

Abstract
Hypertriglyceridemia (HTG) is a heritable risk factor for cardiovascular disease. Investigating the genetics of HTG may identify new drug targets. There are ∼35 known single-nucleotide variants (SNVs) that explain only ∼10% of variation in triglyceride (TG) level. Because of the genetic heterogeneity of HTG, a family study design is optimal for identification of rare genetic variants with large effect size because the same mutation can be observed in many relatives and cosegregation with TG can be tested. We considered HTG in a five-generation family of European American descent (n = 121), ascertained for familial combined hyperlipidemia. By using Bayesian Markov chain Monte Carlo joint oligogenic linkage and association analysis, we detected linkage to chromosomes 7 and 17. Whole-exome sequence data revealed shared, highly conserved, private missense SNVs in both SLC25A40 on chr7 and PLD2 on chr17. Jointly, these SNVs explained 49% of the genetic variance in TG; however, only the SLC25A40 SNV was significantly associated with TG (p = 0.0001). This SNV, c.374A>G, causes a highly disruptive p.Tyr125Cys substitution just outside the second helical transmembrane region of the SLC25A40 inner mitochondrial membrane transport protein. Whole-gene testing in subjects from the Exome Sequencing Project confirmed the association between TG and SLC25A40 rare, highly conserved, coding variants (p = 0.03). These results suggest a previously undescribed pathway for HTG and illustrate the power of large pedigrees in the search for rare, causal variants.
Introduction
Cardiovascular disease (CVD) is the leading cause of death in the United States and poses a significant morbidity and cost for treatment after cardiac events. CVD is associated with the correlated traits of high LDL, low HDL, high total cholesterol, high triglyceride (TG) (defined as 200 ≤ TG < 500 mg/dl in adults1), hypertension, diabetes, and metabolic syndrome. Furthermore, CVD is associated with environmental variables that can be confounded with lipid levels, such as obesity, poor diet, lack of exercise, and smoking.
Hypertriglyceridemia (HTG), defined as TG > 500 mg/dl in adults,1 is a risk factor for CVD, independent of high LDL and low HDL.2–7 Although HDL and TG levels are highly correlated, an independent role of HDL level in CVD etiology has been challenged by recent Mendelian randomization studies and the failure of cholesteryl ester transfer protein inhibitors to reduce vascular events.8,9 Conversely, Mendelian randomization suggests a causal role of TG in CVD.10 Elevated TG has been implicated in both microvascular and macrovascular endothelial damage with associated atherosclerosis.6 Within the United States, ∼16% of adults of European origin have high TG levels, indicating a need for further intervention.7 However, studies of TG level and lipid metabolism have been difficult.6,7 One reason for this difficulty is the existence of high within-individual variation of TG measurement that expands with increasing TG. High TG is also associated with high LDL and low HDL, making it difficult to tease apart the effect of specific lipids on CVD risk within studies.
There are currently few pharmacological treatments for elevated TG. The most common treatment, fibrates, effectively reduces elevated TG and reduces the risk for cardiovascular events.11,12 Unfortunately, some 5% of individuals stop using fibrates because of side effects.13 Other potential drugs, targeting different parts of the metabolic pathway, have been found to have intolerable complications such as fatty liver or to actually raise the risk of cardiovascular events.13
In order to find additional effective treatments, studies of TG need to be undertaken. Focusing on the genetic control of elevated TG may remove some of the confounding with LDL and HDL and lead to new drug targets. TG is known to be heritable and there are several known genetic mutations that influence TG levels, most notably those in the structural loci for ApoA5 and ApoC3.14–21 In mice, expression of both Apoa5 and Apoc3 are associated with TG levels.22–25 Whereas circulating levels of ApoA5 are negatively associated with TG levels, ApoC3 levels are positively correlated with TG. However, there is conflicting evidence in humans for an association between CVD and single-nucleotide variants (SNVs) within APOC3 (MIM 606368) and APOA5 (MIM 107720).26–30 These and other known genetic variants explain only ∼10% of the genetic variation in TG,20,21 which may explain the conflicting evidence indicating a relationship between regulatory SNVs and CVD.
The genetic heterogeneity in the etiology of high TG makes large family studies the optimal design for identification of novel TG loci with large effect sizes.31 This design allows for the study of numerous people with an identical mutation and the ability to study cosegregation as well as association. For these reasons, we set out to discover genes underlying elevated TG in a single large family, described as familial combined hyperlipidemia (FCHL [MIM 144250]) in the 1980s and resampled in the 2000s, yielding five generations, four of which have phenotype and marker data. FCHL is genetically heterogeneous and characterized by variable atherogenic lipoprotein levels in multiple family members,32–35 making it an ideal diagnosis to identify novel lipid level genes. This family includes four related individuals, including the proband, with TG measurement in the top 1/2 percentile of adults in the United States,36 and previously reported genetic variation associated with TG does not explain the elevated levels. Segregation analyses supported at least one major gene for high TG in this family.
By using linkage analysis and whole-exome sequence data, we detected two candidate genes of interest. Although whole-exome sequencing has proven powerful in discovering underlying loci for simple Mendelian traits and de novo mutations, it has not yet been as successful in uncovering major contributing loci to complex traits. With family data, we have the opportunity to observe the same very rare variant in multiple family members and to evaluate cosegregation of a single variant with a phenotype. In this paper, we show that reduction of the search space by using linkage analysis in a large family, and then focusing on rare exome variants, can be used to successfully discover multiple major contributing loci for a heterogeneous, complex quantitative trait. This allows single gene burden testing in a second cohort, eliminating the need for multiple gene tests, which increases the power to detect an association.
Subjects and Methods
Subjects and Phenotype
The family presented here was ascertained in the 1980s as part of a cohort of four families of European American descent that had the diagnosis of FCHL. Observed TG in these families was previously shown to be associated with increased risk of CVD mortality.37 The proband had fasting triglycerides >2,900 mg/dl38 and normal LDL-C levels. The family was subsequently ascertained and diagnosed with FCHL, based on hypertriglyceridemia and hypercholesterolemia in first-degree relatives. The proband was originally hypothesized to represent a homozygote for FCHL, thought to be a Mendelian trait at that time. Re-collection of samples as well as sampling of additional family members, particularly in the youngest generation, took place in the 2000s. The final pedigree contains 5 generations of 121 individuals and includes five large sibships (n = 5, 5, 6, 7, and 9). Fasting TG was measured on 85 individuals (74 in the 1980s and 41 in the 2000s) via standard enzymatic methods, and reported in mg/dl.39 The same methods were employed in the 1980s and 2000s. TG ranged between 22 and 2,926 mg/dl, with a median of 111 mg/dl for individuals not taking statin drugs. Twenty-six family members had TG > 200 mg/dl and 8 had TG > 500 mg/dl. Four related subjects in the second and third generations had TG > 1,000 mg/dl, making this family a good candidate for linkage analysis. In addition, none of the subjects were taking lipid-lowering medications in the 1980s and only nine were taking such medication in the 2000s. All subjects, or their representative, gave written, informed consent and this study was approved by the University of Washington Human Subject Review Board (FWA #00006878).
TG Adjustment
Fasting TG level is a highly right-skewed phenotype that is associated with age, sex, and body mass index in typical European American populations. Because of the ascertainment, the TG in this data set does not follow typical distributions and adjusting the TG via a linear model with the family data would not result in a reliable phenotype. To increase the power to detect linkage, we corrected for this ascertainment by adjusting TG (adjTG) by using the age- and sex-based means and standard deviations from The Lipid Research Clinics Population Studies Data Book, Volume 1,40 added the constant 8 to avoid negative values, and then log transformed the data (log-adjTG) to reduce skew.
