

Over the years, small molecules that can modulate functions of specific cellular proteins have been used more frequently as molecular probes for the exploration of complex biological processes. This small-molecule approach is complementary to a classical genetic approach and thus is fittingly termed “chemical genetics.”[1, 2] Typically, small molecule modulators exert their activity by binding target proteins and blocking their functions, thereby perturbing cellular signaling pathways of interest. While small molecules have proven useful for probing the function of proteins in the etiology of a specific disease, they have also provided promising therapeutic agents. Thus, a great deal of efforts has been made to identify novel small molecules that modulate protein functions.[3]
The ubiquitin-proteasome system (UPS) is the principle conduit for protein turnover in all eukaryotic cells. This ubiquitin-dependent proteolysis involves the attachment of polyubiquitin chains to a substrate via the actions of three enzymes, E1, E2, and E3, marking this substrate for degradation by the 26S proteasome.[4] Given that the regulation of protein homeostasis via the UPS leads to the destruction of targeted proteins, this protein targeting process needs to be carefully regulated. The E3 ubiquitin ligases appear to be the primary source of substrate specificity in the ubiquitination cascade, as they have been shown to bind directly and specifically to the substrate.[5, 6]
Exploiting the UPS, we and others have developed a novel class of small-molecules, the PROTACs (Proteolysis Targeting Chimeric) molecules, designed to induce ubiquitination and degradation of targeted proteins.[7-13] Typically, a PROTAC molecule is comprised of a residue for E3 ubiquitin ligase recognition and a ligand that can be recognized by a targeted protein,[8] thereby recruiting the targeted protein to the E3 ligase for ubiquitination. Unlike gene knockout or siRNA approaches, which knockout or knock-down target proteins at the DNA or RNA level, the PROTAC approach destroys targeted proteins at the posttranslational level, thereby avoiding many issues inherant in a classical genetic approach. For this reason, the PROTAC technology has been increasingly employed as an emerging chemical genetic tool to provide temporal and spatial control.[14]
Thus far, several PROTACs have been successfully developed to target various proteins, such as the estrogen receptor-α (ERα),[11-13, 15] methionine aminopeptidase-2 (MetAP-2),[10] androgen receptor (AR)[7] and cellular retinoic acid-binding proteins (CRABPs).[16] Similarly, Danishefsky et al. have also shown that a natural product geldanamycin (GM)-based small molecule induces degradation of estrogen and androgen receptors.[17-19] While this GM approach offers an additional option for specific degradation, the system appears to be limited to proteins associated with heat shock proteins.
Despite the potential of PROTAC technology as an investigative tool, the relatively high PROTAC concentrations (1-50μM) required for efficient induction of target protein degradation limit their widespread application. We and others have shown that monomeric estradiol (E2)-based PROTACs induce degradation of the estrogen receptor (ER).[11-13] As PROTACs are composed of three parts (small molecule ligand of targeted protein, E3 ubiquitin ligase recognition motif, and linker) optimization of each may afford PROTACs with greater efficacy. Typical ER-targeting PROTACs are composed of a von Hippel-Lindau (pVHL) E3 ubiquitin ligase recognition motif and estradiol (E2), a natural ligand of the ER. These PROTACs are easily screened via western blotting for ER degradation, thus we chose the ER-targeting PROTACs as a model for further optimization of the PROTAC approach.
The new class of PROTACs reported herein utilize a chimeric molecule that consists of two E2s (1) linked to the pVHL targeting pentapeptide mentioned above. Since the PROTAC functions as a bridging molecule that promotes the recruitment of a target protein to an E3 ligase complex, we envisioned that the introduction of two ligands may recruit the binding protein to an E3 ubiquitin ligase more effectively than the one-ligand PROTAC system (Fig. 1). Thus, the use of two E2s is expected to result in more effective ubiquitination and degradation of the target protein.
Figure 1.
