

Abstract
blind sterile (bs) is a spontaneous autosomal-recessive mouse mutation discovered more than 30 years ago. Phenotypically, bs mice exhibit nuclear cataracts and male infertility; genetic analyses assigned the bs locus to mouse chromosome 2. In this study, we first positionally cloned the bs locus and identified a putative causative mutation in the Tbc1d20 gene. Functional analysis established the mouse TBC1D20 protein as a GTPase-activating protein (GAP) for RAB1 and RAB2, and bs as a TBC1D20 loss-of-function mutation. Evaluation of bs mouse embryonic fibroblasts (mEFs) identified enlarged Golgi morphology and aberrant lipid droplet (LD) formation. Based on the function of TBC1D20 as a RABGAP and the bs cataract and testicular phenotypes, we hypothesized that mutations in TBC1D20 may contribute to Warburg micro syndrome (WARBM); WARBM constitutes a spectrum of disorders characterized by eye, brain, and endocrine abnormalities caused by mutations in RAB3GAP1, RAB3GAP2, and RAB18. Sequence analysis of a cohort of 77 families affected by WARBM identified five distinct TBC1D20 loss-of-function mutations, thereby establishing these mutations as causative of WARBM. Evaluation of human fibroblasts deficient in TBC1D20 function identified aberrant LDs similar to those identified in the bs mEFs. Additionally, our results show that human fibroblasts deficient in RAB18 and RAB3GAP1 function also exhibit aberrant LD formation. These findings collectively indicate that a defect in LD formation/metabolism may be a common cellular abnormality associated with WARBM, although it remains unclear whether abnormalities in LD metabolism are contributing to WARBM disease pathology.
Introduction
Warburg micro syndrome (WARBM1 [MIM 60011], WARBM2 [MIM 614225], WARBM3 [MIM 614222]) is a heterogeneous autosomal-recessive disorder characterized by eye, brain, and endocrine abnormalities.1 Causative loss-of-function mutations have been identified in three genes: RAB3GAP12–5 (MIM 602536), RAB3GAP25,6 (MIM 609275), and RAB185,7 (MIM 602207), which all result in clinically indistinguishable WARBM phenotypes. Ophthalmological findings in WARBM children are characterized by bilateral congenital cataracts, microphthalmia, microcornea, small atonic pupils, progressive optic atrophy, and severe cortical visual impairment that results in very poor visual prognosis despite early cataract surgery.2–7 Neurological features include postnatal microcephaly, profound mental retardation, severe truncal hypotonia, and progressive limb spasticity that leads to spastic quadriplegia.2–7 Nerve conduction studies have shown evidence of a progressive axonal peripheral neuropathy.7 Brain MRI scans show bilateral polymicrogyria with hypogenesis of the corpus callosum and cerebellar and cerebellar vermis hypoplasia.2–7 Boys present with micropenis and cryptorchidism resulting from hypothalamic hypogonadism and girls have hypoplastic labia minora, clitoral hypoplasia, and small introitus.2–7 Martsolf syndrome (MS [MIM 212720]), a clinically overlapping yet milder disorder, has been attributed to mutations in RAB3GAP15 and RAB3GAP2.2,8 Functional studies have established that WARBM and MS represent a phenotypic spectrum where the clinical outcome is related to the severity of the mutational effects on protein function. Thus, mutations that result in residual protein function cause MS whereas loss-of-function mutations cause WARBM.2–7,9–16 Currently mutations in RAB3GAP1, RAB3GAP2, and RAB18 account for about 50%5 of WARBM cases, suggesting that mutations in additional genes also cause WARBM.
The blind sterile (bs) mouse was identified in 1983 as a spontaneous autosomal-recessive mutation;17 the initial report showed that bs mice exhibit embryonic nonprogressive nuclear cataracts and spermatid abnormalities associated with bs male infertility.17 Additional morphological studies of bs seminiferous tubules identified the failure of acrosome formation in spermatids.18,19 Although spermatozoa were absent in the majority of bs males, a few nonmotile spermatozoa with severely misshaped heads were seen; thus, it was proposed that the bs gene plays a role in acrosome formation and spermatozoa head shaping.18 Another study examined bs seminiferous tubules and identified round spermatids as the most advanced spermatogenic cell present, but did not identify the presence of either elongating spermatids or spermatozoa.20 This discrepancy was attributed to individual variations among animals or the difference in age of the animals evaluated.20 Initial linkage analysis established bs as an autosomal-recessive locus that maps to the distal region of mouse chromosome 2 near the agouti locus.17 Subsequent genetic analysis narrowed it to a ∼10 cM region between the hydroxyl acid oxidase 1 (Hao1) and non-agouti (a) loci syntenic with human HSA20q11–q13.21 Although the bs eye and testis phenotypes were inherited collectively as a single autosomal-recessive trait,17,21 it remained unclear whether the bs phenotypes were associated with a mutation in a single gene or mutations affecting the function of several tightly linked genes.
The initial goal of this study was to further characterize the bs phenotypes and to positionally clone the bs locus. We identified a loss-of-function mutation in the Tbc1d20 gene as responsible for the bs phenotypes. TBC1D20 is a member of a superfamily of highly evolutionarily conserved proteins containing TBC (Tre-2/Bub2/Cdc16) domains. TBC family members have a role in the regulation of RAB GTPases. All RAB GTPases function as molecular switches alternating between a GTP-bound active state and a GDP-bound inactive state.22–24 The activation of RAB GTPases is mediated by guanine nucleotide exchange factors (GEFs) that facilitate GDP dissociation; GTP-activating proteins (GAPs) accelerate the slow intrinsic RAB GTP hydrolysis from the GTP-bound “active” form to the GDP-bound “inactive” form.22–25 The role of TBC1D20 has been established as a GAP for RAB1 that facilitates COPII-mediated ER-to-Golgi trafficking.26–28
Our findings in bs mice prompted us to a screen a cohort of individuals presenting with clinical manifestations consistent with WARBM for mutations in TBC1D20. We identified five distinct TBC1D20 loss-of-function mutations. Molecular evaluations of mouse and human cells deficient for TBC1D20 function and of human cells deficient for RAB3GAP1 and RAB18 function provided evidence that the aberrant formation of lipid droplets (LDs) is a common cellular phenotype in all these cell lines. Although these findings further imply that WARBM proteins may function in a common yet unknown pathway, it still remains unclear whether observed abnormalities in LD are associated with WARBM disease pathology or whether the disease pathology is a consequence of another still unidentified cellular deficit.
Subjects and Methods
Animals, Phenotypic Evaluation, and Positional Cloning of the bs Locus
CAST/EiJ, AKR/J, and bs mice as well as genomic DNA for C57BL/6J, C3H/HeJ, RIIIS/J, and DBA/2J mouse strains were obtained from The Jackson Laboratory. All mice were used with strict adherence to the guidelines set forth by the Institutional Animal Care and Use Committee (IACUC) at the Medical College of Wisconsin. Mouse eyes were examined with a Topcon SL-D8Z slit lamp biomicroscope with a Nikon SLR-based Photo Slit Lamp imaging system after mydriasis with 1% atropine sulfate (Bausch & Lomb). WT and bs testes weights were measured in age-matched pairs between 4 and 8 weeks of age. Significance was calculated via a t test (GraphPad), where p < 0.05 was considered significant.
For linkage studies, bs/bs female mice, on the congenic AKR/J background, were outcrossed to CAST/EiJ; the resulting F1 progeny were subsequently intercrossed to generate 1,177 F2 progeny. At 4 weeks of age, F2 progeny were clinically evaluated for the presence of cataracts, euthanized, and genotyped as previously described.29 Linkage data were analyzed with MapManager QTX software. cDNA and genomic DNA sequencing from bs/bs and bs/+ mice was utilized to screen open reading frames and intron/exon junctions of candidate genes listed in Table S1 (available online). Primers were designed with Primer3 software and comparative sequence analysis was performed with DNAStar software. Primer sequences are listed in Table S2.
Histology and Immunohistochemistry
Tissues were collected, paraffin embedded, and H&E stained as previously described.30,31 For immunohistochemistry we used E-cadherin (Cell Signaling), MIP (Milipore), DAZL (Abcam), and TRA54 (B-Bridge) as primary antibodies and DyLight 488 goat anti-rat or goat anti-rabbit (Abcam) as secondary antibodies according to the manufacturers’ recommendations. TUNEL and PNA staining was performed with the ApopTag Plus In Situ Apoptosis Fluorescein Detection Kit (Chemicon) and Lectin PNA-Alexa-488 conjugate (Life Technologies), respectively, according to the manufacturers’ recommendations. For proliferation studies, EdU was injected intraperitoneally at a concentration of 100 mg/kg 3 hr prior to euthanizing the mice; EdU detection was performed with the Click iT EdU Alexa Fluor 488 Imaging Kit and counterstained with Hoechst 33342 or DAPI according to the manufacturer’s recommendations (Life Technologies). All cell-counting measurements were performed on sections from a minimum of three separate genotypes with at least ten sections per genotype. Significance was calculated via a Student’s t test (GraphPad), where p < 0.05 was considered significant. All slides were mounted with Vectashield and imaged with a Nikon DS-Fi1 camera on a Nikon Eclipse 80i microscope with NIS-Elements software (Nikon).