Because TG was measured in the 1980s and 2000s for 30 individuals, we generated three overlapping phenotype data sets. Data set “1980only” contains phenotypes from the 1980s only (n = 74, all statin-free). Data set “1980plus” contains phenotypes from the 1980s and phenotypes for only the newly sampled individuals from the 2000s who are not taking lipid-lowering medications (n = 85 statin-free). Data set “2000plus” contains all phenotypes from the 2000s for individuals not taking lipid-lowering medications and phenotypes from the 1980s for individuals who only have phenotype at this time point or who were taking statin drugs in the 2000s (n = 85 statin-free). Phenotypes in data sets 1980plus and 2000plus differ for 21 individuals.
Genotyping
The family was extensively genotyped via highly polymorphic markers, common SNVs, and next-generation exome sequencing. Quality control on all genotype data as well as alignment to the Rutgers Build 35 map was performed as described previously.41,42 In brief, STR markers from Marshfield Panel 9 (1980s), Prevention Genetics Set 13 (2000s), and selected regions from Decode (1980s and 2000s) were genotyped in the family (n = 82). In addition, ∼50K SNVs from the Illumina HumanCVD Bead chip were genotyped in the family (2000s, n = 64).43 Finally, the exome was sequenced on the Solexa platform for 16 individuals selected for their phenotypes and informativeness for imputing the genotypes of others.41,44 Novel SNVs of interest, detected with exome sequencing, were genotyped in the entire pedigree via custom TaqMan Genotyping Assays with the Applied Biosystems 7900HT real-time PCR system45 or Veracode 384-plex Bead Plates.46
Joint Segregation and Linkage Analysis
Given that log-adjTG is a complex trait with an unknown mode of inheritance in these data, we used an iterative Bayesian oligogenic joint segregation and linkage analysis approach from the package Loki2.4.7.47 This approach has many advantages for these data. First, Loki is capable of handling multipoint analysis on a large pedigree. Second, the user does not supply a trait model; rather, a prior distribution on the genetic effects is supplied, allowing the trait model, including the number of QTLs, to vary within a plausible model space. Third, missing trait-locus genotypes can be sampled at each iteration, given the marker segregation in the pedigree. Fourth, genotype covariates can be included in the model, allowing for estimation of the genetic variation explained by a SNV at each iteration, including the information from the estimated missing genotypes. Over an entire run, the average total variation (Vtot) and genetic variation (Vg) explained can then be used to estimate the percent Vtot and Vg explained by that SNV. In addition to modeling the possibility of multiple QTLs, variance resulting from any polygenic effects is captured by Vg. Because calculating p values is time consuming with this method, we calculated the Bayes factor (BF) for 2 cM intervals across the genome, which compares the posterior odds to the prior odds that a QTL is located in the region. We consider a maxBF > 25 as suggestive of linkage.41,48 All identified linkage regions were jointly analyzed, allowing the regions to compete for the modeled QTLs.
Because the trait under study is skewed, it is important to specify a prior distribution that is expansive enough to include a likely model but not so broad that the space cannot be adequately sampled. Furthermore, the detected regions of linkage should be consistent given differing prior distributions. By using methods described previously,49–51 we used several values for the underlying additive genetic effects, ranging between 1 and 8 times the standard deviation (SD) of log-adjTG in order to verify consistency of detected linkage regions. Here we report the results for the prior distribution with mean additive genetic effect = 2.7 times the SD of log-adjTG. All runs contained 1,000 burn-in iterations followed by 200,000 iterations (1 chromosome) or 500,000 iterations (2 chromosomes jointly), of which every 10th iteration was saved. The prior distribution on the number of QTLs followed a curtailed Poisson(1) distribution with a maximum of 15. The total map length was assumed to be 3,000 cM. The prior distribution on the allele frequency for all QTLs is set to Uniform(0,1) and cannot be changed by the user.
Candidate Locus Analysis
Candidate loci were assessed for the estimated percent Vtot and Vg that they explained in the family. These candidate variants were chosen from two sources (Tables 1 and 2 and Table S1 available online). First, candidate SNVs known or thought to be associated with TG,21,52 including APOA5 variant rs3135506, were assessed within the family primarily via the CVD chip data. If the reported SNV was not on the CVD chip, we attempted to find a correlated proxy SNV (r2 > 0.8) within 500 bases of the reported SNV.53,54 APOA5 SNV rs3135506 was genotyped separately. A Bonferroni adjustment was used for the testing of these known candidate loci.
Table 1.
Percent Variance of log-adjTG Explained in the Family, by Previously Reported SNVs
| Chr | Gene (SNV, Proxy) | MIM # | SNV | Proxy | % Vtot (SD) | % Vg (SD) | p Value |
|---|---|---|---|---|---|---|---|
| 1 | DOCK7 | 604774 | rs2131925 | rs1748195 | 3 (3) | 14 (16) | 0.84 |
| 1 | GALNT2 | 602274 | rs1321257 | rs4846914 | 4 (3) | 23 (20) | 0.15 |
| 2 | APOB | 107730 | rs1042034 | NA | 4 (3) | 16 (17) | 0.73 |
| 2 | GCKR | 600842 | rs1260326 | NA | 4 (3) | 18 (19) | 0.09 |
| 3 | MSL2,aPCCB | 614802, 232050 | rs645040 | rs3821445 | 3 (2) | 17 (19) | 0.34 |
| 4 | AFF1 | 159557 | rs442177 | rs3775214 | 2 (2) | 13 (17) | 0.68 |
| 7 | TBL2 | 605842 | rs17145738 | NA | 5 (4) | 20 (18) | 0.04 |
| 8 | LPL,aLPL | 609708 | rs12678919 | rs12679834 | 2 (2) | 14 (19) | 0.64 |
| 8 | TRIB1a | 609461 | rs2954029 | rs17321515 | 3 (2) | 13 (16) | 0.60 |
| 10 | CYP26A1a | 602239 | rs2068888 | rs4418728 | 4 (3) | 24 (21) | 0.25 |
| 11 | FADS1, FADS2 | 606148, 606149 | rs174546 | rs1535 | 3 (2) | 14 (18) | 0.82 |
| 11 | APOA5 | 606368 | rs3135506 | NA | 2 (2) | 14 (18) | 0.67 |
| 15 | LIPC | 151670 | rs6074 | rs871804 | 4 (3) | 18 (17) | 0.17 |
| 15 | LIPC | 151670 | rs261342 | NA | 2 (2) | 15 (19) | 0.94 |
| 16 | CTF1a | 600435 | rs11649653 | NA | 3 (3) | 14 (16) | 0.47 |
| 16 | CETP | 118470 | rs7205804 | rs1532625 | 3 (3) | 21 (21) | 0.99 |
| 19 | APOE,aAPOC1a | 107741, 107710 | rs439401 | NA | 5 (3) | 20 (18) | 0.12 |
| 19 | SUGP1, GATAD2A | 607992, 614997 | rs10401969 | rs3794991 | 1 (2) | 10 (17) | 0.70 |
| 20 | PLTP | 172425 | rs4810479 | NA | 7 (5) | 30 (18) | 0.56 |
Estimated percent total variance (Vtot), percent genetic variance (Vg), and p value for pedigree adjusted association of log-adjTG and known or suspected pathogenic SNVs in the family. Proxy SNVs from the CVD chip were used in place of the reported SNVs that did not exist on the CVD chip. Proxy SNVs are correlated with the original SNV (r2 > 0.8). APOA5 (rs3135506) was genotyped separately in the family. The second column from left gives the gene name for the SNV and its proxy, if necessary. Gene names are from NCBI and aliases are as follows: DOCK7 = ANGPTL3, AFF1 = KHL8, TBL2 = MLXIPL, SUGP1 = CILP2. SD indicates standard deviation.
aSNV is near but not within a gene.
