

Adducts in nuclear DNA are the major cause of cancer cell death triggered by platinum-based anticancer drugs.[1] Thus, cellular uptake and accumulation, distribution and trafficking between subcellular compartments and, ultimately, localization to the nucleus are crucial parameters in the mechanism of these agents. Several techniques have been used to monitor intracellular platinum. These include element-specific analytical methods and nondestructive absorption or emission-based imaging techniques, as well as electron microscopy.[2,3] Fluorophore-tagged derivatives have provided insight into uptake, distribution, and intracellular transformation of platinum.[2] Such an approach has to take into consideration the organelle selectivity of the fluorophore, which may vary widely depending on parameters such as molecular weight, partition coefficient (log P), amphiphilic character, and pKa value.[4] Thus, one drawback of modifying platinum drugs with organic fluorophores is that such conjugates may, at least in part, mimic the properties of the reporter molecule.[5] This would be an undesired feature unless the fluorescent group itself is a functionally important part of the bioactive molecule. Likewise, bulky fluorophores may interfere with the DNA binding mechanism of platinum. To circumvent these problems, we have developed a method based on bioorthogonal ligation chemistry, which allowed us to fluorescently label platinum–acridine hybrid agents in lung cancer cells. Here, we report the development of this technique and demonstrate, for the first time, that post-labeling is a powerful tool for detecting DNA-targeted platinum in subcellular structures.
Platinum–acridines, represented by the prototypical compound 1 (Figure 1), are up to 500-times more cytotoxic to non-small cell lung cancer cells than the drug cisplatin.[6] Studies in NCI-H460 cells suggest that the cytotoxicity produced by compound 1 is triggered by high levels of monofunctional–intercalative hybrid adducts in cellular DNA,[7] which is supported by recent results from chemogenomic screening in S. cerevisiae.[8] Unfortunately, direct fluorescent imaging of platinum–acridines in cancer cells is difficult because the fluorescence of the 9-aminoacridine is severely quenched when intercalated into DNA[9] (Supporting Information). We therefore embarked on developing methodology that allows the labeling of platinum–acridines, in particular their DNA adducts, intracellularly using a high-performance fluorescent dye in conjunction with copper-catalyzed azide–alkyne cycloaddition (CuAAC) (click) chemistry.[10,11] This strategy required modification of compound 1 with an alkyne or azide group in a way that would not interfere with the DNA binding mode of the agent. Molecular models derived from the NMR solution structure of the hybrid adduct[12] (Supporting Information) suggest that installation of a clickable group at the amidine N1-methyl group in 1 (Figure 1), which is protruding out of the DNA major groove, should satisfy this requirement. While direct attachment of either functional group to this position via polymethylene linkers was incompatible with the metal and resulted in complex decomposition, our search identified a stable amide-linked azide derivative (compound 2, Figure 1), which also showed excellent robustness in aqueous media (see Supporting Information). Using gel-based DNA unwinding experiments and CD spectropolarimetry we confirmed that the azide-containing linker does not affect the geometry of the hybrid adduct (Supporting Information). Thus, compound 2 was chosen for the development of the labeling technique in combination with the alkyne-modified form of the bright green-fluorescent dye Alexa Fluor 488 (Figure 1).
Figure 1.

Structures of compounds and reaction scheme for bioorthogonal fluorescent labeling of DNA-targeted platinum–acridine hybrid agents.
First, we established conditions for labeling the DNA adducts of compound 2 using CuAAC chemistry in a randomly platinated linearized plasmid. The procedure involved incubation of the DNA with compound 2 at varying platinum-to-nucleotide ratios (rb) and reaction of the platinated DNA with Alexa Fluor 488-alkyne in the presence of copper-based click reaction buffer, followed by purification of the samples and analysis of the labeled DNA on agarose gels (for details see Supporting Information). The in-gel fluorescence detection of the DNA along with the ethidium-stained gel is shown in Figure 2A. Bands on the gel show a pronounced increase in Alexa Fluor 488 fluorescence intensity with increasing platinum-to-nucleotide ratio (lanes 3–5, top gel). In further support of the notion that Alexa Fluor 488-alkyne was ligated to covalently bound platinum–acridine in the plasmid, a sample of fluorescently labeled DNA was incubated with NaCN, which reverses Pt–DNA adducts.[13] The complete disappearance of Alexa Fluor fluorescence in lane 6 (Figure 2A, top gel) and conversion of the platinated DNA into unmodified plasmid (Figure 2A, bottom gel, band of highest mobility) confirm that, indeed, platinum adducts were modified by the green-fluorescent dye.
