.
Wash the membrane again 3 times with 1× TBST for ~10 min.
Incubate the membrane with anti-ER antibody (1:200) diluted in 3% (w/v) bovine serum albumin in TBST (see Note 18).
Wash the membrane 3 times with 1× TBST for ~10 min.
Incubate the membranes with anti-IgG horseradish peroxidase in 5% (w/v) nonfat dry milk in TBST (see Notes 19-20).
Wash the membranes 3 times with 1× TBST for ~10 min with vigorous agitation.
Visualize the target protein on X-ray film using Pierce ECL Western Blotting Substrate.
Wash the membrane 2--3 times with 1× TBST for ~10 min.
Incubate the membranes with anti-β-actin antibody (1:5,000) in 5% (w/v) nonfat dry milk in TBST (see Notes 20-21).
Repeat steps 19-22 to visualize β-actin.
3.3. Confirmation of Target Protein Ubiquitination
Immuoprecipitation can be performed to confirm the interaction between ubiquitin and the target protein, indicating that the target protein becomes ubiquitinated. The following procedure can be used to pull down ubiquitin and detect the target protein via western blotting. These results can be cross-checked by pulling down the target protein and detecting ubiquitin via western blotting.
Plate the cells (see Note 4) in 100 mm cell culture dishes at a density of ~5.7×105 cells/well (see Note 5) in 10 mL of medium/dish.
Incubate the cells at 37°C, 95% humidity, and 5% carbon dioxide for 24 h.
Change the cell culture medium to medium supplemented with 5% charcoal/dextran treated fetal bovine serum and incubate the cells for another 24 h prior to treatment (see Note 6).
In the same medium used in step 3, treat 2 dishes of cells with vehicle control. Additionally, treat the remaining dishes with increasing concentrations of the PROTAC (see Note 7) or with the (hydroxyproline→norleucine) control PROTAC (see Note 8) in combination with 1 μM of epoxomicin.
Incubate the cells for ~6 h.
Remove the medium from each dish and wash the cells 2 times with 3 mL of ice-cold PBS (see Note 22).
Add 1 mL of lysis buffer to each dish and incubate on ice for 30 min.
Mix the lysis buffer on each dish using a pipette to ensure that all cells are in contact with the buffer.
Incubate the dishes on ice for at least another 30 min and collect the lysates in separate centrifuge tubes.
Pulse sonicate the lysates 4 times for 5 seconds each.
Centrifuge the lysates at 20,000 × g at 4°C for 10 min. Collect the supernatants in new separate centrifuge tubes and discard the pellets.
Determine the protein concentration of each supernatant (see Note 13).
For each lysate, add 50 μL of the Protein G bead slurry to 2 new centrifuge tubes.
Add 500 μL of cold lysis buffer to the tubes containing the bead slurry. Centrifuge at 20,000 × g for 30 sec and discard the supernatants. Repeat this step once more. For each lysate, use one of these tubes in the next step and save one tube for use in step 18.
Add 500 μL of each lysate to separate tubes containing the lysis buffer-rinsed bead slurry and resuspend the beads. Incubate the tubes on a rotating platform at 4°C for 30 min.
Centrifuge the samples at 20,000 × g at 4°C for 10 min and collect the supernatants.
Transfer 400 μL of each cold precleared lysate into a separate centrifuge tube. Add ~1μg of antibody specific for the target protein to each of these tubes except one tube containing a vehicle-treated lysate (see Note 23). To the tube containing this vehicle-treated lysate, add the same amount of non-specific antibody (see Note 24) to serve as a control. All remaining lysate can be saved and used in western blotting as input controls to determine protein levels prior to immunoprecipitation.
Add 50 μL of lysis buffer-rinsed protein G bead slurry to each tube containing antibody and incubate overnight at 4°C on a rotating platform.
Centrifuge at 20,000 × g and 4°C for 5 min. Collect the supernatants to be used as controls.
Wash the beads 5 times with ice-cold PBS. Centrifuge at 20,000 × g for 30 sec and discard the supernatant after each wash. 21. Resuspend the bead pellets in ~50μL of 2× Laemmli sample buffer. 22. To the remaining lysate obtained in step 16 and supernatants obtained in step 19, add an equivalent volume of 2x Laemmli sample buffer. Mix well and incubate at 100°C for 10 min. These samples are ready for use in western blotting.
Centrifuge the tubes containing the immunoprecipitated samples at 20,000 × g for 5 min and collect the supernatants to use for western blotting. The samples may be stored at 4°C for ~2 weeks or at −20°C for longer time periods.
Refer to the western blotting protocol in section 3.2. If immunoprecipitation was performed using the anti-ERα antibody, use the anti-ubiquitin antibody during western blotting. Conversely, if immunoprecipitation was performed using the anti-ubiquitin antibody, use the anti-ERα antibody during western blotting (see Note 25).