Protein Expression and RAB GTP Hydrolysis Assays
RAB family GTPases were cloned into a hexahistidine-GST tag bacterial expression vector pFAT2 expressed in BL21(DE3) cells and proteins were purified with nickel-NTA as described previously.32 WT or bs Tbc1d20 cDNA was cloned in the hexahistidine tag bacterial expression vector pQE32, expressed in JM109 cells, and purified with nickel-NTA agarose. For mammalian expression, WT or bs Tbc1d20 cDNA was cloned into the pcDNA4 vector containing an eGFP tag. GTP-hydrolysis assays were performed as previously described.26,32
Cell Culture and Immunocytochemistry
HeLa and HEK293 cells were cultured in DMEM containing 10% fetal bovine serum at 37°C and 5% CO2. mEFs were isolated from E13.5 mouse embryos and maintained as previously described.30,31 Human fibroblasts were cultured in DMEM supplemented with 20% fetal calf serum and 1% penicillin/streptomycin at 37°C and 3% O2, 5% CO2.
Plasmid transfection of HeLa cells was done with Mirus LT1 transfection reagent (Mirus Bio LLC) and immunocytochemistry was done with primary antibodies (EEA1 [Cell Signaling], SEC31 [BD Biosciences], GM130 [BD Biosciences]) and secondary antibodies, raised in donkey, to mouse, rabbit, and sheep/goat conjugated to HRP, Alexa Fluor 488, Alexa Fluor 555, Alexa Fluor 568, and Alexa Fluor 647 (all from Life Technologies) according to manufacturers’ recommendations. Imaging was done on an Olympus BX61 upright microscope system with a camera (CoolSNAP HQ2; Roper Industries) and MetaMorph imaging software (Molecular Dynamics). Illumination was provided by a Lumen 200 Watt metal halide light source (Prior Scientific Instruments).
WT and bs mEFs were immunostained with GM130 (Abcam), Golgin-97 (Abcam), or ERp72 (Cell Signaling) primary antibodies and Alexa 488-conjugated goat-anti-rabbit secondary antibody (Invitrogen) according to manufacturers’ recommendations. LDs were evaluated as described previously33 utilizing media supplemented with 400 μM oleic acid (Sigma Aldrich) for 6, 18, or 24 hr and stained with 1 μg/μl BODIPY 493/503 (Life Technologies). All slides were mounted via Vectashield with DAPI (Vector Labs). Imaging was done with a Nikon DS-Fi1 camera on a Nikon Eclipse 80i microscope with NIS-Elements software (Nikon). Quantification of lipid droplets was performed as previously described34 with ImageJ (National Institutes of Health) and NIS-Elements software. For each analysis at least 30 cells per genotype were evaluated.
Human fibroblasts cells were fixed and stained as described previously.35 We used primary antibodies to PDI (Abcam), GM130 (BD Biosciences), Golgin-97 (Transduction Laboratories), p115 (Abcam), or EEA1 (New England Biolabs) and Alexa Fluor 488-conjugated donkey anti-mouse or anti-rabbit secondary antibody as appropriate (Life Technologies) and costained with DAPI. For LD analysis, human fibroblasts were treated with 400 μM oleate for 6, 18, or 24 hr and stained with 1 μg/ml BODIPY 493/503 (Life Technologies) as previously described.33 In order to analyze LDs, five frames/slide were imaged under identical microscope settings and LD area was quantified with the “Analyze particles” function on ImageJ Fiji software.36 Imaging was carried out on a Nikon A1R confocal microscope with a 60× oil immersion objective with a 1.4 numerical aperture. The pinhole was set to airy1. Data sets shown are representative of at least three independent experiments.
Protein Stability Assay and Immunoblotting
Tbc1d20 mouse full-length clone (MGC: 25843/IMAGE: 4192736) was obtained from Open Biosystems and cloned into pFLAG-CMV-2 (Sigma) to generate an N-terminal FLAG-tagged Tbc1d20 clone. Mutagenesis to introduce the bs mutation was performed in a two-step process utilizing the QuikChange Site-Directed Mutagenesis Kit (Stratagene) and the Phusion Site-Directed Mutagenesis Kit (Finnzymes) with primers in Table S2. For protein stability studies, HEK293 cells were transfected with either FLAG-Tbc1d20WT or FLAG-Tbc1d20bs treated with cycloheximide (Sigma) according to manufacturers’ recommendations.
For immunoblot analyses, primary antibodies anti-FLAG (Sigma), GM130 (Abcam), HRP-conjugated β-actin (Abcam), Syntaxin VI (New England Biolabs), RAB5 (New England Biolabs), and α-Tubulin (Abcam) and secondary antibodies peroxidase-conjugated AffiniPure Donkey Anti-Rabbit IgG (H+L) (Jackson ImmunoResearch), peroxidase-conjugated anti-mouse, anti-Rabbit (Amersham), and anti-Goat (Life Technologies) were used as previously described.31 Each lane on the blots shown corresponds to an individual lysate sample, and each blot is representative of at least three independent experiments. For the protein stability assays, immunoblots were quantified with ImageJ software, normalizing FLAG to β-actin, and each time point represents at least three independent experiments done in triplicate.
Subjects and Mutation Screening
Our cohort consists of 77 individuals with a spectrum of WARBM disorders including “typical” WARBM (59 cases), MS (5 cases), and 13 atypical cases as previously described.5 These individuals do not carry mutations in the coding sequences of previously analyzed genes, specifically RAB3GAP1, RAB3GAP2, and RAB18. Informed consent was obtained from all participating families, and the studies were approved by the Scottish Multicenter Research Ethics Committee (04:MRE00/19). Permission to publish photographs from WARBM individuals was obtained from the parents. Mutation screening of the complete coding region of TBC1D20, RAB1A, RAB1B, and RAB2A was performed by direct sequence analysis in both directions by using genomic DNA extracted from venous blood according to standard procedures. Primers with M13 tags (Table S3) were designed from the genomic sequences to flank all coding exons and include all intron-exon boundaries by means of ExonPrimer software. Sequencing data was analyzed with Mutation Surveyor software (SoftGenetics). qPCR analysis of genomic DNA from individual 5 was carried out on a LightCycler 480 (Roche) with primers designed to amplify coding regions of TBC1D20 as well as the 5′ and 3′ genes RBCK1 (RefSeq accession number NM_031229.2) and CSNK2A1 (RefSeq NM_177559.2). PCR amplification was quantified through binding of specific monocolor hydrolysis probes (Roche) and analyzed with LightCycler 480 software (Roche). Primers were designed with the Universal Probe Library Assay Design Center and are listed in Table S3.
Results
The bs Lens and Testes Phenotypes
As a starting point, we expanded on the previous characterization of the bs cataract and testes phenotypes. Examination of bs eyes at P14 identified nuclear opacities that progressed by P28 to total opacities with irregularly patterned vacuoles (Figure 1A). An initial report on bs stated that cataracts could be identified at E16 through the eyelids of bs embryos,17 and therefore we expected to observe bs lens morphological abnormalities at E15.5. Histological analysis of E15.5 bs lenses did not detect any morphological abnormalities (not shown), and immunostaining with E-cadherin and MIP did not identify any abnormalities in bs lens epithelial and lens fiber cells; no difference in the number of proliferating cells or in the number of TUNEL(+) cells were noted between WT and bs lenses at E15.5 (Figure S1A). In contrast, at E17.5, bs lenses appeared smaller in size and exhibited degenerated nuclear fibers with small vacuoles between cortical fibers (Figure 1B). By P10, the bs lens phenotype was characterized by severely degenerated TUNEL(+) nuclear fiber cells (Figure 1C). By P28, severe lens degeneration with large vacuoles was present throughout the lens body accompanied by rupture of the lens capsule and lenticular material in the vitreous cavity (Figure 1D). At later time points (12–24 months), the bs lens phenotype did not progress beyond that observed at P28 (not shown). These findings indicate that bs cataracts are associated with a defect in lens fiber cell maturation with an embryonic onset similar to that initially reported.17
Figure 1.
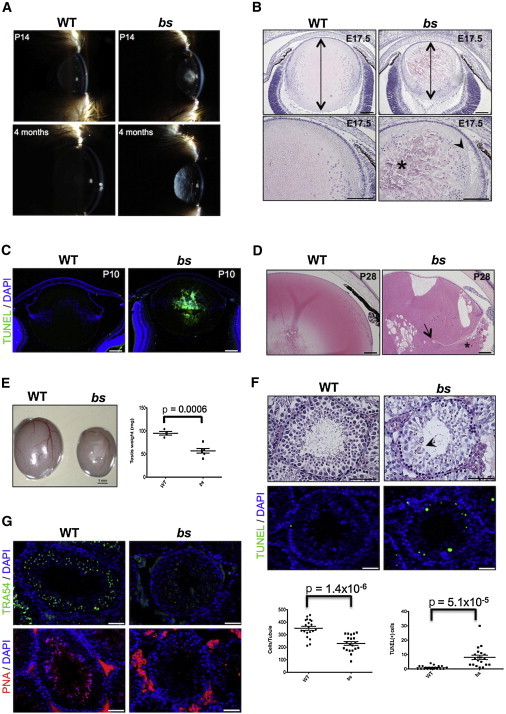
bs Eye and Testes Phenotypes
(A) Clinical image of nuclear cataracts in bs evident at P14 (top) that progress to severe vacuolated cataracts (bottom).