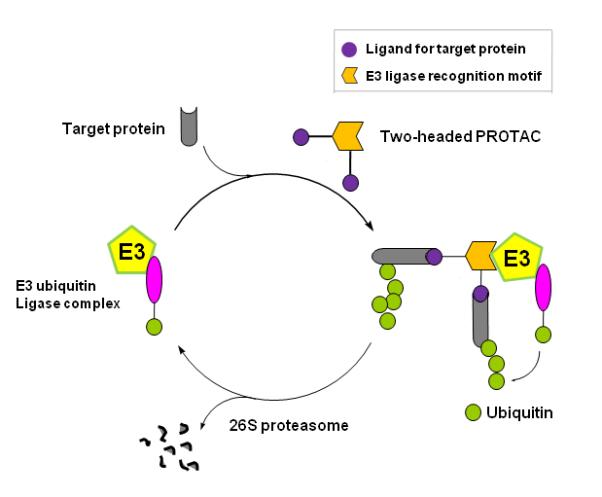
Overall strategy of chemical knock-out by a dimeric ligand based PROTAC. The new class of PROTAC is a chimeric molecule that consists of two ligands linked to a small peptide that corresponds to the recognition site of the pVHL E3 ligase. Upon entry into cells, the two ligands will be recognized by target protein, allowing for recruitment of the target protein to pVHL and its subsequent degradation by the proteasome. Use of two ligands is expected to enhance the efficiency of target protein recruitment to the E3 ligase complex.
To test this hypothesis, we prepared two monomeric E2-based PROTACs (3 & 4) to use as controls and a two-headed PROTAC (5) in which two E2s were attached, one at the N-terminus and the other at the C-terminus of the HIF-1α pentapeptide (Scheme 1). For details on the synthesis of (3 & 4), refer to the supporting information. It has been shown that the C-7 position of E2 can be derivatized while maintaining the ER-binding affinity of the parent compound, E2 (1). For synthesis of the two-headed PROTAC, we first introduced a free amine functional group at the C-7 position of E2 to yield a key intermediate (2). Using the free amine of (2), addition of the linker was achieved using Fmoc-caprylic acid and subsequent coupling with the C-terminus of the HIF-1α pentapeptide to give an intermediate monomeric PROTAC. Following deprotection of the Z-group on leucine the resulting free amine was activated with disuccinimidyl suberate (DSS) and then coupled to compound (2) to yield the fully assembled two-headed PROTAC, (5). The final product was confirmed by 1H NMR and mass spectrometry.
Scheme 1.
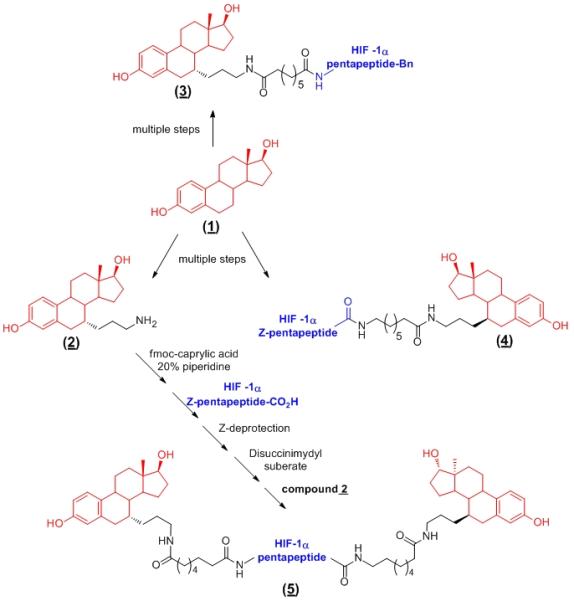
Synthetic scheme of the two-headed ER ligand based PROTAC (5) and structures of control monomeric PROTACs (3 & 4). A protein ligand (E2, 1) is attached to either the N-terminus (3) or C-terminus (4) of the E3 ligase recognition peptide. HIF-1α pentapeptide = H2N-Leu-Ala-ProOH-Tyr-Ile-Bn (for N-terminal attachment) or Z-Leu-Ala-ProOH-Tyr-Ile-CO2H (for C-terminal attachment).