3.4 Confirmation of specific E3-ligase-dependent target protein degradation
Renal carcinoma cell lines 786-O, which does not express VHL, and 786-O/VHL, which is stably transfected with wild-type pVHL (17), can be used to confirm that target protein degradation is dependent on the E3-ligase pVHL. Degradation should occur in 786-O/VHL cells but not in 786-O cells if degradation is pVHL-dependent, suggesting that the PROTAC is functional.
Follow the procedure described in section 3.2 using 786-O and 786-O/VHL cell lines.
3.5 Confirmation of Proteasome-Dependent Target Protein Degradation
Cells can be treated with an optimal dose of the PROTAC in combination with increasing concentrations of a proteasome inhibitor to confirm that degradation of the target protein is proteasome-dependent. Increasing target protein accumulation with increasing inhibitor concentration provides evidence indicating that the PROTAC is functioning as expected.
Acknowledgement
This work was supported by NIH grants R01 CA128903 and R01 ES014849 to KBK.
Footnotes
Following attachment of the handle to the small molecule ligand, it is important to ensure that the binding affinity of the ligand to the target protein is maintained. This can be done by conducting a competitive ligand binding assay (Invitrogen).
The length of the handle must be optimized. It is therefore recommended to synthesize a small library of PROTACs that differ in handle lengths and determine the optimum length via the assay described in Section 3.2.
For the detailed procedure describing PROTAC synthesis, see ref. (2).
When selecting a cell line for PROTAC characterization, it is important to ensure that the chosen cell line expresses the E3 ligase targeted by the PROTAC. Additionally, it is important to ensure that the cell line does not abnormally overexpress proteins known to interact with the protein from which the chosen E3 ligase recognition motif was derived (e.g. HIF-1α or IκBα).
Plate the cells at a density that will allow them to reach ~70% confluence by the time of treatment.
This step is required when working with hormone-dependent cell lines. Charcoal/dextran treated FBS reduces hormones present in the cell culture medium, which ensures that expression of the PROTAC-targeting hormone receptor (e.g. estrogen receptor) is maximized.
The concentrations of PROTAC required to induce target protein degradation will vary based on the cell line and particular PROTAC employed. For the ER-targeting PROTAC, effective concentrations are 10-50 μM.
This mutant PROTAC has an amino acid substitution in the E3 ligase-recognition motif to abolish the interaction between these two moieties. This should prevent PROTAC-induced degradation of the target protein.
The incubation period must be optimized, depending on the cell line and the PROTAC used. For the ER-targeting PROTAC, effective incubation times range from 24-96 h. For PROTACs targeting essential proteins, treatment for greater than 48 hrs may cause cytotoxicity.
Alternatively, the cells may be washed with ice-cold PBS and lysed directly in the cell culture wells. This method yields a less concentrated lysate, and is therefore only recommended when assaying for highly abundant cellular proteins.
Keep the cells on ice whenever possible throughout the entire lysis procedure.
Add the minimum volume of lysis buffer sufficient to dissolve all particulates once the pellet has been resuspended.
The Bradford method of protein quantification is recommended for use with the specified lysis buffer. We suggest the Bio-Rad Protein Assay.
Typical concentrations for gel loading range from 10--25 μg.
The gel percentage must be optimized to resolve the target protein and the loading control. This may allow you to cut the membrane to simultaneously probe for the target protein and loading control. Running a standard containing the purified target protein where possible is highly recommended.
For a detailed electrophoresis protocol, see (18).
For a detailed electrotransfer protocol, see (19)
This concentration can be adjusted to optimize the signal detected. Typical incubation times are ~1 h at room temperature with gentle agitation or overnight at 4°C without agitation. Refer to the manufacturer's instructions when using other antibodies to determine incubation conditions.
For regular-strength ECL substrate, the typical concentration is (1:5,000). However, this concentration may be adjusted to optimize the signal detected. It is important that this antibody be specific for the species in which the primary antibody was raised. For example, the recommended ERα antibody was obtained from a rabbit donor, and therefore anti-rabbit IgG horseradish peroxidase should be used.
Typical incubation times are 1 h at room temperature with gentle agitation or overnight at 4°C without agitation. Refer to the manufacturer's instructions when using other antibodies to determine incubation conditions.
β-actin serves as a loading control.
This procedure applies specifically to cells treated in a 100 mm dish. ~107 cells are required for immunoprecipitation.
This concentration may vary depending on the antibody used. Refer to the manufacturer's instructions when using other antibodies to determine the optimum concentration.
The non-specific antibody selected should be raised in the same species as the specific antibody. Because the antibodies recommended in this protocol were obtained from rabbit (anti-ERα) and mouse (anti-ubiquitin), anti-rabbit and anti-mouse IgG are recommended as controls.
Polyubiquitination of some proteins, e.g. ERα, may interfere with antibody binding. If the target protein is not detected, perform the western blot again using a polyclonal antibody raised against the entire protein.