(B) At E17.5, H&E staining showed smaller bs lens axial lengths (top, arrows); at higher magnification (bottom), disorganized lens fiber cells (asterisk) and cortical vacuoles (arrowhead) were noted. Scale bars represent 100 μm.
(C) At P10 bs lenses exhibited severely degenerated TUNEL(+) nuclear fibers. Scale bars represent 100 μm.
(D) At P28, H&E staining revealed severely degenerated bs lenses, large vacuoles, and ruptured lens capsule (arrow) with lenticular material present in the vitreous cavity (asterisk). Scale bars represent 100 μm.
(E) Adult bs testes were significantly smaller when compared to WT (n = 6). Scale bar represents 1 mm.
(F) H&E analysis (top) of adult bs seminiferous tubules (n = 20) identified significant germ cell depletion and some tubules contained multinucleate cell clusters (arrowhead) consistent with previous reports.18 Scale bars represent 50 μm. A significantly greater number of TUNEL(+) cells (bottom) were present in bs than in WT tubules (n = 15). Scale bars represent 25 μm.
(G) TRA54 immunostaining in WT tubules revealed small punctae and crescent-shaped staining consistent with spermatocytes and round spermatids, respectively, and in bs only TRA54-positive small punctae were present, consistent with spermatocytes (top). PNA staining identified the presence of acrosomes in WT tubules, whereas no PNA-positive cells were noted in bs tubules (bottom). Scale bars represent 25 μm. DNA was stained with DAPI (blue). p values were determined by Student’s t test and error bars represent SEM.
Adult bs testes were significantly smaller than WT testes (Figure 1E) and exhibited a significant depletion of germ cells (Figure 1F). However, the numbers of spermatogonia or Sertoli cells determined after immunostaining with DAZL did not differ between WT and bs nor was there a difference in the number of proliferating spermatogonia (Figure S1B). A significantly greater number of TUNEL(+) cells were observed in bs seminiferous epithelia (Figure 1F), though TUNEL(+) cells did not appear restricted to specific cell types. Two previous studies have reported that bs testes contain elongated spermatids and very small numbers of deformed spermatozoa,18,19 although a third study identified round spermatids as the most advanced bs spermatogenic cells.20 Immunostaining for TRA54 identified large TRA54-positive granules in the second and third layers of WT seminiferous epithelium, consistent with the staining of spermatocytes and round and elongating spermatids.37 In contrast, TRA54 immunostaining in bs showed only discrete punctae consistent with the expected staining of spermatocytes37 (Figures 1G and S1B). RT-PCR and subsequent sequence analysis identified the presence of Tnp1 and Prm2 transcripts in both WT and bs testes (not shown), indicating the presence of post stage 7 spermatids in bs.38 PNA staining (Figure 1G) confirmed the previously reported failure of acrosome formation in bs spermatids18,19
Positional Cloning of the bs Locus
By using recombination analysis of 1,177 F2 (bs × CAST/EiJ) intercross progeny, we narrowed the bs critical region to 416 kb between rs27385663 and rs27343710 (Figures S2A–S2D). Evaluation of the mouse reference genome sequence (GRCm38/mm10) identified 16 RefSeq candidate genes (Table S1). Sequence analysis of the candidate genes identified a c.[691T>A; 692_703del] mutation in Tbc1d20 exon 6 (Figure 2A). No other mutations were identified in bs. The identified Tbc1d20 mutation segregated with all bs F2 intercross progeny and was absent in the mouse dbSNP database. In addition, Tbc1d20 sequences were independently evaluated in the C57BL/6J, C3H/HeJ, CAST/EiJ, RIIIS/J, and DBA/2J strains and were identical to the Tbc1d20 mouse reference sequence (RefSeq NM_024196.3). The bs mutant Tbc1d20 transcript encodes the mutant p.[Phe231Met; p.Arg232 _Val235del] TBC1D20-bs protein with the mutation residing within the evolutionarily highly conserved TBC domain24 (Figure 2B). These findings together suggested that the identified Tbc1d20 mutation in bs is most likely not a rare polymorphism.
Figure 2.
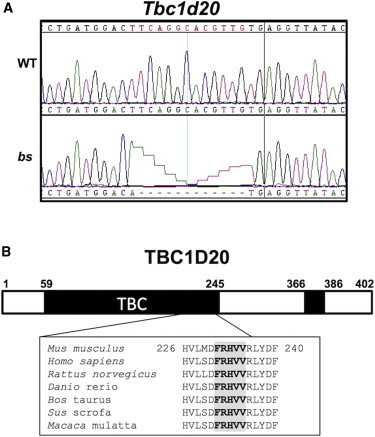
Positional Cloning of the bs Mutation
(A) c.691T>A substitution and subsequent c.692_703del deletion in exon 6 of Tbc1d20 was identified in bs (bottom); WT sequence matched the Tbc1d20 reference sequence (top).
(B) The bs mutation resulting in p.Phe231Met substitution followed by an in-frame p.Arg232 _Val235 deletion affects five evolutionarily highly conserved amino acids within the TBC domain (bold gray shaded). Numbers on top of the figure refer to the amino acids from the mouse TBC1D20 protein (RefSeq NP_077158.1).
Functional Analysis of the bs Mutation
WT mouse TBC1D20 has high GAP activity in the presence of RAB1 and RAB2, consistent with previous reports26,27 (Figure S3). The TBC1D20-bs mutant protein showed strongly reduced activity in the presence of both RAB1 and RAB2, but was more active in comparison to a catalytically inactive TBC1D20 p.Arg105Ala (RA)26 mutant (Figure 3A). To investigate stability of the TBC1D20-bs mutant protein, HEK293 cells were transfected with WT or bs mutant FLAG-tagged mouse Tbc1d20 clones and protein stability was evaluated through a cycloheximide chase study. The stability of TBC1D20-bs mutant protein was significantly lower than that of the WT protein at 6 hr after cycloheximide treatment (Figure S4). Similar to WT human TBC1D20,26 overexpression of the WT mouse TBC1D20 caused the disruption of COPII ER-Golgi transport vesicles; in contrast, the overexpression of TBC1D20-bs mutant protein had little effect on the COPII vesicles resembling the COPII vesicular phenotypes observed after overexpression of catalytically inactive TBC1D20-RA mutant (Figure 3B). Consistent with these findings, overexpression of the WT mouse TBC1D20 also led to a disruption of the Golgi morphology without disruption of endosomes, whereas overexpression of the TBC1D20-bs mutant protein, similar to the overexpression of the catalytically inactive TBC1D20-RA mutant, had little effect on Golgi or endosomal markers (Figure 3C). These data collectively indicate that bs is a Tbc1d20 loss-of-function mutation.
Figure 3.
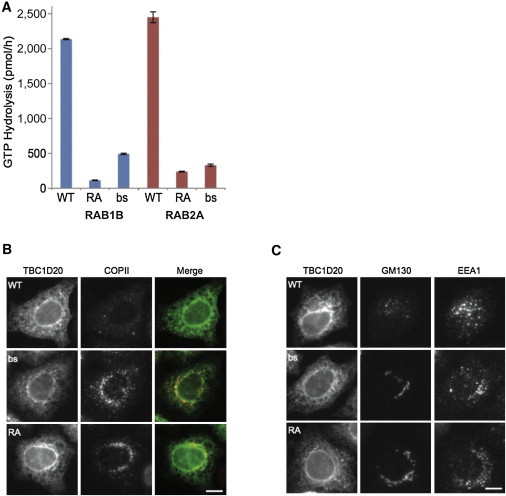
Functional Analysis of the bs Mutation
(A) In the presence of RAB1B or RAB2A, mouse TBC1D20 protein has a high rate of GTP hydrolysis; bs mutant protein results in much lower GTP hydrolysis rate when compared to WT and slightly higher rate than a catalytically inactive RA mutant. Each point on the graph represents the mean values from three independent experiments and error bars indicate SD.
(B and C) Overexpression of mouse WT TBC1D20 caused disruption of COPII ER-Golgi transport vesicles as evident after immunostaining with SEC31 marker (B) and disruption of cis-Golgi as evident after immunostaining with GM130 without disruption of endosomes as evident after immunostaining with EEA1 (C). The TBC1D20-bs mutant protein, like the catalytically inactive RA mutant protein, had little effect on COPII, Golgi, or endosomal markers. Scale bars represent 10 μm.
Cellular Phenotypes of bs Mouse Embryonic Fibroblasts
In HeLa cells, siRNA-mediated depletion of TBC1D20 results in alteration of the Golgi complex;26 thus, we proceeded to evaluate Golgi morphology in the bs mouse mEFs. Immunostaining for the cis-Golgi marker GM130 (Figure 4A) and trans-Golgi marker golgin 97 (Figure S5A) identified expanded Golgi morphology in bs mouse embryonic fibroblasts (mEFs) when compared to WT. However, the Golgi phenotypes observed in bs mEFs differed from those observed in TBC1D20-depleted HeLa cells,26 possibly because of the complete absence of functional TBC1D20 protein in bs mEFs. Immunoblot analysis of WT and bs mEF cell lysates confirmed higher levels of GM130 in bs mEFs lysates (Figure 4B).