Table 2.
Distribution of Novel SNVs under the Linkage Signals on Chr7 and Chr17
| Chr. | 7 | 17 |
|---|---|---|
| # novel sites | 53 | 20 |
| Intergenic | 2 | 1 |
| Intronic | 4 | 1 |
| 3′ UTR | 1 | 1 |
| 5′ UTR | 1 | 0 |
| Synonymous | 23 | 2 |
| Splice | 0 | 1 |
| Missense | 22 | 14 |
| GERP > 3 | 12 | 6 |
| Shared | 1 | 4 |
| Liver expressed | 1 | 2 |
The final row indicates the number of novel SNVs that are missense or splice sites, have Genomic Evolutionary Rate Profiling score (GERP) > 3, and are shared by at least two relatives with high log-adjTG. Novel is defined as not existing in dbSNP134.
Candidate rare SNVs were chosen from the exome data in the linkage regions. Exome sites were filtered based on novelty, coding effect, conservation across mammals, and sharing of the rare allele among the four individuals with TG > 1,000. Because the genetic basis of TG in this family had yet to be discovered, we hypothesized that the high levels of TG were due to novel variation. We defined a variant as novel if it did not have an rsID in dbSNP134 at the time of exome sequencing (April 2012)55 and occurred at most once in the NHLBI Exome Sequencing Project (ESP) data on 6,500 exomes. Only sites that had an effect on the coding sequence (nonsense, missense, or splice) were kept. Conservation across mammals was calculated by the Genomic Evolutionary Rate Profiling (GERP) score;56 positive values indicate conservation and negative values indicate lack of conservation. Only sites with a GERP > 3 were considered because we expect only mutations at locations with high conservation across mammals would be pathogenic for high TG. Finally, the rare allele had to exist in at least two of the four individuals with TG > 1,000. All pedigree members were genotyped for the novel sites selected with these criteria to confirm their estimated effect. Variance components analysis in the package SOLAR57 was used to calculate p values for association, in a mixed model analysis that adjusted for correlation among related individuals through the kinship matrix, which captures any underlying shared polygenic effect.
Validation of Novel Gene Association
We sought confirmation of association with TG for any gene containing a novel SNV that remained associated with the trait after genotyping of the full pedigree. By using TG data from unrelated individuals in the PennCATH, TRIUMPH, Cleveland Clinic, ARIC, CARDIA, CHS, FHS, JHS, MESA, and WHI cohorts in the dbGaP posted ESP data, we combined all rare (MAF < 0.5%) missense, nonsense, and splice SNVs with GERP > 4.8 into a single genotype factor (1 = presence of a minor allele and 0 = absence of all minor alleles) in individuals with measured TG. We used a GERP cutoff of 4.8 because this is the 75th percentile for GERP of all identifiable LDL-raising pathogenic SNVs in LDLR (MIM 606945)55,56,58,59 (data not shown). We assumed that individuals missing genotypes at these rare SNVs were noncarriers. We performed this two-sided whole-gene test via a linear model for log(TG) on the {0, 1} genotype factor, using the package R with and without adjusting for race (first three principal components or ethnic group), age, and sex. Statin and fibrate usage is not contained in this data. However, because these medications reduce TG levels, their absence from the model serves to make the test more conservative.
Results
Phenotype
The adjTG is a stable phenotype over time in this data set. The correlation of adjTG values ∼20 years apart, in the 21 individuals with data at both time points and not on statins, is highly significant (r2 = 0.78, p = 2.7 × 10−5) with positive slope coefficient (β = 0.5). adjTG level appears to have larger variance at higher levels, as expected. The adjusted values were independent of age and sex, as expected.
Known TG SNVs
There were 18 SNVs, or their proxies, on the CVD chip of the total 35 (Table 1) that were reported to have an effect on TG.21,52 These 18 SNVs and the APOA5 SNV, rs3135506, explained at most 7% Vtot in log-adjTG in this family (Table 1). In addition, there was no association with the cumulative genetic risk score21 and log-adjTG (p = 0.20). rs4810479, in the 5′ region of PLTP (MIM 172425), explained the most Vtot in log-adjTG, 7% (SD = 5%), but was not significantly associated with log-adjTG (p = 0.56), adjusting for pedigree structure. The APOA5 SNV, rs3135506, explained 2.1% Vtot (SD = 2%). SNV rs17145738 in TBL2 ([MIM 605842] alias MLXIPL) and near one of our linkage signals (see below), explained 5% Vtot in log-adjTG (SD = 4%) and had the lowest p value for association, p = 0.04, adjusting for pedigree structure. This result is not significant given the Bonferroni adjustment for multiple testing (Bonferonni cutoff of 0.003). Furthermore, the minor allele is associated with lower log-adjTG, indicating that this common SNV is unlikely to explain the high TG observed in this family. Because these known SNVs did not explain much of the Vtot in log-adjTG and were not significantly associated with log-adjTG, we performed linkage analysis to identify regions of interest.
Joint Segregation and Linkage Analysis Results
Segregation analysis, both alone and in conjunction with linkage analysis, shows evidence of at least one QTL underlying log-adjTG. Given the prior mean additive genetic effect = 2.7 times the SD of log-adjTG, the posterior probability of at least one QTL is 0.97, 0.88, and 0.998 for data sets 1980only, 1980plus, and 2000plus. The posterior probability of exactly one QTL is 0.83, 0.54, and 0.23 for data sets 1980only, 1980plus, and 2000plus. In addition, the three overlapping data sets show support for log-adjTG linkage to chr7 and chr17 (Figure 1). Data sets 1980only and 1980plus support linkage to both chromosomes. Data set 2000plus shows support for only chr17. Using differing prior distributions on the genetic effects did not change the locations of the linkage signals (data not shown). Because data set 1980plus shows support for both regions and contains more phenotype data than does 1980only, we continued the analysis with this data set. The posterior predicted model for 1980plus is a recessive mode of inheritance.
Figure 1.
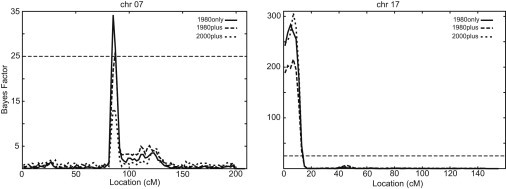
Evidence of Linkage, as Measured by the Bayes Factor, on Chromosomes 7 and 17 for the Three Data Sets 1980only, 1980plus, and 2000plus
Location is given in cM and spans the entire length of the chromosomes. The horizontal dashed line at 25 indicates the cutoff for evidence for linkage.
