

Abstract
Background
In Saccharomyces cerevisiae, Rad59 is required for multiple homologous recombination mechanisms and viability in DNA replication-defective rad27 mutant cells. Recently, four rad59 missense alleles were found to have distinct effects on homologous recombination that are consistent with separation-of-function mutations. The rad59-K166A allele alters an amino acid in a conserved α-helical domain, and, like the rad59 null allele diminishes association of Rad52 with double-strand breaks. The rad59-K174A and rad59-F180A alleles alter amino acids in the same domain and have genetically similar effects on homologous recombination. The rad59-Y92A allele alters a conserved amino acid in a separate domain, has genetically distinct effects on homologous recombination, and does not diminish association of Rad52 with double-strand breaks.
Results
In this study, rad59 mutant strains were crossed with a rad27 null mutant to examine the effects of the rad59 alleles on the link between viability, growth and the stimulation of homologous recombination in replication-defective cells. Like the rad59 null allele, rad59-K166A was synthetically lethal in combination with rad27. The rad59-K174A and rad59-F180A alleles were not synthetically lethal in combination with rad27, had effects on growth that coincided with decreased ectopic gene conversion, but did not affect mutation, unequal sister-chromatid recombination, or loss of heterozygosity. The rad59-Y92A allele was not synthetically lethal when combined with rad27, stimulated ectopic gene conversion and heteroallelic recombination independently from rad27, and was mutually epistatic with srs2. Unlike rad27, the stimulatory effect of rad59-Y92A on homologous recombination was not accompanied by effects on growth rate, cell cycle distribution, mutation, unequal sister-chromatid recombination, or loss of heterozygosity.
Conclusions
The synthetic lethality conferred by rad59 null and rad59-K166A alleles correlates with their inhibitory effect on association of Rad52 with double-strand breaks, suggesting that this may be essential for rescuing replication lesions in rad27 mutant cells. The rad59-K174A and rad59-F180A alleles may fractionally reduce this same function, which proportionally reduced repair of replication lesions by homologous recombination and growth rate. In contrast, rad59-Y92A stimulates homologous recombination, perhaps by affecting association of replication lesions with the Rad51 recombinase. This suggests that Rad59 influences the rescue of replication lesions by multiple recombination factors.
Keywords: Homologous recombination, Saccharomyces cerevisiae, DNA replication, Genome stability, Loss of heterozygosity
Background
In Saccharomyces cerevisiae, defective DNA replication stimulates homologous recombination (HR), suggesting that the lesions that accumulate following replication failure are substrates for HR [1-11]. Rad27 is a structure-specific endonuclease [12] required for completion of lagging strand synthesis [13], and has also been implicated in base excision repair [14], and double-strand break repair by non-homologous end joining [15]. Loss of Rad27 leads to accumulation of single-stranded gaps or nicks on daughter DNA strands [2,16]. Collision of replication forks with these lesions results in fork collapse and generation of double-strand breaks (DSB) [8,17] that can stimulate HR. Importantly, concomitant loss of Rad27 and components of the HR apparatus leads to synthetic lethality [18-20]. These observations implicate HR in repair of DSBs that accumulate in the absence of Rad27. Failure to repair DSBs leads to chromosome loss [21] that is greatly stimulated in rad27 null mutant cells [8], suggesting that the essential role for the HR apparatus in rad27 mutants may be prevention of lethal levels of chromosome loss.
RAD59 encodes a protein that augments the ability of Rad52, the central HR protein in yeast [22,23], to anneal complementary DNA strands in vitro[24], and both are required for viability in rad27 null mutant cells [19,20]. RAD59 and RAD52 are also required to repair DSBs by single-strand annealing (SSA) [21,25-28], and HR between inverted repeats by an annealing-dependent template switch at stalled replication forks [29-31]. Since RAD59 exerts much of its effect on HR with RAD52[21,32,33], the function of RAD59 required in the absence of RAD27 may be in collaboration with RAD52.