Figure 4.
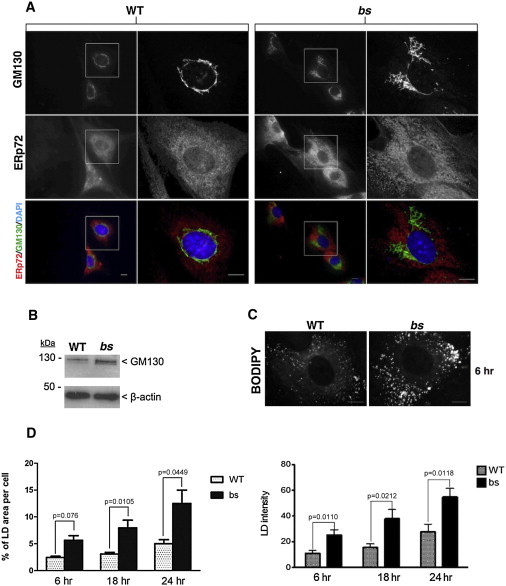
bs mEFs Cellular Phenotypes
(A) GM130 immunostaining (green) revealed enlarged cis-Golgi in bs mEFs, when compared to WT mEFs. ER immunostaining with ERp72 (red) did not identify any differences between WT and bs mEFs. DNA was stained with DAPI (blue).
(B) Immunoblot analysis revealed greater levels of GM130 protein present in bs than in WT mEF cell lysates relative to β-actin.
(C) Oleic acid treatment for 6 hr after staining with the neutral lipid dye BODIPY 493/503 revealed expanded LD structures in bs when compared to WT mEFs.
(D) Quantification analyses after oleic acid treatment for 6, 18, and 24 hr confirmed significantly greater size and fluorescence intensity of LDs in bs mEFs when compared to WT mEFs. Shown in the graphs are mean values per cell (>30 cells). p values shown on top of each graph were determined by Student’s t test and error bars represent SEM.
Scale bars represent 5 μm.
Recently it was shown that after hepatitis C infection, TBC1D20 was recruited to LDs;39 additionally, in Drosophila RAB1 was shown to influence LD size.40 These findings implicated TBC1D20 in LD metabolism, so we proceeded to evaluate LD formation in bs mEFs. Staining of WT and bs mEFs with the neutral lipid dye BODIPY 493/503 did not identify any LDs (Figure S5B). However, after treatment with oleic acid for 6 hr and subsequent staining with BODIPY 493/503, bs mEFs exhibit a significantly (p = 0.0126) greater number of LDs (n = 255 ± 12.68 SEM; >30 cells) when compared to number of LDs in WT mEFs (n = 158 ± 27.68 SEM; > 30 cells). Additionally, 6 hr after oleic acid treatment, LDs in bs mEFs exhibited a significantly greater total area per cells as well as significantly increased LD fluorescence intensity when compared to WT mEFs (Figure 4C). Oleic acid treatment for 18 hr or 24 hr resulted in further expansion and merging of LDs in both WT and bs mEFs (Figure S5B); a significantly greater LD area per cell and significantly increased LD fluorescence was noted in bs when compared to WT mEFs (Figure 4C).
Mutations in TBC1D20 Cause WARBM
Congenital cataracts and testicular abnormalities observed in bs mice resemble the cataracts and testicular abnormalities observed in individuals affected with WARBM. WARBM is considered a monogenic disorder resulting from RAB GTPase aberrations.41 Given that bs mouse lens and testicular phenotypes were due to a mutation in TBC1D20, a protein that is involved in regulation of RAB GTPases, we wanted to explore whether mutations in TBC1D20 (RefSeq NM_144628.2) could cause WARBM. Therefore, we proceeded to screen a cohort of 77 families diagnosed with a spectrum of WARBM disorders. We identified five distinct germline homozygous TBC1D20 loss-of-function mutations (Figure 5A). Two affected siblings from a Polish family (individuals 1.1 and 1.2) had a homozygous c.199C>T mutation in exon 2 leading to a premature stop at amino acid 67 (p.Arg67∗). In an affected girl from the Netherlands (individual 2), a homozygous c.292C>T mutation in exon 3 was identified, resulting in a putative protein with a premature stop at amino acid 98 (p.Gln98∗). Two affected sisters from a Pakistani family (individuals 3.1 and 3.2) had a homozygous CA deletion (c.352_353delCA) in exon 4 resulting in a putative protein with a frame shift at amino acid 118, the addition of nine novel amino acids, and a premature stop (p.Gln118Glufs∗9). In an affected boy from Egypt (individual 4), a homozygous c.672G>A mutation was identified in exon 6 resulting in a putative protein with a premature stop at position 224 (p.Trp224∗). Finally, an affected girl from Pakistan (individual 5) had a homozygous microdeletion, c.?−175_1113+?del, indicating a loss of exons 2–8, which was further verified by quantitative PCR (not shown), most probably leading to a complete protein loss. Parents for all individuals except individual 5 were heterozygous for the respective mutations in keeping with their carrier status; parental DNA from individual 5 was not available for sequence analysis. None of the identified TBC1D20 mutations were present in Exome Variant Server (ESP6500SI-V2) or in our in-house panel of 200 controls. Because both human26 and mouse TBC1D20 (Figure 3A) regulate RAB1 and RAB2, we expanded our screening to include RAB1A (RefSeq NM_004161.4), RAB1B (RefSeq NM_030981.2), and RAB2A (RefSeq NM_002865.2). However, no pathogenic mutations were identified.
Figure 5.
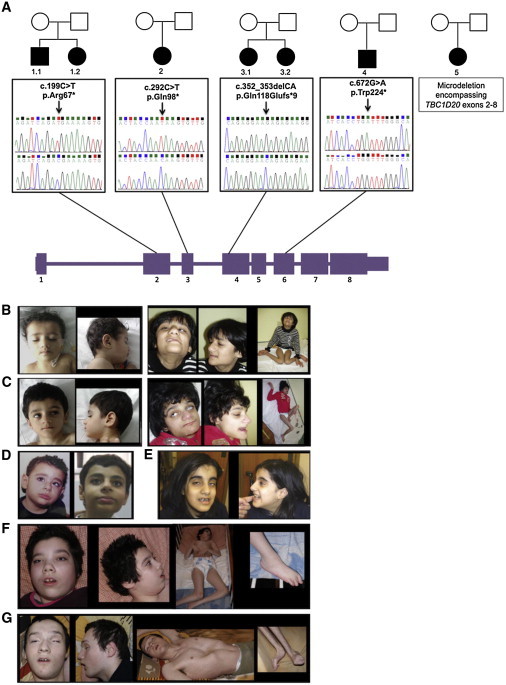
Pedigrees, TBC1D20 Mutations, and Clinical Features of Individuals with WARBM
(A) Filled symbols indicate individuals with WARBM and numbers represent individual identifiers. Chromatograms of germline TBC1D20 mutations in exons 2, 3, 4, and 6 are shown below each pedigree (top) and controls (bottom). The TBC1D20 mutation in individual 5 is a microdeletion encompassing exons 2 thorough 8 confirmed by qPCR.
(B–G) Predominant clinical features of individuals with WARBM with TBC1D20 mutations included microcephaly, low anterior hairline, broad dense laterally descending eyebrows, microphthalmia, low anterior hairline, prominent subnasal region and chin, kyphoscoliosis, severe spastic quadriplegia with contractures, and diminished muscle bulk. Permission was obtained from parents of individuals with WARBM for publication of these images.
(B) Individual 3.1 shown at age 3 (first two panels) and age 13 (last three panels).
(C) Individual 3.2 (sister of individual 3.1 shown in B) at age 1 (first two panels) and at age 11 (last three panels).
(D) Individual 4 is shown at age 3 (first panel) and age 14 (second panel).
(E) Individual 5 is 15 years old.
(F) Individual 1.1 is 21 years old.
(G) Individual 1.2 (brother of individual 1.1 shown in F) is 16 years old.
All individuals with TBC1D20 mutations show the same range of clinical features (Figures 5B–5G and Table S4) as do WARBM-affected individuals with RAB3GAP1, RAB3GAP2, or RAB18 mutations.2–7 Shortly after birth, individuals with TBC1D20 mutations presented with congenital cataracts and microphthalmia. Despite cataract surgery they have poor vision resulting from optic atrophy and several affected children have also developed glaucoma. All individuals with TBC1D20 mutations have severe to profound developmental delay (not having learned to walk and speaking 4–5 words at most) with autistic features. Although their head circumferences were normal at birth, they later developed postnatal microcephaly. Facial features included deep-set eyes, ptosis, wide nasal bridge, relatively narrow mouth, low anterior hairline, and prominent subnasal region. Boys with TBC1D20 mutations had both micropenis and cryptorchidism. In the first year of life, all individuals with TBC1D20 mutations had severe axial hypotonia then gradually developed lower and upper limb spasticity leading to spastic quadriplegia.