Figure 2.

(A) Electrophoretic analysis of copper-catalyzed ligation reactions between linearized (BamH I), platinated pUC19 plasmid DNA and Alexa Fluor 488-alkyne with reaction conditions indicated for each incubation (for details see Supporting Information). The top gel shows Alexa Fluor fluorescence monitored at 520 nm. Note the increase in band intensities and mobility shift with increasing platinum content. The latter band shift is caused by accumulation of positive charge on the plasmid. The bottom panel depicts the same gel stained with ethidium bromide. Saturation of DNA binding sites by Alexa Fluor-tagged platinum–acridine causes inefficient staining by ethidium at rb = 0.2. (B) CD spectra recorded in the ICD region for unmodified DNA (dotted trace), DNA modified with compound 2 (dashed trace), and DNA modified with compound 2 after ligation with Alexa Fluor 488-alkyne (solid trace).
CD spectra were recorded of native DNA globally modified with compound 2 before and after undergoing ligation chemistry with Alexa Fluor dye (Figure 2B). The adducts formed by compound 2 give rise to positive Cotton effects in the 300–500 nm range characteristic of the induced circular dichroism of the intercalated acridine chromophore.[14] After labeling with Alexa Fluor 488-alkyne, the CD spectrum shows an additional negative ICD band at 498 nm, which mimics the absorbance spectrum of the dye, a consequence of the DNA chirality experienced by the ligated fluorophore. The hypsochromic shift of 4 nm observed for the acridine-based long-wavelength ICD bands in the labeled adduct relative to the unlabeled adduct of 2 suggests that ligation of Alexa Fluor dye perturbs the hybrid binding mode leading to partial unstacking of the acridine moiety.[12] By contrast, a comparison of the ICD signatures of DNA-bound compounds 1 and 2 shows that addition of the azide-containing side chain does not compromise acridine intercalation. This observation corroborates the notion that compound 2, but not its fluorophore-tagged form, is a faithful mimic of compound 1, which validates the post-labeling approach.
Next, we tested if the labeling strategy developed in cell-free DNA can be applied to whole cancer cells. For confocal fluorescence microscopy studies, NCI-H460 cells were incubated with azide-modified platinum under conditions that were expected to produce high levels of intracellular drug without killing the cells, as confirmed by the absence of the morphological hallmarks of apoptotic cell death.[15] Briefly, cultured cells were treated with three different doses (1, 5, or 25 μM) of compound 2 for 3 h, washed, formalin-fixed, permeabilized, and incubated with CuAAC reaction buffer in the presence of Alexa Fluor 488-alkyne. No-platinum controls treated with fluorescent dye in the presence or absence of copper were also prepared. Following exhaustive washing to remove excess labeling mix and co-staining with nuclear dye, cells were imaged by confocal fluorescence microscopy.
Images of platinum-treated NCI-H460 cells incubated with Alexa Fluor 488-alkyne/copper clearly demonstrate that the labeling technology can be applied to whole cells (Figure 3A). Cells treated with compound 2 show a pronounced intensity increase in fluorescence emission relative to condition matched controls as a result of selective intracellular azide–alkyne ligation chemistry. Alexa Fluor 488 emission intensities in the nuclei of treated cells (n > 50) show a clear correlation with increasing incubation concentrations of compound 2, confirming that intracellular modification of platinum-tethered azide is the source of the fluorescence (see Supporting Information).
Figure 3.

Imaging of compound 2 (5 μM) in fixed NCI-H460 lung cancer cells using Alexa Fluor 488-alkyne. Cells were co-stained with Hoechst 33342. (A) Single confocal image planes of Alexa Fluor 488 (green), Hoechst 33342 (blue), the green and blue channels merged, and corresponding bright field image are shown for compound 2 treatment and control conditions. Control groups were treated with Alexa Fluor 488-alkyne and copper-based ligation buffer. No-copper controls were treated with Alexa Fluor 488-alkyne in the absence of copper. (B) Single confocal image plane of the cellular distribution of platinum–azide-related fluorescence in NCI-H460 cells during mitosis (labeled a) and interphase (example labeled b).