The purpose of the current study was to explore the function of RAD59 required for the viability of rad27 null mutant cells. We investigated how four rad59 mutations previously characterized with respect to their effects on SSA [21,27], affected survivorship when combined with a rad27 null mutation. We found that rad59-K166A, which alters an amino acid in a conserved, putative α-helical domain [27,34,35], was synthetically lethal in combination with rad27. Because rad59-K166A diminishes association of Rad52 with DSBs [21], this may be a function required for the viability of rad27 null mutant cells. The rad59-K174A and rad59-F180A mutations, which alter amino acids in the same α-helical domain, and have genetically similar effects on SSA [21], were not synthetically lethal with rad27, but resulted in distinct effects on growth that correlated with their degree of inhibition of HR. This strongly implicates RAD59-dependent HR as a requirement for viability in rad27 null mutant cells. The rad59-Y92A mutation, which alters an amino acid in a separate, conserved loop domain and confers genetically distinct effects on SSA [27,34] was not synthetically lethal with rad27, and had a stimulatory effect on HR. This effect was genetically equivalent to that of a null allele of SRS2, which encodes a helicase that disassembles Rad51-DNA filaments [36,37], suggesting that Rad59 may affect association of Rad51 with replication lesions. The distinct effects of the rad59 alleles suggest that Rad59 possesses multiple, discrete roles in responding to the consequences of dysfunctional replication.
Results
The rad59 mutant alleles display distinct effects on survival and growth in cells defective for lagging strand synthesis
To further explore the function of RAD59 required for viability in rad27 null mutant cells, the effects of combining the rad27::LEU2 allele with the various rad59 alleles were determined by examining their ability to yield viable spores upon co-segregation in genetic crosses. The various RAD27/rad27::LEU2 RAD59/rad59 double heterozygotes were sporulated and tetrads dissected onto rich medium (Figure 1). As observed previously, the rad27::LEU2 and rad59::LEU2 alleles did not appear together in any of the colonies arising from the spores, consistent with synthetic lethality [19,20]. The rad59-K166A allele, which alters a conserved lysine in the region of Rad59 that corresponds to the α-helical domain of the β − β − β − α motif of human Rad52 (Additional file 1: Figure S1) [27,34,35] displayed the same failure to appear with the rad27::LEU2 allele, indicative of synthetic lethality.
Figure 1.
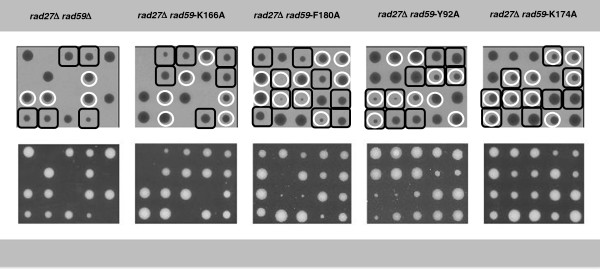
The rad59 mutant alleles have distinct effects on survival in cells that are defective for lagging strand synthesis. Diploid strains heterozygous at the RAD27 (rad27::LEU2/RAD27) and RAD59 (rad59/RAD59) loci were sporulated and tetrads dissected onto YPD medium. The resulting colonies were examined after 72 h of growth at 30°. Colonies from five representative tetrads from each strain are displayed. The genotype of each colony was determined by PCR as described in the Methods. In the inverted image, colonies possessing a rad27::LEU2 allele are boxed in black, and those possessing a rad59 allele are circled in white.
The rad59-K174A and rad59-F180A alleles alter conserved amino acids in the same putative α-helical domain as rad59-K166A but were able to form viable spores upon segregation with rad27::LEU2 (Figure 1). Doubling time of the rad27::LEU2 rad59-F180A double mutant was a statistically significant (p = 0.045) 24% longer than that observed for the rad27 single mutant, which correlated with a ratio of G1 to S + G2/M cells that was a statistically significant (p = 0.0031) 2.6-fold lower (Figure 2; Additional file 1: Table S2). In contrast, doubling time of the rad27::LEU2 rad59-K174A double mutant was not significantly different from that of the rad27::LEU2 single mutant (p = 0.71) (Table 1; Additional file 1: Table S2).
Figure 2.

The rad59 mutant alleles have distinct effects on cell cycle distribution in rad27::LEU2 mutant cells. Wild-type, single and double mutant strains were grown to mid-log phase at 30°, fixed, stained with propidium iodide, and submitted to flow cytometric analysis as described in the Methods. (A) Cell cycle profiles for wild-type and rad59 mutant strains. (B) Cell cycle profiles for rad27 single and rad27 rad59 double mutants. The distribution of cells with 1n and 2n DNA content in representative cultures of each strain are depicted. (C) Cell cycle distribution for wild-type and mutant strains. Median ratios of G1 to S + G2/M cells from a minimum of five independent cultures are indicated for each strain, and 95% confidence intervals are plotted.