Brain Phenotypes in Individuals with WARBM and in bs Mice with TBC1D20 Mutations
Cranial MRIs were available from four individuals with TBC1D20 mutations (Figure 6). The MRI analysis revealed predominantly frontal polymicrogyria (Figure 6A), corpus callosum hypogenesis particularly affecting the splenium (Figures 6B and 6M), widened lateral ventricles, and megacisterna magna as a consequence of cerebellar vermis hypoplasia (Figures 6B, 6G, and 6M). Follow-up MRIs on individuals 3.1 and 3.2 revealed atrophy of the cerebellar vermis and hemispheres (Figures 6C and 6E). Given the severe brain abnormalities observed in individuals with TBC1D20 mutations, we also evaluated brains from bs mice. However, no obvious morphological abnormalities were identified (Figure S6).
Figure 6.
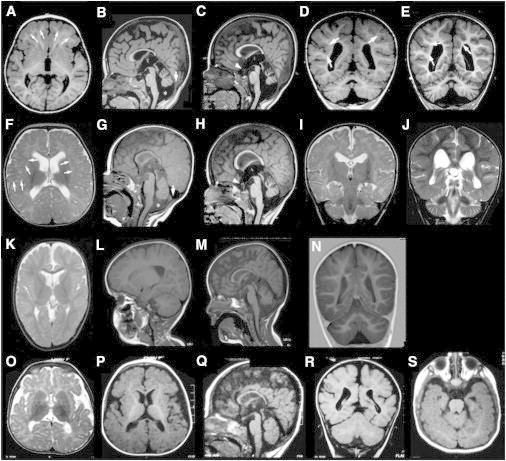
Brain MRIs from Individuals with TBC1D20 Mutations
(A–E) Individual 3.1 at age 2 years (A–C) and at age 4 years and 11 months (D, E).
(F–J) Individual 3.2 at age 6 months (F–I) and at 2 years and 8 months (H, J).
(K–N) Individual 2 at age 2 years and 6 months.
(O–S) Individual 4 at age 7 months.
The general pattern is similar in all individuals: predominantly frontal polymicrogyria (arrows in A), which sometimes extends to the Sylvian fissures and temporal and occipital lobes (E–G), corpus callosum hypogenesis, particularly of the splenium (B, G, M, Q), and enlarged cisterna magna (asterisks in B and G and also shown in C, H, M, and Q) due to cerebellar vermis hypoplasia. Follow-up of the two sisters 3.1 (C and E) and 3.2 (H and J) showed clear atrophy of the cerebellar vermis and hemispheres in both individuals. The optic chiasm was also hypoplastic in both these individuals (arrowheads in C and H).
Cellular Phenotypes of Fibroblasts from Individuals with WARBM
We proceeded to evaluate fibroblasts from an individual with a TBC1D20 mutation (p.Gln98∗) to determine whether these cells exhibited Golgi and LD abnormalities like those observed in bs mEFs (Figure 4C). Without a treatment with oleic acid, we didn’t observe any LDs in control or TBC1D20 (p.Gln98∗) fibroblasts after staining with BODIPY 493/503 (Figure S7). However, a significant increase in the size of LDs was observed in TBC1D20 (p.Gln98∗) cells compared to control fibroblasts after treatment for 18 or 24 hr with oleic acid (Figures 7A and 7B). To determine whether altered LDs are a common WARBM feature, we additionally evaluated LD formation in fibroblasts from individuals with RAB18 (p.Leu24Gln) or RAB3GAP1 (c.649−2A>G) mutations.2,7 After oleic acid treatment for 18 and 24 hr, these cell lines also exhibited greater size of LDs when compared to controls (Figure 7B). Of note, TBC1D20 (p.Gln98∗), RAB18 (p.Leu24Gln), and RAB3GAP1 (c.649−2A>G) fibroblasts did not exhibit any differences in Golgi, ER, or endosomal structures from control cell lines after immunocytochemical and immunoblot evaluation (Figures 7C and 7D). It is particularly interesting that the altered Golgi phenotypes observed in HeLa cells after siRNA-mediated depletion of TBC1D2026 and in bs mEFs (Figure 4A) were not observed in the human TBC1D20 (p.Gln98∗) cells (Figure 7). Because the phenotype is shared by human- and mouse-derived cells, it is possible that its absence in the TBC1D20-deficient fibroblasts reflects compensatory mechanism acting in these cells rather than species-specific differences. Notwithstanding this, we identify aberrant LD formation as a cellular phenotype common to WARBM fibroblasts with mutations in TBC1D20, RAB18, and RAB3GAP1 and in bs mEFs. Thus, the aberrant LD phenotype is seen in multiple genotypes in two species and is unaffected by any compensatory mechanism.
Figure 7.
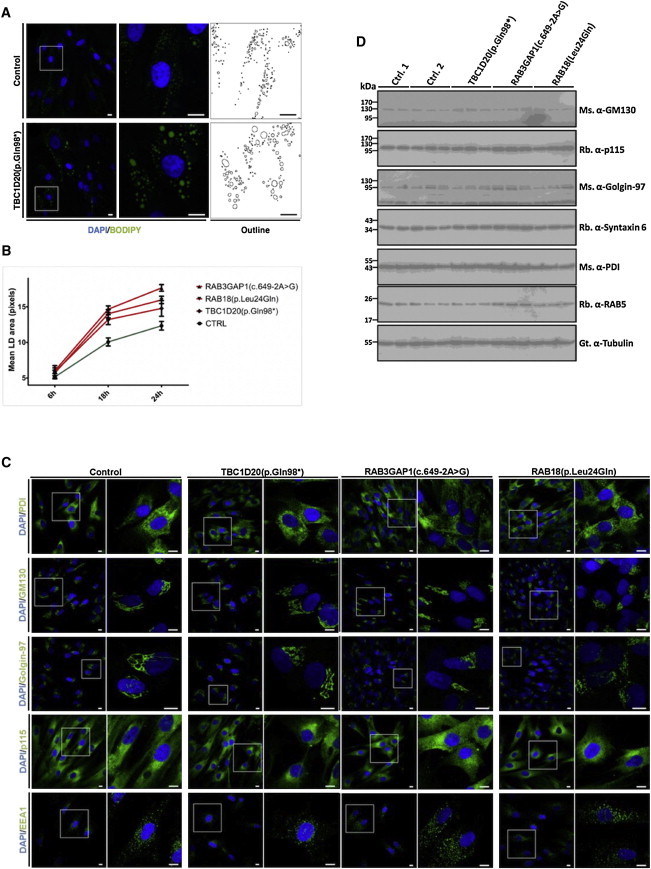
WARBM Fibroblasts Cellular Phenotypes
(A) Treatment of TBC1D20 (p.Gln98∗) fibroblasts with 400 μM oleic acid for 18 hr resulted in a significantly greater size of LDs when compared to identically treated controls. LDs were stained with the neutral lipid dye BODIPY 493/503 (green) and DNA was stained with DAPI (blue).
(B) Oleic acid treatment of RAB18 (p.Leu24Gln) and RAB3GAP1 (c.649−2A>G) fibroblasts for 18 or 24 hr also resulted in significantly larger LDs when compared to controls (>30 cells); the error bars indicate SEM.
(C) Immunofluorescence analysis with PDI as an ER marker; GM130, Golgin-97, and p115 as Golgi markers; and EEA1 as an endosomal marker did not identify any difference between TBC1D20 (p.Gln98∗), RAB18 (p.Leu24Gln), RAB3GAP1 (c.649−2A>G), and control fibroblasts. Scale bars represent 10 μm.
(D) Immunoblot analysis of cell lysates from TBC1D20 (p.Gln98∗), RAB18 (p.Leu24Gln), RAB3GAP1 (c.649−2A>G), and control fibroblasts did not identify any differences in expression of GM130, Golgin-97, p115, Syntaxin 6, PDI, and RAB5 proteins.
Discussion
In this study, we report that loss-of-function mutations in TBC1D20 are responsible for lens and testes phenotypes in bs mice and cause WARBM in humans. Clinical evaluation of individuals carrying pathogenic TBC1D20 mutations is sufficient to conclude that they cause a disease phenotype indistinguishable to that caused by mutations in RAB3GAP1, RAB3GAP2, or RAB18.4–6 As in these cases, congenital cataracts were apparent shortly after birth and was accompanied by microphthalmia, microcornea, and small atonic pupils. Despite early cataract surgery, vision in these individuals remains poor. They also exhibit a general pattern of brain abnormalities including postnatal microcephaly, bilateral predominantly frontal polymicrogyria with hypogenesis of the corpus callosum, and cerebellar vermis hypoplasia. Additionally, they suffer from profound developmental delay and have not learned to crawl, pull up to a standing position, walk, or talk. Characteristically, these individuals showed congenital hypotonia from about 8–12 months and then progressive spasticity that leads to contractures and spastic quadriplegia later in life. Furthermore, WARBM boys with mutations in TBC1D20 exhibited micropenis and cryptorchidism. It was reported recently that mutations in RAB3GAP1, RAB3GAP2, and RAB18 collectively contribute to more than 50% of all WARBM cases.5 The identified human TBC1D20 mutations account for approximately an additional 5%, but a sizable proportion of WARBM cases still remain unexplained at the genetic level. This suggests that WARBM may be associated with yet further genetic heterogeneity.
