

Abstract
Anti-diabetic drugs that activate the protein PPARγ had a bright start but soon lost appeal due to undesirable side effects. Subtle modifications may once again make them suitable for treating diabetes.
Obesity and its associated disorders (diabetes, cancer and cardiovascular disease among others) have reached epidemic proportions worldwide. Understanding the mechanisms of metabolic control to prevent and treat these metabolic disorders is therefore a top research priority. Spiegelman and colleagues (Choi et al.1) make a significant contribution showing that phosphorylation of one protein, PPARγ, plays an important role in insulin resistance and obesity.
The story of PPARγ begins in 1987, when the Spiegelman group2 caught a ‘big fish’ by identifying PPARγ as a fat cell (adipocyte)-specific regulatory element. This seminal discovery led to the determination of PPARγ’s key role in adipocyte differentiation, and fuelled over two decades of intensive research that attributed an ever-expanding list of functions to PPARγ, ranging from roles in metabolism3, to immune homeostasis4 to longevity5.
Perhaps the most clinically relevant finding has been the now well-established link between PPARγ activity and insulin sensitivity6. This association justified a rigorous search for PPARγ activators, which subsequently found their way into the clinic as potent and effective insulin sensitizers. The initial clinical success, however, was soon overshadowed by reports of several insidious side effects that include weight gain, osteoporosis and heart failure7. What’s more, the negative attitude was sustained by the difficulty to rationally design PPARγ drugs, mainly as it was not clear how to tease out efficacy from side effects. Consequently, the initial enthusiasm surrounding PPARγ activators evolved into scepticism. Choi and co-workers’ data could revert the negative swing of the PPARγ pendulum.
In their search for PPARγ regulators, the authors1 find that the enzyme cyclin-dependent kinase 5 (CDK5) phosphorylates PPARγ on serine residue 273. Activation of CDK5 itself involves truncation of the p35 protein to p25, possibly in response to cytokines or other pro-inflammatory signals (Fig. 1). p25 then translocates to the nucleus, where it associates with, and activates, CDK5 in a way reminiscent of the activation of other CDK enzymes.
Figure 1. Regulation of PPARγ activity.
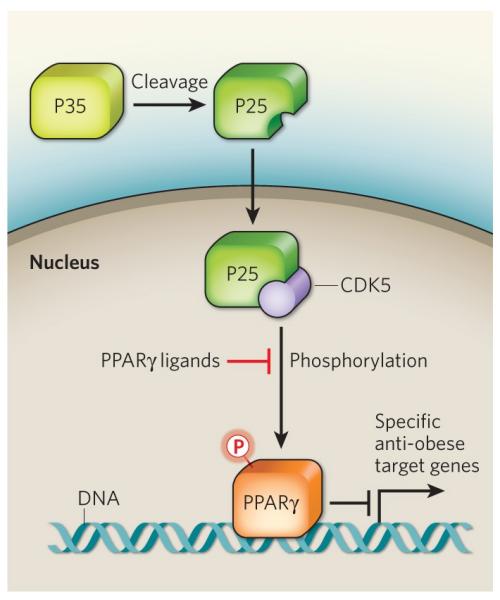
In the obese state, pro-inflammatory signals lead to the cleavage of the p35 protein to p25. p25 then translocates to the nucleus where it binds to CDK5 and activates it. Choi et al.1 show that, CDK5 in turn phosphorylates PPARγ on serine residue 273, thereby preventing the transcription of specific PPARγ targets that have anti-obesity effects. Anti-diabetic PPARγ ligands prevent serine 273 phosphorylation.
High cytokines levels are commonly observed in obesity. The authors therefore asked whether cytokine-mediated CDK5 activation regulates PPARγ in the obese state. Indeed, they find that CDK5 phosphorylates PPARγ when mice are fed a high-fat diet. As well as affecting adipocyte differentiation, PPARγ also regulates the expression of some genes associated with the metabolic disorders. Choi et al. show that CDK5-mediated phosphorylation of this nuclear receptor does not affect its role in adipocyte differentiation, but selectively inhibits the expression of a specific subset of its gene targets; the mechanism remains unknown.
Intriguingly, the anti-diabetic PPARγ ligands that were previously considered to act solely by activating PPARγ potently inhibit its CDK5-mediated phosphorylation1, probably by inducing a conformational change in PPARγ. This alternative way of modifying PPARγ activity, could clarify the longstanding paradox: why PPARγ activation by a wide range of ligands does not always correlate with the ligands’ in vivo efficacy. Also, different PPARγ activators — whether potent (such as rosiglitazone) or weak (MRL24) — may have similar insulin-sensitizing effects in vivo, because of their similar capacity to revert CDK5-mediated phosphorylation. In strong support of these hypotheses, Choi et al.1 show that, in human patients receiving rosiglitazone, the levels of phosphorylated PPARγ in fat tissue correlates clearly with clinical parameters of glucose tolerance, and may therefore serve as an indicator for diabetes.