Use a PROTAC concentration that yields nearly complete target protein degradation at the selected incubation time.
Another broadly-acting proteasome inhibitor, such as bortezomib, may also be used.
References
- 1.Li W, et al. Genome-wide and functional annotation of human E3 ubiquitin ligases identifies MULAN, a mitochondrial E3 that regulates the organelle's dynamics and signaling. PLoS One. 2008;3:e1487. doi: 10.1371/journal.pone.0001487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jang ER, Lee W, Kim KB. Targeted Degradation of Proteins by PROTACs. Curr. Protoc. Chem Biol. 2010;2:71–87. doi: 10.1002/9780470559277.ch090242. [DOI] [PubMed] [Google Scholar]
- 3.Sakamoto KM. Protacs for treatment of cancer. Pediatr Res. 2010;67:505–508. doi: 10.1203/PDR.0b013e3181d35017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sakamoto KM, et al. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A. 2001;98:8554–8559. doi: 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang D, et al. Degradation of target protein in living cells by small-molecule proteolysis inducer. Bioorg Med Chem Lett. 2004;14:645–648. doi: 10.1016/j.bmcl.2003.11.042. [DOI] [PubMed] [Google Scholar]
- 6.Zhang D, et al. Targeted Degradation of Proteins by Small Molecules: A Novel Tool for Functional Proteomics. Combinatorial Chemistry & High Throughput Screening. 2004;7:691–699. doi: 10.2174/1386207043328364. [DOI] [PubMed] [Google Scholar]
- 7.Schneekloth JS, Jr., et al. Chemical Genetic Control of Protein Levels: Selective in Vivo Targeted Degradation. J Am Chem Soc. 2004;126:3748–3754. doi: 10.1021/ja039025z. [DOI] [PubMed] [Google Scholar]
- 8.Rodriguez-Gonzalez A, et al. Targeting steroid hormone receptors for ubiquitination and degradation in breast and prostate cancer. Oncogene. 2008;27:7201–7211. doi: 10.1038/onc.2008.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cyrus K, et al. Impact of linker length on the activity of PROTACs. Mol Biosyst. 2010 doi: 10.1039/c0mb00074d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cyrus K, et al. Jostling for position: optimizing linker location in the design of estrogen receptor-targeting PROTACs. ChemMedChem. 2010;5:979–985. doi: 10.1002/cmdc.201000146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cyrus K, et al. Two-headed PROTAC: an effective new tool for targeted protein degradation. Chembiochem. 2010;11:1531–1534. doi: 10.1002/cbic.201000222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sakamoto KM. Chimeric molecules to target proteins for ubiquitination and degradation. Methods Enzymol. 2005;399:833–847. doi: 10.1016/S0076-6879(05)99054-X. [DOI] [PubMed] [Google Scholar]
- 13.Sakamoto KM, et al. Development of Protacs to target cancer-promoting proteins for ubiquitination and degradation. Mol Cell Proteomics. 2003;2:1350–1358. doi: 10.1074/mcp.T300009-MCP200. [DOI] [PubMed] [Google Scholar]
- 14.Lee H, et al. Targeted degradation of the aryl hydrocarbon receptor by the PROTAC approach: a useful chemical genetic tool. Chembiochem. 2007;8:2058–2062. doi: 10.1002/cbic.200700438. [DOI] [PubMed] [Google Scholar]
- 15.Puppala D, et al. Development of an aryl hydrocarbon receptor antagonist using the proteolysis-targeting chimeric molecules approach: a potential tool for chemoprevention. Mol Pharmacol. 2008;73:1064–1071. doi: 10.1124/mol.107.040840. [DOI] [PubMed] [Google Scholar]
- 16.Itoh Y, et al. Protein knockdown using methyl bestatin-ligand hybrid molecules: design and synthesis of inducers of ubiquitination-mediated degradation of cellular retinoic acid-binding proteins. J Am Chem Soc. 2010;132:5820–5826. doi: 10.1021/ja100691p. [DOI] [PubMed] [Google Scholar]
- 17.Baba M, et al. Loss of von Hippel-Lindau protein causes cell density dependent deregulation of CyclinD1 expression through hypoxia-inducible factor. Oncogene. 2003;22:2728–2738. doi: 10.1038/sj.onc.1206373. [DOI] [PubMed] [Google Scholar]
- 18.Gallagher SR. One-dimensional SDS gel electrophoresis of proteins. Curr Protoc Toxicol. 2007 doi: 10.1002/0471140856.txa03fs32. Appendix 3, Appendix 3F. [DOI] [PubMed] [Google Scholar]
- 19.Ursitti JA, Mozdzanowski J, Speicher DW. Electroblotting from polyacrylamide gels. Curr Protoc Protein Sci. 2001 doi: 10.1002/0471140864.ps1007s00. Chapter 10, Unit 10 17. [DOI] [PubMed] [Google Scholar]