This study has also established bs as a WARBM mouse model. The bs mice recapitulate the lens and testes phenotypes observed in individuals with WARBM although cryptorchidism was not observed in bs mice. Additionally, no obvious bs brain or neuromuscular abnormalities were identified in these animals. The embryonic bs lenses undergo normal development and differentiation, suggesting that maturation of fiber cells is the underlying defect associated with bs cataracts. Similarly, the bs testes undergo normal development and differentiation, with the failure of acrosome formation in bs spermatids ultimately resulting in bs male infertility. The role of TBC1D20 in the lens or testis has never been investigated. In general, the role of RAB GTPases is to regulate vesicular trafficking and organelle remodeling.22 As a part of their maturation processes, both lens and testis cells undergo a unique organelle-remodeling process that is mechanistically poorly understood. In lens fiber cells, organelle degradation is essential for the development of the organelle-free zone and lens transparency.42,43 In spermatogenesis, the acrosome is a Golgi-derived organelle with an important role at fertilization.44,45 Therefore, it is possible that TBC1D20 has a critical role in organelle remodeling in maturing lens and testis cells in both humans and mice as evident by the lens and testicular phenotypes observed in bs mice and individuals with WARBM resulting from a loss of TBC1D20 function. However, Rab3gap1-deficient mice do not exhibit any gross eye, brain, or genital abnormalities, but exhibit abnormal release of synaptic vesicles and altered hippocampal synaptic plasticity.46 This phenotypic difference between mice and individuals with WARBM resulting from RAB3GAP1 mutations was previously attributed to the differential species-specific pathway redundancy.5 Collectively these findings suggest that additional factors such as genetic modifiers may have a role in regulating the onset and severity of the disease phenotype.
An association between the molecular function of TBC1D20 and that of RAB3GAP1, RAB3GAP2, and RAB18 is implied by the indistinguishable effects of their functional loss in humans. However, functional studies have not linked these genes to a common molecular pathway. RAB3GAP1 and RAB3GAP2 form a complex known to regulate RAB3 function in neurotransmitter release.46,47 Indeed, RAB3GAP1 is thought to be essential for modulation of neurotransmitter release via a process known as synaptic homeostasis.48 RAB18 has been linked to regulation of diverse processes including LD metabolism,49–54 endocytosis,55 exocytosis of secretory granules,56,57 and ER-to-Golgi trafficking.58 TBC1D20 is an ER protein that, via its GAP regulation of RAB1, facilitates ER-to-Golgi trafficking and organization of the Golgi complex in both yeast and HeLa cells.26–28 Altered Golgi morphology observed in bs mEFs is consistent with these findings, although the aberrant Golgi morphology is more severe in bs mEFs when compared to the aberrant Golgi morphology observed in HeLa cells after siRNA-mediated TBC1D20 depletion.26 In contrast, we observed no Golgi abnormalities in the human fibroblasts deficient for the TBC1D20 function. In yeast, both Gyp5p and Gyp8p RABGAPs can act on the RAB1 ortholog Ypt1.28 Therefore, it is possible that differentially expressed additional RAB1GAPs may be responsible for full or partial compensation of the TBC1D20 function, facilitating Golgi organization in some TBC1D20-deficient cell lines.
Although differentially expressed RAB1GAPs may be compensating for the role of TBC1D20 in Golgi organization, our results show that TBC1D20 is indispensable for proper LD formation. Here, we show that a defect in LD formation is a common cellular feature not only in bs mEFs and human fibroblasts deficient in TBC1D20 but also in fibroblasts deficient in RAB3GAP1 and RAB18. Thus, we provide evidence for a common WARBM cellular pathway. The role of RAB18 as an LD protein involved in lipid metabolism has already been established.49–54 In a study in Drosophila, overexpression of “dominant-negative” and “constitutively active” RAB1 mutants resulted in increased LD size,40 additionally supporting a role of TBC1D20 in LD formation and/or metabolism. However, in the same study overexpression of dominant-negative and constitutively active RAB18 mutants did not influence the LD size.40 The discrepancy between these findings and those of this study where LD size was increased in RAB18 (p.Leu24Gln) cells may be explained by the fact that WT RAB18 protein is completely absent in these cells. Further studies will be required to determine whether the role of TBC1D20 as a regulator of RAB1 can explain its involvement in LD formation or whether in vivo regulatory activity of TBC1D20 toward RAB18 is the underlying mechanism. Unlike TBC1D20 and RAB18, the RAB3GAP complex has never been directly associated with a role in LD formation and/or metabolism. Interestingly though, SNPs in linkage disequilibrium with RAB3GAP1 have been associated with total cholesterol and high-density lipoprotein cholesterol levels in a genome-wide association study.59 Therefore, additional studies are needed to determine the molecular relationships between TBC1D20, RAB18, and the RAB3GAP complex during LD formation and/or metabolism.
LDs are ER-associated organelles and store triacylglycerols and cholesteryl esters used in metabolism and membrane and steroid synthesis.60–63 Though the roles of LDs are incompletely understood, a number of findings are suggestive of their involvement in disease. The accumulation of LDs has been associated with two different forms of complicated hereditary spastic paraplegias (HSPs). Mutations in BSCL2 (MIM 606158) cause congenital generalized lipodystrophy type 2 (CGL2 [MIM 269700]) and spastic paraplegia 17 (SPG17 [MIM 270685]) whereas mutations in SPG20 (MIM 607111) cause spastic paraplegia 20 (SPG20 [MIM 275900]).64 Hereditary spastic paraplegias share some resemblance to WARBM in that they are characterized by lower limb spasticity and progress to affect the upper limbs.64 At this point it is not certain whether the LD phenotype in WARBM cell lines underlies the associated pathology or whether it is in turn the consequence of another underlying yet unidentified cellular deficit. Nevertheless, it may provide a context for future investigations at the molecular level. Further, the potential identification of additional unknown genes and mutations resulting in WARBM could inform their characterization. In combination, these approaches could provide additional valuable information about the molecular mechanisms underlying WARBM disease pathology.
Acknowledgments
We would like to thank the WARBM subjects, their families, and many health care professionals for referring affected families and the West Midlands Regional Genetics laboratory. We are grateful to A. Gallacher, S. McKay, and M.A. Maier for technical assistance. The study was supported by research grants from the UK Newlife Charity (I.A.A.), Program Leader Development Track Fellowship from the Medical Research Council (I.A.A.), Healthier Wisconsin Grant from the Medical College of Wisconsin (A.D.E.), and the National Eye Institute at the National Institutes of Health (EY019943 to B.C. and EY018872 to D.J.S.).
Contributor Information
Irene A. Aligianis, Email: irene.aligianis@igmm.ed.ac.uk.