To determine whether the two-headed PROTAC (5) has a higher ER-binding affinity than its monomeric counterparts, a competitive ligand binding affinity assay was performed (Fig. 2A). The binding assay results clearly indicated that PROTAC (5) had at least three times better ER binding affinity than PROTACs (3) or (4). Contrary to the control compounds, 3 & 4, the two-headed PROTAC (5) appears to have poor solubility, causing the loss of binding affinity at high concentrations. Next, the two-head PROTAC (5) was tested for its ER degradation efficiacy and compared to its monomeric counterparts via western blotting. After a 48 hour incubation with the PROTACs, (5) showed a superior ability to degrade the ER when compared to (4) and (3). While there were no obvious differences between dimeric and monomeric PROTACs at 1 μM, a dramatic difference was observed at 10 μM. However, a negative control PROTAC (NC), where the critical hydroxyproline residue of (3) is replace with norleucine, did not cause ER degradation (Fig. 2B). This implies that the PROTAC-induced ER degradation seen is E3 ligase-dependent. The results also suggest that the second E2 on (5) affords it a greater than five times advantage in ER degradation, as 10 μM of (5) gave superior ER degradation when compared to 50 μM of (3) and (4).
Figure 2.
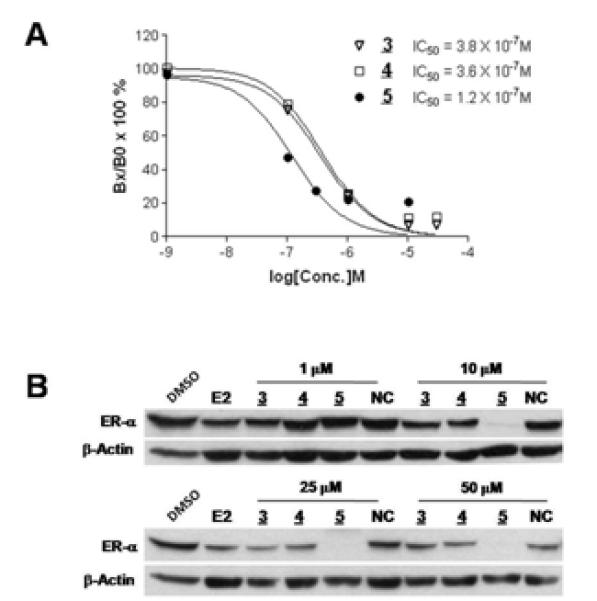
Dimeric ligand based PROTAC are more effective than their monomeric counterparts. A. While the monomeric PROTACs (3 & 4) have similar binding affinity to the ER, the dimeric PROTAC (5) has a three times greater affinity for the ER. The ordinate is Bx/Bo, specifically bound radioligand in the presence of a given amount of competitor (Bx) divided by specifically bound radioligand in the absence of competitor (Bo). B. The dimeric E2 based PROTAC (5) efficientially induces degradation of the ER in a concentration-dependent manner. Unlike its monomeric counterparts (3 & 4), the two-headed PROTAC (5) induced complete destruction of the ER. NC is a negative control in which the critical hydroxyproline is replaced with norleucine.
We then examined whether the two-headed PROTAC-mediated ER degradation is proteasome-dependent using western blotting and immunofluorescence studies. As shown in Fig. 3A, the ER degradation by (5) was quickly blocked by treatment with epoxomicin, a proteasome-specific natural product inhibitor, indicating that PROTAC (5)-mediated ER degradation is proteasome-dependent. Similarly, immunofluorescence data showed the dramatic attenuation of signal (green) in cells with the treatment of the two-headed PROTAC (5). An accumulation of the green signal was seen when (5) was co-treated with epoxomicin, indicating that PROTAC (5)-mediated ER degradation is blocked by the proteasome inhibitor. Thus, it appears that the two-headed PROTAC utilizes the UPS to cause ER degradation.
Figure 3.
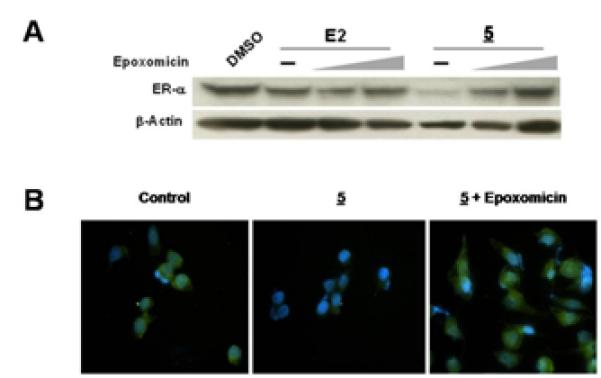
The two-headed PROTAC (5) induces degradation of the ER in a proteasome-dependent manner. A. Co-treatment of cells with the PROTAC (5, 10 μM)and a proteasome inhibitor (0, 100 and 500 nM of epoxomicin) over 48 hours blocks the PROTAC-mediated degradation of the ER. E2 (10 nM) plus inhibitor is included as a positive control. B. Immunofluorescent images of MCF-7 cells treated as in 3A where the green color indicates the ER and the blue color (DAPI) indicates the nucleus. ER degradation is seen in (5) when compared to control, while co-treatment of (5) with epoxomicin (100 nM) caused accumulation of the ER.