Novel SNVs
Within and near these linkage regions, we found five novel SNVs that met our criteria for candidacy, labeled N7, N17a, N17b, N17c, and N17d, that were private to this family (Tables 2 and 3). Variant N7 is shared by all four IDs with TG > 1,000. Although chr17 contained four SNVs of interest, they were shared by three different pairs of IDs with TG > 1,000: N17a at one pair, N17b at another, and N17c and N17d at a different pair. These five SNVs explain 19%, 7.8%, 4.8%, 5.3%, and 5.3% of Vtot, respectively, in data set 1980plus. It is expected that the estimated %Vtot explained will decrease upon full genotyping in the family, because the most informative people have measured or imputable genotypes and the branch of the family without variability of the SNV of interest will lower the Vtot explained. Therefore, we chose to pursue full genotyping of N7 and N17a only because they explain >7% of Vtot with just the exome genotypes, which is more than the known SNVs in PLTP, APOA5, and TBL2 noted above. These SNVs were in SLC25A40 (MIM 610821) and PLD2 (MIM 602384), respectively. Additionally, no known pathogenic or novel likely pathogenic variation was found in the known TG genes LPL (MIM 09708), APOC2 (MIM 608083), APOA5, GPIHBP1 (MIM 612757), and LMF1 (MIM 611761) or in the lipid-related genes CETP (MIM 118470) and LDLR in the 16 exomes.
Table 3.
Percent Variance of log-adjTG Explained in the Family, by Candidate Novel SNVs
| Variant | N7 | N17a | N17b | N17c | N17d |
|---|---|---|---|---|---|
| Chr | 7 | 17 | 17 | 17 | 17 |
| Pos. | 87477251 | 4711152 | 6021379 | 2290581 | 4496467 |
| Gene | SLC25A40 | PLD2 | WSCD1 | MNT | SMTNL2 |
| RefSeq | NM_018843.3 | NM_002663.4 | NM_015253.1 | NM_020310.2 | NM_001114974.1; NM_198501.2 |
| NT change | c.374A>G | c.85A>T | c.1246A>G | c.1363G>C | c.730+1G>T; c.298+1G>T |
| Protein change | p.Tyr125Cys | p.Thr29Ser | p.Arg416Gly | p.Val455Leu | NA |
| Coding effect | missense | missense | missense | missense | splice |
| GERP | 5.1 | 5.1 | 4.3 | 3.9 | 5.0 |
| %Vtot as exome variant | 19 | 7.8 | 4.8 | 5.3 | 5.3 |
| %Vg as exome variant | 49 | 23 | 22 | 27 | 27 |
| maxBF | 1.8 | 48 | 103 | 72 | 66 |
Estimated percent total variance (Vtot) and genetic variance (Vg) in log-adjTG explained by each of the candidate novel SNVs within the linkage regions, using only individuals that have exome data. MaxBF are given for when the novel variant is included as a marker and covariate in the joint linkage and association analysis. Missing genotype data are imputed for other family members. Physical positions are from build 37. In dbSNP137, rsIDs have been assigned to SNVs N17b, N17c, and N17d as follows: N17b = rs200724890, N17c = rs201365025, N17d = rs202160684. There are two RefSeq numbers for SMTNL2 because it has two isoforms.
Jointly analyzing the chromosomes and their associated novel SNVs, genotyped in the full family, clarifies the relationship among these two regions (Figures 2 and S1). As expected, given the complex model, adjustment for each SNV impacts the linkage evidence for the other region. When both chromosomes are analyzed jointly, the linkage evidence on chr7 was reduced (maxBF = 8), but the linkage to chr17 remained (maxBF = 207). Linkage results are similar when including the fully genotyped SNVs N7 and N17a as markers only, in the analysis, indicating that when neither of the SNVs is included as a genotype covariate, the posterior distribution favors chr17 over chr7. However, only SNV N7 explained all evidence for linkage (chr17 maxBF reduced from 207 to 1, and chr7 maxBF reduced from 8 to 0.5) when included as a genotype covariate (Figure 2). When included as a genotype covariate, N17a explained some of the signal on chr17 (maxBF decreases from 207 to 69) and chr7 (maxBF reduces from 8 to 3.5).
Figure 2.
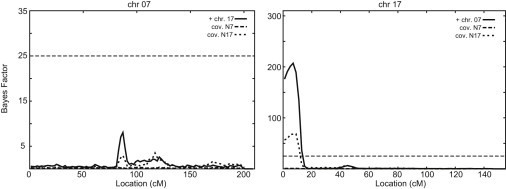
Evidence of Linkage in Data Set 1980plus, as Measured by the Bayes Factor, on Chromosomes 7 and 17 when Analyzed Jointly, and with either SNV N7 or N17 as a Genotype Covariate
Location is given in cM and spans the entire length of the chromosomes. The horizontal dashed line at 25 indicates the original cutoff for evidence for linkage.
The relative strength of each candidate SNV is reflected in the percent Vg explained by each, as well as the significance of the pedigree-adjusted association with log-adjTG. N7 (Ngt = 40) explained 19% of log-adjTG Vtot and 49% of Vg. N17a (Ngt = 78) explained 7% of log-adjTG Vtot and 25% Vg. When both variants are included jointly, they explained 49% of log-adjTG Vg; with N7 and N17a explaining 40% (p = 0.0001) and 8.6% (p = 0.31) of log-adjTG Vg, respectively. We also explored the significance of the known TG SNVs while adjusting for N7 genotype. In this case, each SNV explained <5% of Vtot and were not statistically significant (unadjusted p > 0.08). The TBL2 SNV remained insignificant (unadjusted p = 0.08). Furthermore, this SNV is carried by individuals with log-adjTG below the mean, and these are not the same individuals as those that carry the N7 variant. Although N7 is associated with log-adjTG, it is not associated with LDL-C in this family (p = 0.83, Figure S2).
Confirmation of SLC25A40 Association
SLC25A40 was significantly associated with log(TG) in a separate cohort by using ESP data (Table 4). There were five rare missense SNVs (Table 5) in SLC25A40 with GERP > 4.8 identified in ESP subjects with TG data. Notably, the SLC25A40 SNV in the linkage family had a GERP of 5.1 and each of these five SNVs had GERPs > 5.1; two were predicted to be “probably damaging” by PolyPhen.60 Six subjects of European American (EA) descent and two of African American (AA) descent carried one of these variants. The two of AA descent carried the same SNV; this SNV did not vary in EA in the entire ESP data. The SNVs found in the EA-descent subjects were also specific to that ancestry group. No subject carried more than one of these SLC25A40 SNVs. After pooling carrier status for each of these SNVs into a single factor, the whole-gene test gives a significant positive association between these high GERP variants at SLC25A40 and log(TG) (β = 0.45, two-sided p = 0.02), adjusted for age and sex. This whole-gene association with log(TG) remains significant and positive when adjusted for race, age, and sex (β = 0.42, two-sided p = 0.03). Results are similar when applied to individuals older than 25 and when removing individuals with a rare SNV in the gene, whose GERP score ranges between 2 and 4.8. Although the sample size is too small for stratified testing, the coefficient is positive in both ancestry groups (β = 0.18 and 1.1 for EA and AA descent, respectively). Although we focused on SLC25A40 as the best candidate from this region, we sought to rule out associations of other regional genes and log(TG) in the ESP cohort by similar analyses (Table S2). Carrier status at PLD2 (p = 0.48), WSCD1 (p = 0.19), MNT (p = 0.81 [MIM 603039]), and SMTNL2 (p = 0.87) did not predict log(TG).
Table 4.