Duska J. Sidjanin, Email: dsidjani@mcw.edu.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
DNA star, http://www.dnastar.com/
ExonPrimer, http://ihg.gsf.de/ihg/ExonPrimer.html
GraphPad, http://graphpad.com/
Human Genome Variation Society, http://www.hgvs.org/mutnomen/
ImageJ, http://rsbweb.nih.gov/ij/
ImageJ Fiji, http://fiji.sc/Fiji
LightCycler 480, http://www.roche-applied-science.com/shop/products/lightcycler14301-480-gene-scanning-software
MapManager QTX, http://www.mapmanager.org
Metamoroph, http://www.moleculardevices.com/products/software/meta-imaging-series/metamorph.html
Mouse Genome Browser, http://genome.ucsc.edu/cgi-bin/hgGateway?hgsid=336780771&clade=mammal&org=Mouse&db=0
Mutation Surveyor, http://www.softgenetics.com/mutationSurveyor.html
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Universal ProbeLibrary Assay Design Center, http://www.roche-applied-science.com/webapp/wcs/stores/servlet/CategoryDisplay?catalogId=10001&tab=Assay+Design+Center&identifier=Universal+Probe+Library&langId=-1
References
- 1.Warburg M., Sjö O., Fledelius H.C., Pedersen S.A. Autosomal recessive microcephaly, microcornea, congenital cataract, mental retardation, optic atrophy, and hypogenitalism. Micro syndrome. Am. J. Dis. Child. 1993;147:1309–1312. doi: 10.1001/archpedi.1993.02160360051017. [DOI] [PubMed] [Google Scholar]
- 2.Aligianis I.A., Johnson C.A., Gissen P., Chen D., Hampshire D., Hoffmann K., Maina E.N., Morgan N.V., Tee L., Morton J. Mutations of the catalytic subunit of RAB3GAP cause Warburg Micro syndrome. Nat. Genet. 2005;37:221–223. doi: 10.1038/ng1517. [DOI] [PubMed] [Google Scholar]
- 3.Abdel-Salam G.M., Hassan N.A., Kayed H.F., Aligianis I.A. Phenotypic variability in Micro syndrome: report of new cases. Genet. Couns. 2007;18:423–435. [PubMed] [Google Scholar]
- 4.Morris-Rosendahl D.J., Segel R., Born A.P., Conrad C., Loeys B., Brooks S.S., Müller L., Zeschnigk C., Botti C., Rabinowitz R. New RAB3GAP1 mutations in patients with Warburg Micro Syndrome from different ethnic backgrounds and a possible founder effect in the Danish. Eur. J. Hum. Genet. 2010;18:1100–1106. doi: 10.1038/ejhg.2010.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Handley M.T., Morris-Rosendahl D.J., Brown S., Macdonald F., Hardy C., Bem D., Carpanini S.M., Borck G., Martorell L., Izzi C. Mutation spectrum in RAB3GAP1, RAB3GAP2, and RAB18 and genotype-phenotype correlations in warburg micro syndrome and Martsolf syndrome. Hum. Mutat. 2013;34:686–696. doi: 10.1002/humu.22296. [DOI] [PubMed] [Google Scholar]
- 6.Borck G., Wunram H., Steiert A., Volk A.E., Körber F., Roters S., Herkenrath P., Wollnik B., Morris-Rosendahl D.J., Kubisch C. A homozygous RAB3GAP2 mutation causes Warburg Micro syndrome. Hum. Genet. 2011;129:45–50. doi: 10.1007/s00439-010-0896-2. [DOI] [PubMed] [Google Scholar]
- 7.Bem D., Yoshimura S., Nunes-Bastos R., Bond F.C., Kurian M.A., Rahman F., Handley M.T., Hadzhiev Y., Masood I., Straatman-Iwanowska A.A. Loss-of-function mutations in RAB18 cause Warburg micro syndrome. Am. J. Hum. Genet. 2011;88:499–507. doi: 10.1016/j.ajhg.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Handley M.T., Aligianis I.A. RAB3GAP1, RAB3GAP2 and RAB18: disease genes in Micro and Martsolf syndromes. Biochem. Soc. Trans. 2012;40:1394–1397. doi: 10.1042/BST20120169. [DOI] [PubMed] [Google Scholar]
- 9.Ainsworth J.R., Morton J.E., Good P., Woods C.G., George N.D., Shield J.P., Bradbury J., Henderson M.J., Chhina J. Micro syndrome in Muslim Pakistan children. Ophthalmology. 2001;108:491–497. doi: 10.1016/s0161-6420(00)00540-6. [DOI] [PubMed] [Google Scholar]
- 10.Derbent M., Agras P.I., Gedik S., Oto S., Alehan F., Saatçi U. Congenital cataract, microphthalmia, hypoplasia of corpus callosum and hypogenitalism: report and review of Micro syndrome. Am. J. Med. Genet. A. 2004;128A:232–234. doi: 10.1002/ajmg.a.30109. [DOI] [PubMed] [Google Scholar]
- 11.Dursun F., Güven A., Morris-Rosendahl D. Warburg Micro syndrome. J. Pediatr. Endocrinol. Metab. 2012;25:379–382. doi: 10.1515/jpem-2011-0459. [DOI] [PubMed] [Google Scholar]
- 12.Graham J.M., Jr., Hennekam R., Dobyns W.B., Roeder E., Busch D. MICRO syndrome: an entity distinct from COFS syndrome. Am. J. Med. Genet. A. 2004;128A:235–245. doi: 10.1002/ajmg.a.30060. [DOI] [PubMed] [Google Scholar]
- 13.Mégarbané A., Choueiri R., Bleik J., Mezzina M., Caillaud C. Microcephaly, microphthalmia, congenital cataract, optic atrophy, short stature, hypotonia, severe psychomotor retardation, and cerebral malformations: a second family with micro syndrome or a new syndrome? J. Med. Genet. 1999;36:637–640. [PMC free article] [PubMed] [Google Scholar]
- 14.Rodríguez Criado G., Rufo M., Gómez de Terreros I. A second family with Micro syndrome. Clin. Dysmorphol. 1999;8:241–245. [PubMed] [Google Scholar]
- 15.Yildirim M.S., Zamani A.G., Bozkurt B. Warburg micro syndrome in two children from a highly inbred Turkish family. Genet. Couns. 2012;23:169–174. [PubMed] [Google Scholar]
- 16.Yüksel A., Yesil G., Aras C., Seven M. Warburg Micro syndrome in a Turkish boy. Clin. Dysmorphol. 2007;16:89–93. doi: 10.1097/MCD.0b013e328054c404. [DOI] [PubMed] [Google Scholar]
- 17.Varnum D.S. Blind-sterile: a new mutation on chromosome 2 of the house mouse. J. Hered. 1983;74:206–207. doi: 10.1093/oxfordjournals.jhered.a109768. [DOI] [PubMed] [Google Scholar]
- 18.Sotomayor R.E., Handel M.A. Failure of acrosome assembly in a male sterile mouse mutant. Biol. Reprod. 1986;34:171–182. doi: 10.1095/biolreprod34.1.171. [DOI] [PubMed] [Google Scholar]
- 19.Fouquet J.P., Valentin A., Kann M.L. Perinuclear cytoskeleton of acrosome-less spermatids in the blind sterile mutant mouse. Tissue Cell. 1992;24:655–665. doi: 10.1016/0040-8166(92)90036-7. [DOI] [PubMed] [Google Scholar]
- 20.Yanagimachi R., Wakayama T., Kishikawa H., Fimia G.M., Monaco L., Sassone-Corsi P. Production of fertile offspring from genetically infertile male mice. Proc. Natl. Acad. Sci. USA. 2004;101:1691–1695. doi: 10.1073/pnas.0307832100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spence S.E., Gilbert D.J., Harris B.S., Davisson M.T., Copeland N.G., Jenkins N.A. Genetic localization of Hao-1, blind-sterile (bs), and Emv-13 on mouse chromosome 2. Genomics. 1992;12:403–404. doi: 10.1016/0888-7543(92)90392-6. [DOI] [PubMed] [Google Scholar]
- 22.Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 2009;10:513–525. doi: 10.1038/nrm2728. [DOI] [PubMed] [Google Scholar]
- 23.Barr F., Lambright D.G. Rab GEFs and GAPs. Curr. Opin. Cell Biol. 2010;22:461–470. doi: 10.1016/j.ceb.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frasa M.A., Koessmeier K.T., Ahmadian M.R., Braga V.M. Illuminating the functional and structural repertoire of human TBC/RABGAPs. Nat. Rev. Mol. Cell Biol. 2012;13:67–73. doi: 10.1038/nrm3267. [DOI] [PubMed] [Google Scholar]
- 25.Cherfils J., Zeghouf M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol. Rev. 2013;93:269–309. doi: 10.1152/physrev.00003.2012. [DOI] [PubMed] [Google Scholar]
- 26.Haas A.K., Yoshimura S., Stephens D.J., Preisinger C., Fuchs E., Barr F.A. Analysis of GTPase-activating proteins: Rab1 and Rab43 are key Rabs required to maintain a functional Golgi complex in human cells. J. Cell Sci. 2007;120:2997–3010. doi: 10.1242/jcs.014225. [DOI] [PubMed] [Google Scholar]
- 27.Sklan E.H., Serrano R.L., Einav S., Pfeffer S.R., Lambright D.G., Glenn J.S. TBC1D20 is a Rab1 GTPase-activating protein that mediates hepatitis C virus replication. J. Biol. Chem. 2007;282:36354–36361. doi: 10.1074/jbc.M705221200. [DOI] [PubMed] [Google Scholar]
- 28.De Antoni A., Schmitzová J., Trepte H.H., Gallwitz D., Albert S. Significance of GTP hydrolysis in Ypt1p-regulated endoplasmic reticulum to Golgi transport revealed by the analysis of two novel Ypt1-GAPs. J. Biol. Chem. 2002;277:41023–41031. doi: 10.1074/jbc.M205783200. [DOI] [PubMed] [Google Scholar]