Platinum–azide-related fluorescence is observed at high intensity within nuclei and at much lower intensity within the cytoplasm. (For a detailed analysis of distinct cellular regions, see Supporting Information.). Figure 3B shows colocalization images of Alexa Fluor 488 and the nuclear stain Hoechst 33342 within NCI-H460 cells during different stages of the cell cycle. During mitosis, the highest levels of green fluorescence are observed in the condensed, replicated chromatin, whereas cells in interphase show the strongest fluorescence signal across the entire nucleus. Most strikingly, the highest fluorescence intensity in the latter organelle is observed in the nucleoli, the sites of ribosomal RNA (rRNA) transcription in cells in interphase.[16] An NCI-H460 cell imaged at high magnification shows the relative distribution of Alexa Fluor within the cell and confirms that compound 2 accumulates in the nucleus and nucleolus (Figure 4). Analysis of a large number of cells (n = 40–50) shows that the emission in the 488 nm excitation channel has a 50% higher relative intensity within the nucleolar regions compared to the surrounding chromatin (see Supporting Information).
Figure 4.
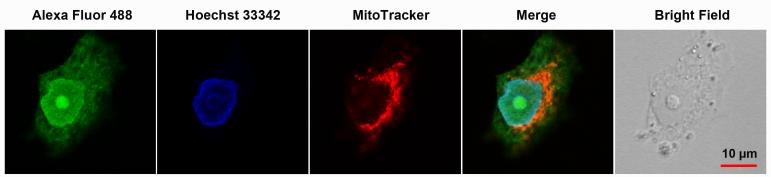
Intracellular distribution of compound 2 in a NCI-H460 cell in interphase. Cells were co-stained with nuclear (Hoechst 33342) and mitochondrial (MitoTracker Deep Red) dyes. The nucleolus, which is readily identified in the bright-field image and as a nuclear region unstained by Hoechst dye, appears as an area of high fluorescent intensity in the Alexa Fluor 488 and merged channel images.
In conclusion, the current study demonstrates that copper-mediated click chemistry is a powerful imaging tool for mapping the subcellular localization of a platinum-containing pharmacophore in fixed cancer cells. This was achieved by turning a potent platinum–acridine anticancer agent into a fluorophore-ligatable cellular probe. Compound 2 represents the first example of a platinum-based anticancer agent compatible with alkyne–azide ligation chemistry. The rapid accumulation of compound 2 in chromatin in the nucleus after only 3 h of incubation observed in this study is consistent with the high platinum–DNA adduct levels in NCI-H460 cells determined previously by ICP-MS.[7] The imaging results support the notion that damage to nuclear DNA is a major trigger of S-phase arrest and apoptosis in NCI-H460 cells.[8,17] The biological consequences of platinum–acridine localization to the nucleoli, on the other hand, remain elusive. Potential targets for platinum adduct formation in this dynamic, non-membrane bound structure include transcriptionally active rRNA genes (rDNA) and (nascent) rRNA itself.[16] Nucleolar rRNA synthesis is an emerging target in anticancer drug development, and certain DNA intercalators have demonstrated selective inhibitory action at the rRNA transcriptional level.[18,19] It is worthwhile to mention that, in deletion strains of S. cerevisiae, platinum–acridines elicit responses from genes involved in RNA metabolism, which suggests that inhibition of the rRNA synthesis machinery may contribute to the cell kill produced by these agents.[8] Finally, it is anticipated that the new imaging platform can be extended to more biocompatible and ultrafast copper-free click chemistry, which would allow the monitoring of platinum levels and localization in real-time within live cells.[20]
Supplementary Material
Footnotes
This work was supported by the US National Institutes of Health (grant CA101880). X. Q. gratefully acknowledges support from the China Scholarship Council (grant #2011694010).
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201210079
Contributor Information
Song Ding, Department of Chemistry, Wake Forest University, Winston-Salem, NC 27109, USA.
Xin Qiao, Department of Chemistry, Wake Forest University, Winston-Salem, NC 27109, USA.
Dr. Jimmy Suryadi, Department of Chemistry, Wake Forest University, Winston-Salem, NC 27109, USA
Dr. Glen S. Marrs, Department of Biology, Wake Forest University, Winston-Salem, NC 27109, USA
Prof. Gregory L. Kucera, Department of Internal Medicine, Section on Hematology and Oncology, Wake Forest University Health Sciences, Winston-Salem, NC 27157, USA
Prof. Ulrich Bierbach, Department of Chemistry, Wake Forest University, Winston-Salem, NC 27109, USA.