Demographic Count Data and Age Distribution for the ESP Cohort, Broken down by Quantile of log(TG)
| Log(TG) quantile | M | F | EA | AA | Mean (Min, Max) Age |
|---|---|---|---|---|---|
| [1.77,4.36] | 341 | 597 | 434 | 504 | 50.15 (12, 85) |
| (4.36,4.74] | 298 | 666 | 461 | 503 | 57.60 (18, 84) |
| (4.74,5.13] | 312 | 596 | 522 | 386 | 58.27 (20, 93) |
| (5.13,6.84] | 343 | 589 | 664 | 268 | 59.69 (21, 90) |
This data set contains 3,770 verified unrelated individuals. Abbreviations are as follows: M, male; F, female; EA, European American; AA, African American.
Table 5.
Rare SNVs with GERP > 4.8 in SLC25A40 Found in Individuals in the ESP Data
| Position | rsID | NT Change | MAF % (EA/AA/All) | AA Change | GERP | PolyPhen | n |
|---|---|---|---|---|---|---|---|
| 87470986 | NA | c.785A>G | 0.0118/0.0/0.0078 | p.Gln262Arg | 5.92 | B | 1 EA |
| 87483601 | NA | c.182G>T | 0.0/0.0454/0.0154 | p.Gly61Val | 5.91 | PD | 2 AA |
| 87473169 | rs145515966 | c.641G>C | 0.0117/0.0/0.0077 | p.Trp214Ser | 5.67 | PD | 1 EA |
| 87473143 | rs140104130 | c.667T>A | 0.0349/0.0/0.0231 | p.Trp223Arg | 5.67 | B | 3 EA |
| 87477276 | NA | c.349G>A | 0.0116/0.0/0.0077 | p.Ala117Thr | 5.13 | B | 1 EA |
MAF % based on 8,600 European (EA) and 4,406 African American (AA), self-reported ancestry subjects; TG were measured in 2,168 EA and 1,793 AA subjects, with ancestry genetically verified. Abbreviations are as follows: B, benign; PD, probably damaging.
Discussion
We identified two genes, SLC25A40 and PLD2, each containing a novel variant that cosegregates with severely high TG levels in a single large family ascertained for segregation of FCHL. Joint analysis of the linkage regions and these variants indicated that the variant in SLC25A40 is the most probable variant for pathogenicity. However, we cannot rule out a possible interaction or additive effect between these and other identified variants. No known common pathogenic variants were found to be associated with TG in this family. We were able to replicate the effects of rare, conserved missense mutations only in SLC25A40 in the separate ESP cohort.
SLC25A40 is solute carrier family 25, member 40, and its product localizes to the mitochondria, where it is involved in membrane transport.61 Although the gene is ubiquitously expressed, its product is found mainly in the adrenal gland.62 The novel variant in this gene (RefSeq accession number NM_018843.3), c.374A>G, is located in the seventh exon (of 12), is highly conserved (GERP = 5.1), and causes a p.Tyr125Cys substitution in the 338 amino acid protein. This change is just outside the second helical transmembrane region of the protein. Although there is no PolyPhen prediction, the tyrosine at amino acid 125 is perfectly conserved across vertebrates.58,63 Furthermore, the Grantham score is 194, indicating a high chemical dissimilarity from the wild-type.64 This particular mutation causes a new cysteine that neighbors a cysteine at position 124 and increases the number of cysteines to 9. Cysteines play a critical role in protein folding because they are the only amino acids that can create disulfide bonds that stabilize the folded form of the protein.65 This change of a single amino acid to a cysteine allows for an increased number of folding possibilities, and possibly a lower entropy stabilization, which can disrupt the protein’s function.
It is possible that this specific variant is not pathogenic, but is in linkage disequilibrium with a causal variant not detected by our exome sequencing. Further gene sequencing, as well as functional studies, are needed to verify the association. However, we found evidence for a pathogenic effect of high TG for rare, conserved (GERP > 5) missense variants in SLC25A40 in a separate, unrelated cohort, supporting the idea that SLC25A40 affects TG levels.
Although our analyses suggest that a PLD2 missense variant may also impact TG, whole-gene testing in a separate cohort does not support pathogenicity for the gene. Further study may be warranted, because this gene may have biological relevance to TG. PLD2 is phospholipase D2, whose product catalyzes the hydrolysis of phosphatidylcholine to phosphatidic acid and choline, and is ubiquitously expressed. The novel variant in this gene (RefSeq NM_002663.4), c.85A>T, causes a p.Thr29Ser (out of 934 amino acids) and is highly conserved across mammals.
Three other candidate genes identified on chr17, WSCD1, MNT, and SMTNL2, also were not significantly associated with TG in this separate cohort. Of these three genes, only SMNTL2 is expressed in liver. SMNTL2 is smoothelin-like 2, of which very little is currently known. Because neither WSCD1 nor MNT are expressed in liver, they are less likely to be involved in TG regulation. Little is known about WSCD1, the WSC domain containing 1 gene. MNT encodes the MAX dimerization protein and is thought to repress transcription by binding to DNA binding proteins.66,67 Reliance on the exome-sequence data rather than full-genome sequencing may have missed other potentially causal variants.
It is worth noting that the size of the linkage signals do not necessarily correlate with the candidate gene with the most evidence for causality. When chr7 and chr17 are analyzed jointly, the posterior distribution prefers a recessive QTL with moderate allele frequency on chr17. This is most probably due to the ascertainment scheme, lack of prior information on the allele frequency, and the observed phenotype segregation. The ascertainment scheme, which is useful for observing a rare allele multiple times, artificially inflates the observed allele frequency. In addition, the uniform prior distribution on the allele frequencies does not allow for a preference for rare alleles. Finally, the skew of the phenotype data in which offspring may have much higher phenotype than their parents results in evidence for a recessive trait. Therefore, segregation analysis showed overwhelming support for a recessive gene with a common allele frequency, which fit the marker and trait segregation at chr17. Further analysis including the exome-sequence data as covariate effects, which included a fixed rare allele frequency in the model, favored a dominant gene as evidenced by the high association of the SNV on chr7 with TG in the family.
The sampling and analysis design used here is a powerful approach for successful identification of novel genes and biological pathways underlying heterogeneous complex traits. Ascertainment of a large family segregating extreme values of a quantitative phenotype reduces the number of putative underlying highly penetrant loci, increasing the chances of finding a variant with biological relevance in multiple family members. The large size of the pedigree, although a fraction of the size of a typical GWAS sample, provides multiple dimensions of information that, when used jointly, substantially increases the power to detect linkage.31 Furthermore, correction for the ascertainment of these extreme phenotypes, as in this study, further increases power to detect linkage. The use of linkage analysis further drastically reduces the search space, limiting the multiple testing problem seen with GWASs or exome-wide burden tests. Finally, joint linkage and association in such pedigrees, as when both chromosomes 7 and 17 compete for the modeled QTL and the candidate novel variants are included as covariates, can help determine which of the identified candidate genes has the most promise. One limitation of this approach is that the same rare variant may not be found in a second family or enough unrelated individuals to provide confirmation; however, a single or limited number of gene-based burden tests can be made in an unrelated sample, again avoiding the penalty of exome-wide multiple comparisons.