This refreshing wind over the PPARγ field also points to some important areas of future research, which would revitalize the once-bustling PPARγ research. First, does CDK5 selectively affect PPARγ function or does it also apply to other pathways? This enzyme’s activation is more likely to be part of a signalling pathway that informs the cell about a potentially harmful metabolic context. Indeed, CDK5 is a potent stimulator of insulin secretion, through phosphorylation of components of the secretory machinery8,9. The search for other CDK5 targets — including other transcriptional regulators and nuclear receptors — is therefore certainly warranted. Second, both a PPARγ phosphatase that reverts the CDK5 effect, and components of the regulatory pathway involving CDK5 and p35/p25 (ref. 10) should be identified; this knowledge would open up another way to modulate PPARγ activity. Third, genetic evidence supporting a role for CDK5-mediated signalling in metabolic disorders would be welcome. For this, data obtained through previous genome-wide association studies should be carefully reanalysed, while taking into account the genomic regions that contain the gene for CDK5 and its upstream regulators. Finally, elucidating how CDK5 phosphorylation alters PPARγ structure might be informative. Potential conformational changes may affect other post-translational modifications of PPARγ and alter recruitment of its other regulators. Of priority should be exploring whether CDK5-mediated PPARγ phosphorylation supports recruitment of PPARγ cofactors that unfavourably affect metabolism, such as TIF2/SRC-2 and RIP140 (refs 11, 12), at the expense of those with a more favourable metabolic connotation, such as SRC-1 and PGC-1 (refs 11,13).
Almost 25 years after their 1987 paper2, the Spiegelman group1 might have once again caught a big ‘fat’ fish by explaining why classical PPARγ drug discovery was poised to fail. The research community was fishing with the wrong bait. Indeed, Choi and colleagues’ results seriously question the screening strategies of the drug industry to identify extremely potent PPARγ activators, which were not necessarily more potent insulin sensitizers.
Targeting PPARγ should now be a rather straightforward strategy and could lead to compounds that induce conformational changes of the PPARγ protein, activate it in moderation, and most importantly fully remove its phosphorylation mark15. Such compounds could still reprogram the expression of crucial metabolic gene sets, but should lack the typical PPARγ side effects caused by full activation of this receptor. Also, although several effective CDK5 inhibitors exist (including roscovitine), we would favour a drug that specifically affects PPARγ; complete CDK5 inhibition would also interfere with its other functions, for instance, in the central nervous system14. Altogether, Choi and colleagues’ work heralds a new era of drug discovery, now able to rationally target PPARγ activity, so to improve diabetes and avoid side effects.
References
- 1.Choi JH, et al. Nature. 2010;466(7305):451–456. doi: 10.1038/nature09291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Distel RJ, Ro HS, Rosen BS, Groves DL, Spiegelman BM. Cell. 1987;49:835–844. doi: 10.1016/0092-8674(87)90621-0. [DOI] [PubMed] [Google Scholar]
- 3.Tontonoz P, Spiegelman BM. Annu. Rev. Biochem. 2008;77:289–312. doi: 10.1146/annurev.biochem.77.061307.091829. [DOI] [PubMed] [Google Scholar]
- 4.Straus DS, Glass CK. Trends Immunol. 2007;28:551–558. doi: 10.1016/j.it.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 5.Argmann C, et al. PLoS Genet. 2009;5:e1000752. doi: 10.1371/journal.pgen.1000752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deeb SS, et al. Nature Genet. 1998;20:284–287. doi: 10.1038/3099. [DOI] [PubMed] [Google Scholar]
- 7.Nissen SE, Wolski KN. Engl. J. Med. 2007;356:2457–2471. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]
- 8.Wei FY, et al. Nature Med. 2005;11:1104–1108. doi: 10.1038/nm1299. [DOI] [PubMed] [Google Scholar]
- 9.Lilja L, et al. J. Biol. Chem. 2001;276:34199–34205. doi: 10.1074/jbc.M103776200. [DOI] [PubMed] [Google Scholar]
- 10.Dhavan R, Tsai LH. Nature Rev. Mol. Cell. Biol. 2001;2:749–759. doi: 10.1038/35096019. [DOI] [PubMed] [Google Scholar]
- 11.Picard F, et al. Cell. 2002;111:931–941. doi: 10.1016/s0092-8674(02)01169-8. [DOI] [PubMed] [Google Scholar]
- 12.Leonardsson G, et al. Proc. Natl Acad. Sci. USA. 2004;101:8437–8442. doi: 10.1073/pnas.0401013101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Puigserver P, et al. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 14.Patrick GN, et al. Nature. 1999;402:615–622. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- 15.Knouff C, Auwerx J. Endocr. Rev. 2004;25:899–918. doi: 10.1210/er.2003-0036. [DOI] [PubMed] [Google Scholar]