- 29.Talamas E., Jackson L., Koeberl M., Jackson T., McElwee J.L., Hawes N.L., Chang B., Jablonski M.M., Sidjanin D.J. Early transposable element insertion in intron 9 of the Hsf4 gene results in autosomal recessive cataracts in lop11 and ldis1 mice. Genomics. 2006;88:44–51. doi: 10.1016/j.ygeno.2006.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hassemer E.L., Le Gall S.M., Liegel R., McNally M., Chang B., Zeiss C.J., Dubielzig R.D., Horiuchi K., Kimura T., Okada Y. The waved with open eyelids (woe) locus is a hypomorphic mouse mutation in Adam17. Genetics. 2010;185:245–255. doi: 10.1534/genetics.109.113167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liegel R., Chang B., Dubielzig R., Sidjanin D.J. Blind sterile 2 (bs2), a hypomorphic mutation in Agps, results in cataracts and male sterility in mice. Mol. Genet. Metab. 2011;103:51–59. doi: 10.1016/j.ymgme.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fuchs E., Haas A.K., Spooner R.A., Yoshimura S., Lord J.M., Barr F.A. Specific Rab GTPase-activating proteins define the Shiga toxin and epidermal growth factor uptake pathways. J. Cell Biol. 2007;177:1133–1143. doi: 10.1083/jcb.200612068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Listenberger L.L., Brown D.A. Fluorescent detection of lipid droplets and associated proteins. Curr. Protoc. Cell Biol. 2007;Chapter 24:2. doi: 10.1002/0471143030.cb2402s35. [DOI] [PubMed] [Google Scholar]
- 34.Li Q., Pène V., Krishnamurthy S., Cha H., Liang T.J. Hepatitis C virus infection activates an innate pathway involving IKK-α in lipogenesis and viral assembly. Nat. Med. 2013;19:722–729. doi: 10.1038/nm.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jadeja S., Mort R.L., Keighren M., Hart A.W., Joynson R., Wells S., Potter P.K., Jackson I.J. A CNS-specific hypomorphic Pdgfr-beta mutant model of diabetic retinopathy. Invest. Ophthalmol. Vis. Sci. 2013;54:3569–3578. doi: 10.1167/iovs.12-11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pereira L.A., Tanaka H., Nagata Y., Sawada K., Mori H., Chimelli L.M., Nishimune Y. Characterization and expression of a stage specific antigen by monoclonal antibody TRA 54 in testicular germ cells. Int. J. Androl. 1998;21:34–40. doi: 10.1046/j.1365-2605.1998.00086.x. [DOI] [PubMed] [Google Scholar]
- 38.Mali P., Kaipia A., Kangasniemi M., Toppari J., Sandberg M., Hecht N.B., Parvinen M. Stage-specific expression of nucleoprotein mRNAs during rat and mouse spermiogenesis. Reprod. Fertil. Dev. 1989;1:369–382. doi: 10.1071/rd9890369. [DOI] [PubMed] [Google Scholar]
- 39.Nevo-Yassaf I., Yaffe Y., Asher M., Ravid O., Eizenberg S., Henis Y.I., Nahmias Y., Hirschberg K., Sklan E.H. Role for TBC1D20 and Rab1 in hepatitis C virus replication via interaction with lipid droplet-bound nonstructural protein 5A. J. Virol. 2012;86:6491–6502. doi: 10.1128/JVI.00496-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang C., Liu Z., Huang X. Rab32 is important for autophagy and lipid storage in Drosophila. PLoS ONE. 2012;7:e32086. doi: 10.1371/journal.pone.0032086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mitra S., Cheng K.W., Mills G.B. Rab GTPases implicated in inherited and acquired disorders. Semin. Cell Dev. Biol. 2011;22:57–68. doi: 10.1016/j.semcdb.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bassnett S., Shi Y., Vrensen G.F. Biological glass: structural determinants of eye lens transparency. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011;366:1250–1264. doi: 10.1098/rstb.2010.0302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vrensen G.F., Graw J., De Wolf A. Nuclear breakdown during terminal differentiation of primary lens fibres in mice: a transmission electron microscopic study. Exp. Eye Res. 1991;52:647–659. doi: 10.1016/0014-4835(91)90017-9. [DOI] [PubMed] [Google Scholar]
- 44.Moreno R.D., Ramalho-Santos J., Sutovsky P., Chan E.K., Schatten G. Vesicular traffic and golgi apparatus dynamics during mammalian spermatogenesis: implications for acrosome architecture. Biol. Reprod. 2000;63:89–98. doi: 10.1095/biolreprod63.1.89. [DOI] [PubMed] [Google Scholar]
- 45.Berruti G., Paiardi C. Acrosome biogenesis: Revisiting old questions to yield new insights. Spermatogenesis. 2011;1:95–98. doi: 10.4161/spmg.1.2.16820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sakane A., Manabe S., Ishizaki H., Tanaka-Okamoto M., Kiyokage E., Toida K., Yoshida T., Miyoshi J., Kamiya H., Takai Y., Sasaki T. Rab3 GTPase-activating protein regulates synaptic transmission and plasticity through the inactivation of Rab3. Proc. Natl. Acad. Sci. USA. 2006;103:10029–10034. doi: 10.1073/pnas.0600304103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sudhof T.C. The synaptic vesicle cycle. Annu. Rev. Neurosci. 2004;27:509–547. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- 48.Müller M., Pym E.C., Tong A., Davis G.W. Rab3-GAP controls the progression of synaptic homeostasis at a late stage of vesicle release. Neuron. 2011;69:749–762. doi: 10.1016/j.neuron.2011.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pulido M.R., Diaz-Ruiz A., Jiménez-Gómez Y., Garcia-Navarro S., Gracia-Navarro F., Tinahones F., López-Miranda J., Frühbeck G., Vázquez-Martínez R., Malagón M.M. Rab18 dynamics in adipocytes in relation to lipogenesis, lipolysis and obesity. PLoS ONE. 2011;6:e22931. doi: 10.1371/journal.pone.0022931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martin S., Driessen K., Nixon S.J., Zerial M., Parton R.G. Regulated localization of Rab18 to lipid droplets: effects of lipolytic stimulation and inhibition of lipid droplet catabolism. J. Biol. Chem. 2005;280:42325–42335. doi: 10.1074/jbc.M506651200. [DOI] [PubMed] [Google Scholar]
- 51.Martin S., Parton R.G. Characterization of Rab18, a lipid droplet-associated small GTPase. Methods Enzymol. 2008;438:109–129. doi: 10.1016/S0076-6879(07)38008-7. [DOI] [PubMed] [Google Scholar]
- 52.Ozeki S., Cheng J., Tauchi-Sato K., Hatano N., Taniguchi H., Fujimoto T. Rab18 localizes to lipid droplets and induces their close apposition to the endoplasmic reticulum-derived membrane. J. Cell Sci. 2005;118:2601–2611. doi: 10.1242/jcs.02401. [DOI] [PubMed] [Google Scholar]
- 53.Zehmer J.K., Huang Y., Peng G., Pu J., Anderson R.G., Liu P. A role for lipid droplets in inter-membrane lipid traffic. Proteomics. 2009;9:914–921. doi: 10.1002/pmic.200800584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goodman J.M. The gregarious lipid droplet. J. Biol. Chem. 2008;283:28005–28009. doi: 10.1074/jbc.R800042200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lütcke A., Parton R.G., Murphy C., Olkkonen V.M., Dupree P., Valencia A., Simons K., Zerial M. Cloning and subcellular localization of novel rab proteins reveals polarized and cell type-specific expression. J. Cell Sci. 1994;107:3437–3448. doi: 10.1242/jcs.107.12.3437. [DOI] [PubMed] [Google Scholar]
- 56.Vazquez-Martinez R., Cruz-Garcia D., Duran-Prado M., Peinado J.R., Castaño J.P., Malagon M.M. Rab18 inhibits secretory activity in neuroendocrine cells by interacting with secretory granules. Traffic. 2007;8:867–882. doi: 10.1111/j.1600-0854.2007.00570.x. [DOI] [PubMed] [Google Scholar]
- 57.Vazquez-Martinez R., Martinez-Fuentes A.J., Pulido M.R., Jimenez-Reina L., Quintero A., Leal-Cerro A., Soto A., Webb S.M., Sucunza N., Bartumeus F. Rab18 is reduced in pituitary tumors causing acromegaly and its overexpression reverts growth hormone hypersecretion. J. Clin. Endocrinol. Metab. 2008;93:2269–2276. doi: 10.1210/jc.2007-1893. [DOI] [PubMed] [Google Scholar]
- 58.Dejgaard S.Y., Murshid A., Erman A., Kizilay O., Verbich D., Lodge R., Dejgaard K., Ly-Hartig T.B., Pepperkok R., Simpson J.C., Presley J.F. Rab18 and Rab43 have key roles in ER-Golgi trafficking. J. Cell Sci. 2008;121:2768–2781. doi: 10.1242/jcs.021808. [DOI] [PubMed] [Google Scholar]
- 59.Ma L., Yang J., Runesha H.B., Tanaka T., Ferrucci L., Bandinelli S., Da Y. Genome-wide association analysis of total cholesterol and high-density lipoprotein cholesterol levels using the Framingham heart study data. BMC Med. Genet. 2010;11:55. doi: 10.1186/1471-2350-11-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Martin S., Parton R.G. Lipid droplets: a unified view of a dynamic organelle. Nat. Rev. Mol. Cell Biol. 2006;7:373–378. doi: 10.1038/nrm1912. [DOI] [PubMed] [Google Scholar]
- 61.Greenberg A.S., Coleman R.A., Kraemer F.B., McManaman J.L., Obin M.S., Puri V., Yan Q.W., Miyoshi H., Mashek D.G. The role of lipid droplets in metabolic disease in rodents and humans. J. Clin. Invest. 2011;121:2102–2110. doi: 10.1172/JCI46069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Reue K. A thematic review series: lipid droplet storage and metabolism: from yeast to man. J. Lipid Res. 2011;52:1865–1868. doi: 10.1194/jlr.E020602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Walther T.C., Farese R.V., Jr. Lipid droplets and cellular lipid metabolism. Annu. Rev. Biochem. 2012;81:687–714. doi: 10.1146/annurev-biochem-061009-102430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Blackstone C. Cellular pathways of hereditary spastic paraplegia. Annu. Rev. Neurosci. 2012;35:25–47. doi: 10.1146/annurev-neuro-062111-150400. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.