Although common alleles have been found to explain a small portion of moderate to severe HTG in unrelated samples, we show that rare variation plays a major role in explaining severe HTG in a family previously diagnosed with FCHL and that this knowledge can be used to identify novel genes and biological pathways of interest. Additionally, our linkage results and the replication of a HTG effect in the ESP data, which is not ascertained on FCHL, lend support to an oligogenic inheritance of the lipid traits contributing to the diagnosis of FCHL in this family. Further evidence for this includes the fact that LDL-C level is not associated with the SLC25A40 variant in these data, the proband had normal LDL-C and apoB levels, we do not detect linkage between LDL-C and this region of chr7 (data not shown) by similar methodology, and we have previously reported linkage between apoB level and chr4 in this family.42 FCHL families may not have a monogenic disorder as earlier described, but rather a confluence of separate traits in the same family. Furthermore, the complex nature of the trait within this family is borne out by the fact that three carriers of the SLC25A40 variant do not have HTG, although two of them have TG near the 95th percentile (Figure S2), and that one individual with TG > 500 does not carry the SLC25A40 variant, indicating that other genetic variants may be influential. However, the polygenic effects are accounted for in the analysis and have a small impact on the trait, relative to the major gene component. Indeed, SLC25A40 may be shown to cause familial hypertriglyceridemia (MIM 145750), given that it does not appear to raise LDL-C. We note that our proband had highly elevated TG for a subject with FCHL, which is why the proband was thought to potentially be a FCHL homozygote; thus, the relationship of this locus to a more typical FCHL family with more modest HTG is not yet clear. Neither SLC25A40 nor PLD2 (or their respective biological pathways) have been previously implicated in triglyceride levels, to our knowledge. Functional studies, in vitro and in vivo, will need to be carried out to verify that they impact TG levels and to discover the mechanism by which they have an effect. Given the relevance of high TG to CVD, TG-lowering treatments targeted to these pathways may be identified.
Acknowledgments
Thanks go to Peter Byers for his helpful comments. Funding for this analysis was provided by National Institutes of Health grants P01 HL030086, T32 GM007454, and R01 HL094976 and the State of Washington Life Sciences Discovery Fund award to the Northwest Institute of Genetic Medicine (grant 265508). The authors wish to acknowledge the support of the National Heart, Lung, and Blood Institute (NHLBI) and the contributions of the research institutions, study investigators, field staff, and study participants in creating this resource for biomedical research. Funding for GO ESP was provided by NHLBI grants RC2 HL-103010 (HeartGO), RC2 HL-102923 (LungGO), and RC2 HL-102924 (WHISP). The exome sequencing was performed through NHLBI grants RC2 HL-102925 (BroadGO) and RC2 HL-102926 (SeattleGO). Genotyping services were provided through the RS&G Service by the Northwest Genomics Center at the University of Washington, Department of Genome Sciences, under US Federal Government contract number HHSN268201100037C from the National Heart, Lung, and Blood Institute.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
R statistical software, http://www.r-project.org/
References
- 1.National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation. 2002;106:3143–3421. [PubMed] [Google Scholar]
- 2.Hokanson J.E., Austin M.A. Plasma triglyceride level is a risk factor for cardiovascular disease independent of high-density lipoprotein cholesterol level: a meta-analysis of population-based prospective studies. J. Cardiovasc. Risk. 1996;3:213–219. [PubMed] [Google Scholar]
- 3.Patel A., Barzi F., Jamrozik K., Lam T.H., Ueshima H., Whitlock G., Woodward M., Asia Pacific Cohort Studies Collaboration Serum triglycerides as a risk factor for cardiovascular diseases in the Asia-Pacific region. Circulation. 2004;110:2678–2686. doi: 10.1161/01.CIR.0000145615.33955.83. [DOI] [PubMed] [Google Scholar]
- 4.Tirosh A., Rudich A., Shochat T., Tekes-Manova D., Israeli E., Henkin Y., Kochba I., Shai I. Changes in triglyceride levels and risk for coronary heart disease in young men. Ann. Intern. Med. 2007;147:377–385. doi: 10.7326/0003-4819-147-6-200709180-00007. [DOI] [PubMed] [Google Scholar]
- 5.Miller M., Cannon C.P., Murphy S.A., Qin J., Ray K.K., Braunwald E., PROVE IT-TIMI 22 Investigators Impact of triglyceride levels beyond low-density lipoprotein cholesterol after acute coronary syndrome in the PROVE IT-TIMI 22 trial. J. Am. Coll. Cardiol. 2008;51:724–730. doi: 10.1016/j.jacc.2007.10.038. [DOI] [PubMed] [Google Scholar]
- 6.Kohli P., Cannon C.P. Triglycerides: how much credit do they deserve? Med. Clin. North Am. 2012;96:39–55. doi: 10.1016/j.mcna.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 7.Miller M., Stone N.J., Ballantyne C., Bittner V., Criqui M.H., Ginsberg H.N., Goldberg A.C., Howard W.J., Jacobson M.S., Kris-Etherton P.M., American Heart Association Clinical Lipidology, Thrombosis, and Prevention Committee of the Council on Nutrition, Physical Activity, and Metabolism. Council on Arteriosclerosis, Thrombosis and Vascular Biology. Council on Cardiovascular Nursing. Council on the Kidney in Cardiovascular Disease Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2011;123:2292–2333. doi: 10.1161/CIR.0b013e3182160726. [DOI] [PubMed] [Google Scholar]
- 8.Voight B.F., Peloso G.M., Orho-Melander M., Frikke-Schmidt R., Barbalic M., Jensen M.K., Hindy G., Hólm H., Ding E.L., Johnson T. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet. 2012;380:572–580. doi: 10.1016/S0140-6736(12)60312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schwartz G.G., Olsson A.G., Abt M., Ballantyne C.M., Barter P.J., Brumm J., Chaitman B.R., Holme I.M., Kallend D., Leiter L.A., dal-OUTCOMES Investigators Effects of dalcetrapib in patients with a recent acute coronary syndrome. N. Engl. J. Med. 2012;367:2089–2099. doi: 10.1056/NEJMoa1206797. [DOI] [PubMed] [Google Scholar]
- 10.Sarwar N., Sandhu M.S., Ricketts S.L., Butterworth A.S., Di Angelantonio E., Boekholdt S.M., Ouwehand W., Watkins H., Samani N.J., Saleheen D., Triglyceride Coronary Disease Genetics Consortium and Emerging Risk Factors Collaboration Triglyceride-mediated pathways and coronary disease: collaborative analysis of 101 studies. Lancet. 2010;375:1634–1639. doi: 10.1016/S0140-6736(10)60545-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee M., Saver J.L., Towfighi A., Chow J., Ovbiagele B. Efficacy of fibrates for cardiovascular risk reduction in persons with atherogenic dyslipidemia: a meta-analysis. Atherosclerosis. 2011;217:492–498. doi: 10.1016/j.atherosclerosis.2011.04.020. [DOI] [PubMed] [Google Scholar]