References
- [1].Wang D, Lippard SJ. Nature Rev. Drug Discov. 2005;4:307–320. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- [2].Klein AV, Hambley TW. Chem. Rev. 2009;109:4911–4920. doi: 10.1021/cr9001066. [DOI] [PubMed] [Google Scholar]
- [3].Wedlock LE, Berners-Price SJ. Aust. J. Chem. 2011;64:692–704. [Google Scholar]
- [4].Trapp S, Horobin RW. Eur. Biophys. J. 2005;34:959–966. doi: 10.1007/s00249-005-0472-1. [DOI] [PubMed] [Google Scholar]
- [5].Liang XJ, Shen DW, Chen KG, Wincovitch SM, Garfield SH, Gottesman MM. J. Cell. Physiol. 2005;202:635–641. doi: 10.1002/jcp.20253. [DOI] [PubMed] [Google Scholar]
- [6].Suryadi J, Bierbach U. Chem. Eur. J. 2012;18:12926–12934. doi: 10.1002/chem.201202050. [DOI] [PubMed] [Google Scholar]
- [7].Qiao X, Zeitany AE, Wright MW, Essader AS, Levine KE, Kucera GL, Bierbach U. Metallomics. 2012;4:645–652. doi: 10.1039/c2mt20031g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cheung-Ong K, Song KT, Ma Z, Shabtai D, Lee AY, Gallo D, Heisler LE, Brown GW, Bierbach U, Giaever G, Nislow C. ACS Chem. Biol. 2012;7:1892–1901. doi: 10.1021/cb300320d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Fornasiero D, Kurucsev T. Biophys. Chem. 1985;23:31–37. doi: 10.1016/0301-4622(85)80061-2. [DOI] [PubMed] [Google Scholar]
- [10].Demko ZP, Sharpless KB. Angew. Chem. Int. Edit. 2002;41:2110–2113. [PubMed] [Google Scholar]
- [11].Buck SB, Bradford J, Gee KR, Agnew BJ, Clarke ST. A. Salic, Biotechniques. 2008;44:927–929. doi: 10.2144/000112812. [DOI] [PubMed] [Google Scholar]
- [12].Baruah H, Wright MW, Bierbach U. Biochemistry. 2005;44:6059–6070. doi: 10.1021/bi050021b. [DOI] [PubMed] [Google Scholar]
- [13].Kane SA, Lippard SJ. Biochemistry. 1996;35:2180–2188. doi: 10.1021/bi952240a. [DOI] [PubMed] [Google Scholar]
- [14].Kostrhunova H, Malina J, Pickard AJ, Stepankova J, Vojtiskova M, Kasparkova J, Muchova T, Rohlfing ML, Bierbach U, Brabec V. Mol. Pharmaceutics. 2011;8:1941–1954. doi: 10.1021/mp200309x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Saraste A, Pulkki K. Cardiovasc. Res. 2000;45:528–537. doi: 10.1016/s0008-6363(99)00384-3. [DOI] [PubMed] [Google Scholar]
- [16].Lam YW, Trinkle-Mulcahy L, Lamond AI. J. Cell. Sci. 2005;118:1335–1337. doi: 10.1242/jcs.01736. [DOI] [PubMed] [Google Scholar]
- [17].Smyre CL, Saluta G, Kute TE, Kucera GL, Bierbach U. ACS Med. Chem. Lett. 2011;2:870–874. doi: 10.1021/ml2001888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Drygin D, Rice WG, Grummt I. Annu. Rev. Pharmacol. 2010;50:131–156. doi: 10.1146/annurev.pharmtox.010909.105844. [DOI] [PubMed] [Google Scholar]
- [19].Drygin D, Lin A, Bliesath J, Ho CB, O’Brien SE, Proffitt C, Omori M, Haddach M, Schwaebe MK, Siddiqui-Jain A, Streiner N, Quin JE, Sanij E, Bywater MJ, Hannan RD, Ryckman D, Anderes K, Rice WG. Cancer Res. 2011;71:1418–1430. doi: 10.1158/0008-5472.CAN-10-1728. [DOI] [PubMed] [Google Scholar]
- [20].Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, Bertozzi CR. Proc. Natl. Acad. Sci. U S A. 2007;104:16793–16797. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.