Recent technological advancements have given researchers a much broader toolbox to use to investigate the complex mechanistic functions and interactions of proteins. Both genetic (knockout, RNAi, etc) and non-genetic (small molecule) approaches have been employed to eludicate these roles in cells. However, innate limitations of the genetic approach have left a significant role for non-genetic technologies. Thus, we have focused on the optimization of the PROTAC technology to overcome the limitations currently seen with these compounds.
In summary, here we reported a novel class of PROTAC containing two small molecule ligands instead of one ligand. Our proof of concept study clearly showed that the two-headed PROTAC gives superior ER degradation when compared to one-headed PROTACs. The two-headed PROTAC also provides improved binding affinity between the PROTAC and the ER. This novel approach may be useful for the design of more effective PROTACs. Current efforts are focused on the synthesis of additional two-headed PROTACs targeting other proteins.
Experimental Section
Chemical Reagents
All chemical reagents were purchased from Aldrich Chemical Co. A negative control PROTAC (NC) was synthesized following a procedure similar to that previously reported.[20] Epoxomicin was synthesized as previously reported.[21] For more detailed information on the chemical synthesis of PROTACs, see supporting information.
Cell Culture
The MCF7 human breast cancer cell line was purchased from the American Type Culture Collection (Manassas, VA). MCF7 cells were maintained in RPMI 1640 (Gibco, Carlsbad, CA) medium containing 10 % (v/v) FBS (Gibco, Carlsbad, CA) and 100 U/mL penicillin-100 ug/mL streptomycin (Gibco, Carlsbad, CA). All experiments were performed when the cells were 70 % confluent and had been maintained in 5% (v/v) charcoal-dextran treated FBS RPMI with antibiotics for at least 24 hours. All compounds were treated in a DMSO vehicle at the appropriate dilutions for 48 hours.
Western Blotting
Whole cell lysates were prepared and denatured prior to electrophoresis. Equal sample concentrations were run on 10% SDS-PAGE and transferred to PVDF. Membranes were probed with anti-ER-α and anti-β-actin antibodies and detected using ECL and film.
ER Binding Affinity Assay
Competitive ligand binding assays were performed according to the manufacturer’s protocol (Invitrogen). For more detail, see the supporting information. The percent specific binding affinity was calculated and IC50 values were obtained using one site competition curve analyses provided by the Graph Pad Prism. Relative binding affinity (RBA) was calculated using the following equation, RBA = {IC50 (E2)/IC50 (sample)} × 100.
Immunofluorescence
Cell were treated on sterilized coverslips in the manner described in the cell culture section and stained with DAPI as well as an anti-ER antibody which was conjugated to FITC. For more detail, see the supporting information. The stained cells were visualized on an inverted fluorescence microscope (Nikon Ti-U microscope) with NIS Element Research image analysis software.
Supplementary Material
Acknowledgements
This work has been supported by Graduate School Academic Year, Kentucky Opportunity, and American Foundation for Pharmaceutical Education Fellowships (M.W.). K. B. K. gratefully acknowledges the support of the Markey Cancer Foundation, NIH (CA131059), and DoD. H. S. thanks the NIH for financial support (ES014849). We thank members of the Swanson and Kim laboratories for their helpful comments on the manuscript. We are grateful to Dr. Wooin Lee for helping with the collection of fluorescent images.