- 12.Bruckert E., Labreuche J., Deplanque D., Touboul P.J., Amarenco P. Fibrates effect on cardiovascular risk is greater in patients with high triglyceride levels or atherogenic dyslipidemia profile: a systematic review and meta-analysis. J. Cardiovasc. Pharmacol. 2011;57:267–272. doi: 10.1097/FJC.0b013e318202709f. [DOI] [PubMed] [Google Scholar]
- 13.Wierzbicki A.S., Hardman T.C., Viljoen A. New lipid-lowering drugs: an update. Int. J. Clin. Pract. 2012;66:270–280. doi: 10.1111/j.1742-1241.2011.02867.x. [DOI] [PubMed] [Google Scholar]
- 14.Pennacchio L.A., Olivier M., Hubacek J.A., Krauss R.M., Rubin E.M., Cohen J.C. Two independent apolipoprotein A5 haplotypes influence human plasma triglyceride levels. Hum. Mol. Genet. 2002;11:3031–3038. doi: 10.1093/hmg/11.24.3031. [DOI] [PubMed] [Google Scholar]
- 15.Talmud P.J., Hawe E., Martin S., Olivier M., Miller G.J., Rubin E.M., Pennacchio L.A., Humphries S.E. Relative contribution of variation within the APOC3/A4/A5 gene cluster in determining plasma triglycerides. Hum. Mol. Genet. 2002;11:3039–3046. doi: 10.1093/hmg/11.24.3039. [DOI] [PubMed] [Google Scholar]
- 16.Romeo S., Pennacchio L.A., Fu Y., Boerwinkle E., Tybjaerg-Hansen A., Hobbs H.H., Cohen J.C. Population-based resequencing of ANGPTL4 uncovers variations that reduce triglycerides and increase HDL. Nat. Genet. 2007;39:513–516. doi: 10.1038/ng1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kathiresan S., Melander O., Guiducci C., Surti A., Burtt N.P., Rieder M.J., Cooper G.M., Roos C., Voight B.F., Havulinna A.S. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat. Genet. 2008;40:189–197. doi: 10.1038/ng.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kooner J.S., Chambers J.C., Aguilar-Salinas C.A., Hinds D.A., Hyde C.L., Warnes G.R., Gómez Pérez F.J., Frazer K.A., Elliott P., Scott J. Genome-wide scan identifies variation in MLXIPL associated with plasma triglycerides. Nat. Genet. 2008;40:149–151. doi: 10.1038/ng.2007.61. [DOI] [PubMed] [Google Scholar]
- 19.Teslovich T.M., Musunuru K., Smith A.V., Edmondson A.C., Stylianou I.M., Koseki M., Pirruccello J.P., Ripatti S., Chasman D.I., Willer C.J. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nilsson S.K., Heeren J., Olivecrona G., Merkel M. Apolipoprotein A-V; a potent triglyceride reducer. Atherosclerosis. 2011;219:15–21. doi: 10.1016/j.atherosclerosis.2011.07.019. [DOI] [PubMed] [Google Scholar]
- 21.Johansen C.T., Kathiresan S., Hegele R.A. Genetic determinants of plasma triglycerides. J. Lipid Res. 2011;52:189–206. doi: 10.1194/jlr.R009720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baroukh N., Bauge E., Akiyama J., Chang J., Afzal V., Fruchart J.C., Rubin E.M., Fruchart-Najib J., Pennacchio L.A. Analysis of apolipoprotein A5, c3, and plasma triglyceride concentrations in genetically engineered mice. Arterioscler. Thromb. Vasc. Biol. 2004;24:1297–1302. doi: 10.1161/01.ATV.0000130463.68272.1d. [DOI] [PubMed] [Google Scholar]
- 23.Ito Y., Azrolan N., O’Connell A., Walsh A., Breslow J.L. Hypertriglyceridemia as a result of human apo CIII gene expression in transgenic mice. Science. 1990;249:790–793. doi: 10.1126/science.2167514. [DOI] [PubMed] [Google Scholar]
- 24.Pennacchio L.A., Olivier M., Hubacek J.A., Cohen J.C., Cox D.R., Fruchart J.C., Krauss R.M., Rubin E.M. An apolipoprotein influencing triglycerides in humans and mice revealed by comparative sequencing. Science. 2001;294:169–173. doi: 10.1126/science.1064852. [DOI] [PubMed] [Google Scholar]
- 25.Maeda N., Li H., Lee D., Oliver P., Quarfordt S.H., Osada J. Targeted disruption of the apolipoprotein C-III gene in mice results in hypotriglyceridemia and protection from postprandial hypertriglyceridemia. J. Biol. Chem. 1994;269:23610–23616. [PubMed] [Google Scholar]
- 26.Russo G.T., Meigs J.B., Cupples L.A., Demissie S., Otvos J.D., Wilson P.W., Lahoz C., Cucinotta D., Couture P., Mallory T. Association of the Sst-I polymorphism at the APOC3 gene locus with variations in lipid levels, lipoprotein subclass profiles and coronary heart disease risk: the Framingham offspring study. Atherosclerosis. 2001;158:173–181. doi: 10.1016/s0021-9150(01)00409-9. [DOI] [PubMed] [Google Scholar]
- 27.Olivieri O., Bassi A., Stranieri C., Trabetti E., Martinelli N., Pizzolo F., Girelli D., Friso S., Pignatti P.F., Corrocher R. Apolipoprotein C-III, metabolic syndrome, and risk of coronary artery disease. J. Lipid Res. 2003;44:2374–2381. doi: 10.1194/jlr.M300253-JLR200. [DOI] [PubMed] [Google Scholar]
- 28.Vaessen S.F., Schaap F.G., Kuivenhoven J.A., Groen A.K., Hutten B.A., Boekholdt S.M., Hattori H., Sandhu M.S., Bingham S.A., Luben R. Apolipoprotein A-V, triglycerides and risk of coronary artery disease: the prospective Epic-Norfolk Population Study. J. Lipid Res. 2006;47:2064–2070. doi: 10.1194/jlr.M600233-JLR200. [DOI] [PubMed] [Google Scholar]
- 29.Bi N., Yan S.K., Li G.P., Yin Z.N., Chen B.S. A single nucleotide polymorphism -1131T>C in the apolipoprotein A5 gene is associated with an increased risk of coronary artery disease and alters triglyceride metabolism in Chinese. Mol. Genet. Metab. 2004;83:280–286. doi: 10.1016/j.ymgme.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 30.Dallongeville J., Cottel D., Montaye M., Codron V., Amouyel P., Helbecque N. Impact of APOA5/A4/C3 genetic polymorphisms on lipid variables and cardiovascular disease risk in French men. Int. J. Cardiol. 2006;106:152–156. doi: 10.1016/j.ijcard.2004.10.065. [DOI] [PubMed] [Google Scholar]
- 31.Wijsman E.M. The role of large pedigrees in an era of high-throughput sequencing. Hum. Genet. 2012;131:1555–1563. doi: 10.1007/s00439-012-1190-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jarvik G.P., Brunzell J.D., Austin M.A., Krauss R.M., Motulsky A.G., Wijsman E. Genetic predictors of FCHL in four large pedigrees. Influence of ApoB level major locus predicted genotype and LDL subclass phenotype. Arterioscler. Thromb. 1994;14:1687–1694. doi: 10.1161/01.atv.14.11.1687. [DOI] [PubMed] [Google Scholar]
- 33.Goldstein J.L., Schrott H.G., Hazzard W.R., Bierman E.L., Motulsky A.G. Hyperlipidemia in coronary heart disease. II. Genetic analysis of lipid levels in 176 families and delineation of a new inherited disorder, combined hyperlipidemia. J. Clin. Invest. 1973;52:1544–1568. doi: 10.1172/JCI107332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goldstein J.L., Dana S.E., Brunschede G.Y., Brown M.S. Genetic heterogeneity in familial hypercholesterolemia: evidence for two different mutations affecting functions of low-density lipoprotein receptor. Proc. Natl. Acad. Sci. USA. 1975;72:1092–1096. doi: 10.1073/pnas.72.3.1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Breslow J.L. Genetics of lipoprotein abnormalities associated with coronary artery disease susceptibility. Annu. Rev. Genet. 2000;34:233–254. doi: 10.1146/annurev.genet.34.1.233. [DOI] [PubMed] [Google Scholar]
- 36.Ford E.S., Li C., Zhao G., Pearson W.S., Mokdad A.H. Hypertriglyceridemia and its pharmacologic treatment among US adults. Arch. Intern. Med. 2009;169:572–578. doi: 10.1001/archinternmed.2008.599. [DOI] [PubMed] [Google Scholar]
- 37.Austin M.A., McKnight B., Edwards K.L., Bradley C.M., McNeely M.J., Psaty B.M., Brunzell J.D., Motulsky A.G. Cardiovascular disease mortality in familial forms of hypertriglyceridemia: A 20-year prospective study. Circulation. 2000;101:2777–2782. doi: 10.1161/01.cir.101.24.2777. [DOI] [PubMed] [Google Scholar]
- 38.Chait A., Brunzell J.D. Chylomicronemia syndrome. Adv. Intern. Med. 1992;37:249–273. [PubMed] [Google Scholar]
- 39.Warnick G.R. Enzymatic methods for quantification of lipoprotein lipids. Methods Enzymol. 1986;129:101–123. doi: 10.1016/0076-6879(86)29064-3. [DOI] [PubMed] [Google Scholar]
- 40.US Dept of Health and Human Services . Volume 1. National Institutes of Health; Washington, DC: 1980. (The Lipid Research Clinics’ Population Studies Data Book). [Google Scholar]
- 41.Rosenthal E.A., Ronald J., Rothstein J., Rajagopalan R., Ranchalis J., Wolfbauer G., Albers J.J., Brunzell J.D., Motulsky A.G., Rieder M.J. Linkage and association of phospholipid transfer protein activity to LASS4. J. Lipid Res. 2011;52:1837–1846. doi: 10.1194/jlr.P016576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wijsman E.M., Rothstein J.H., Igo R.P., Jr., Brunzell J.D., Motulsky A.G., Jarvik G.P. Linkage and association analyses identify a candidate region for apoB level on chromosome 4q32.3 in FCHL families. Hum. Genet. 2010;127:705–719. doi: 10.1007/s00439-010-0819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Keating B.J., Tischfield S., Murray S.S., Bhangale T., Price T.S., Glessner J.T., Galver L., Barrett J.C., Grant S.F.A., Farlow D.N. Concept, design and implementation of a cardiovascular gene-centric 50 k SNP array for large-scale genomic association studies. PLoS ONE. 2008;3:e3583. doi: 10.1371/journal.pone.0003583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ng S.B., Turner E.H., Robertson P.D., Flygare S.D., Bigham A.W., Lee C., Shaffer T., Wong M., Bhattacharjee A., Eichler E.E. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.De La Vega F.M., Dailey D., Ziegle J., Williams J., Madden D., Gilbert D.A. New generation pharmacogenomic tools: a SNP linkage disequilibrium Map, validated SNP assay resource, and high-throughput instrumentation system for large-scale genetic studies. Biotechniques. 2002;(Suppl):48–50. 52, 54. [PubMed] [Google Scholar]
- 46.Lin C.H., Yeakley J.M., McDaniel T.K., Shen R. Medium- to high-throughput SNP genotyping using VeraCode microbeads. Methods Mol. Biol. 2009;496:129–142. doi: 10.1007/978-1-59745-553-4_10. [DOI] [PubMed] [Google Scholar]
- 47.Heath S.C. Markov chain Monte Carlo segregation and linkage analysis for oligogenic models. Am. J. Hum. Genet. 1997;61:748–760. doi: 10.1086/515506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gagnon F., Jarvik G.P., Badzioch M.D., Motulsky A.G., Brunzell J.D., Wijsman E.M. Genome scan for quantitative trait loci influencing HDL levels: evidence for multilocus inheritance in familial combined hyperlipidemia. Hum. Genet. 2005;117:494–505. doi: 10.1007/s00439-005-1338-4. [DOI] [PubMed] [Google Scholar]
- 49.Yu, D. (2003). Testing the robustness of Markov chain Monte Carlo segregation and linkage analysis when normality assumptions are violated. PhD thesis, University of Washington, Seattle, WA.
- 50.Wijsman E.M., Daw E.W., Yu C.E., Payami H., Steinbart E.J., Nochlin D., Conlon E.M., Bird T.D., Schellenberg G.D. Evidence for a novel late-onset Alzheimer disease locus on chromosome 19p13.2. Am. J. Hum. Genet. 2004;75:398–409. doi: 10.1086/423393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gagnon F., Jarvik G.P., Motulsky A.G., Deeb S.S., Brunzell J.D., Wijsman E.M. Evidence of linkage of HDL level variation to APOC3 in two samples with different ascertainment. Hum. Genet. 2003;113:522–533. doi: 10.1007/s00439-003-1006-5. [DOI] [PubMed] [Google Scholar]
- 52.Goldberg I.J., Eckel R.H., McPherson R. Triglycerides and heart disease: still a hypothesis? Arterioscler. Thromb. Vasc. Biol. 2011;31:1716–1725. doi: 10.1161/ATVBAHA.111.226100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Johnson A.D., Handsaker R.E., Pulit S.L., Nizzari M.M., O’Donnell C.J., de Bakker P.I. SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics. 2008;24:2938–2939. doi: 10.1093/bioinformatics/btn564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Abecasis G.R., Altshuler D., Auton A., Brooks L.D., Durbin R.M., Gibbs R.A., Hurles M.E., McVean G.A., 1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sherry S.T., Ward M.H., Kholodov M., Baker J., Phan L., Smigielski E.M., Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cooper G.M., Stone E.A., Asimenos G., Green E.D., Batzoglou S., Sidow A., NISC Comparative Sequencing Program Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15:901–913. doi: 10.1101/gr.3577405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Almasy L., Blangero J. Multipoint quantitative-trait linkage analysis in general pedigrees. Am. J. Hum. Genet. 1998;62:1198–1211. doi: 10.1086/301844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Davydov E.V., Goode D.L., Sirota M., Cooper G.M., Sidow A., Batzoglou S. Identifying a high fraction of the human genome to be under selective constraint using GERP++ PLoS Comput. Biol. 2010;6:e1001025. doi: 10.1371/journal.pcbi.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pagliarini D.J., Calvo S.E., Chang B., Sheth S.A., Vafai S.B., Ong S.E., Walford G.A., Sugiana C., Boneh A., Chen W.K. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pontius J.U., Wagner L., Schuler G.D. UniGene: a unified view of the transcriptome. In: McEntyre J., Ostell J., editors. The NCBI Handbook. National Center for Biotechnology Information; Bethesda, MD: 2003. [Google Scholar]
- 63.Siepel A., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Grantham R. Amino acid difference formula to help explain protein evolution. Science. 1974;185:862–864. doi: 10.1126/science.185.4154.862. [DOI] [PubMed] [Google Scholar]
- 65.Kim P.S., Baldwin R.L. Specific intermediates in the folding reactions of small proteins and the mechanism of protein folding. Annu. Rev. Biochem. 1982;51:459–489. doi: 10.1146/annurev.bi.51.070182.002331. [DOI] [PubMed] [Google Scholar]
- 66.Blackwood E.M., Eisenman R.N. Max: a helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science. 1991;251:1211–1217. doi: 10.1126/science.2006410. [DOI] [PubMed] [Google Scholar]
- 67.Kretzner L., Blackwood E.M., Eisenman R.N. Myc and Max proteins possess distinct transcriptional activities. Nature. 1992;359:426–429. doi: 10.1038/359426a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.