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
References
- [1].Crews CM, Splittgerber U. Trends Biochem Sci. 1999;24:317. doi: 10.1016/s0968-0004(99)01425-5. [DOI] [PubMed] [Google Scholar]
- [2].Schreiber SL. Bioorg Med Chem. 1998;6:1127. doi: 10.1016/s0968-0896(98)00126-6. [DOI] [PubMed] [Google Scholar]
- [3].Strausberg RL, Schreiber SL. Science. 2003;300:294. doi: 10.1126/science.1083395. [DOI] [PubMed] [Google Scholar]
- [4].Hershko A, Ciechanover A. Annu Rev Biochem. 1998;67:425. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- [5].Skowyra D, Craig KL, Tyers M, Elledge SJ, Harper JW. Cell. 1997;91:209. doi: 10.1016/s0092-8674(00)80403-1. [DOI] [PubMed] [Google Scholar]
- [6].Deshaies RJ. Annu Rev Cell Dev Biol. 1999;15:435. doi: 10.1146/annurev.cellbio.15.1.435. [DOI] [PubMed] [Google Scholar]
- [7].Rodriguez-Gonzalez A, Cyrus K, Salcius M, Kim K, Crews CM, Deshaies RJ, Sakamoto KM. Oncogene. 2008;27:7201. doi: 10.1038/onc.2008.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lee H, Puppala D, Choi EY, Swanson H, Kim KB. Chembiochem. 2007;8:2058. doi: 10.1002/cbic.200700438. [DOI] [PubMed] [Google Scholar]
- [9].Puppala D, Lee H, Kim KB, Swanson HI. Mol Pharmacol. 2008;73:1064. doi: 10.1124/mol.107.040840. [DOI] [PubMed] [Google Scholar]
- [10].Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Proc Natl Acad Sci U S A. 2001;98:8554. doi: 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sakamoto KM, Kim KB, Verma R, Ransick A, Stein B, Crews CM, Deshaies RJ. Mol Cell Proteomics. 2003;2:1350. doi: 10.1074/mcp.T300009-MCP200. [DOI] [PubMed] [Google Scholar]
- [12].Zhang D, Baek SH, Ho A, Kim K. Bioorg Med Chem Lett. 2004;14:645. doi: 10.1016/j.bmcl.2003.11.042. [DOI] [PubMed] [Google Scholar]
- [13].Zhang D, Baek S-H, Ho A, Lee H, Jeong YS, Kim K. Combinatorial Chemistry & High Throughput Screening. 2004;7:691. doi: 10.2174/1386207043328364. [DOI] [PubMed] [Google Scholar]
- [14].Verma R, Peters NR, D’Onofrio M, Tochtrop GP, Sakamoto KM, Varadan R, Zhang M, Coffino P, Fushman D, Deshaies RJ, King RW. Science. 2004;306:117. doi: 10.1126/science.1100946. [DOI] [PubMed] [Google Scholar]
- [15].Schneekloth JS, Jr., Fonseca FN, Koldobskiy M, Mandal A, Deshaies R, Sakamoto K, Crews CM. J Am Chem Soc. 2004;126:3748. doi: 10.1021/ja039025z. [DOI] [PubMed] [Google Scholar]
- [16].Itoh Y, Ishikawa M, Naito M, Hashimoto Y. J Am Chem Soc. 132:5820. doi: 10.1021/ja100691p. [DOI] [PubMed] [Google Scholar]
- [17].Zheng FF, Kuduk SD, Chiosis G, Munster PN, Sepp-Lorenzino L, Danishefsky SJ, Rosen N. Cancer Res. 2000;60:2090. [PubMed] [Google Scholar]
- [18].Kudrin AV. J Trace Elem Med Biol. 2000;14:129. doi: 10.1016/s0946-672x(00)80001-2. [DOI] [PubMed] [Google Scholar]
- [19].Kuduk SD, Harris TC, Zheng FF, Sepp-Lorenzino L, Ouerfelli Q, Rosen N, Danishefsky SJ. Bioorg Med Chem Lett. 2000;10:1303. doi: 10.1016/s0960-894x(00)00208-0. [DOI] [PubMed] [Google Scholar]
- [20].Zhang S, Qin C, Safe SH. Environ Health Perspect. 2003;111:1877. doi: 10.1289/ehp.6322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sin N, Kim KB, Elofsson M, Meng L, Auth H, Kwok BH, Crews CM. Bioorg Med Chem Lett. 1999;9:2283. doi: 10.1016/s0960-894x(99)00376-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.