Table 1.
Doubling times in wild-type and mutant haploid cells
| Genotype | Doubling time (min) | 95% confidence interval |
|---|---|---|
| Wild-type |
111 |
99, 120 |
|
rad59-Y92A |
119 |
97, 124 |
|
rad59-K174A |
131 |
111, 147 |
|
rad59-F180A |
112 |
99, 128 |
|
rad27::LEU2 |
164 |
137, 180 |
|
rad27::LEU2 rad59-Y92A |
176 |
136, 195 |
|
rad27::LEU2 rad59-K174A |
153 |
126, 177 |
| rad27::LEU2 rad59-F180A | 205 | 183, 230 |
Doubling times of freshly dissected segregants were determined as described in the Methods. Displayed for each genotype is the median doubling time and 95% confidence interval, determined from at least ten independent cultures.
The rad59-Y92A allele alters a conserved amino acid in another region of extensive conservation with Rad52 (Additional file 1: Figure S1) [27,34], and was observed to yield viable spores upon segregation with rad27::LEU2 (Figure 1). While the colonies derived from the rad27::LEU2 rad59-Y92A double mutant spores sometimes appeared smaller than the rad27::LEU2 single mutant colonies on dissection plates, neither the doubling times (p = 0.707) (Table 1; Additional file 1: Table S2), nor the ratios of G1 to S + G2/M cells (p = 0.60) (Figure 2, Additional file 1: Table S2) were significantly different for the rad27::LEU2 single and rad27::LEU2 rad59-Y92A double mutant strains. This suggests that germination of rad27::LEU2 rad59-Y92A double mutant spores may sometimes take longer than rad27::LEU2 single mutant spores. We did not observe significant effects of the tested rad59 missense alleles on doubling time (p > 0.15) (Table 1; Additional file 1: Table S2), or cell cycle distribution (p > 0.50) (Figure 2; Additional file 1: Table S2) in cells that possessed a wild-type RAD27 gene. Since all four rad59 missense mutations support steady-state levels of Rad59 that are comparable to wild-type [27], their effects on viability and growth when combined with rad27::LEU2 cannot be attributed to changes in the level of Rad59 in the cell. Altogether, these observations suggest that RAD59 plays a critical role in determining the growth characteristics of cells defective for lagging strand synthesis.
The rad59 alleles affect a RAD51-dependent mechanism for repairing replication lesions
The central strand exchange factor, Rad51 [38], is often required for mechanisms of HR that require RAD59, including those involved in spontaneous HR between inverted and unlinked repeat sequences [39,40]. Like RAD59, an intact RAD51 gene is necessary for viability in rad27::LEU2 mutant cells [18-20], suggesting that RAD51-dependent HR plays a critical role in responding to replication lesions. Accordingly, loss of RAD27 results in increases in HR events that require RAD51[18]. We used an assay that measures spontaneous ectopic gene conversion involving unlinked, mutant alleles of the SAM1 gene [41] to examine effects of the rad27::LEU2 mutation on HR in haploid strains (Figure 3A). Loss of RAD27 resulted in a dramatic, 4,700-fold increased rate of ectopic gene conversion (Figure 3B; Additional file 1: Table S2), indicating that accumulation of replication lesions can greatly stimulate HR between unlinked sequences.
Figure 3.

The rad59 mutant alleles have distinct effects on gene conversion between un-linked repetitive elements in haploid strains. (A) The spontaneous ectopic gene conversion system: Haploid strains containing a sam1-∆Bgl II-HOcs allele at the SAM1 locus on chromosome XII, a sam1-∆Sal I allele at the HIS3 locus on chromosome XV, and the sam2::HIS3 allele at the SAM2 locus on chromosome IV (not pictured) were grown to saturation in YPD supplemented with AdoMet, and plated onto medium lacking AdoMet to select for cells in which a recombination event generates a functional SAM1 gene and an AdoMet prototrophic cell. The opposite orientations of the sam1 alleles relative to their centromeres prevents the isolation of single crossovers. Only conversions of the sam1-∆Bgl II-HOcs allele to wild-type are observed due to the absence of a promoter for the sam1-∆Sal I allele. The sam2::HIS3 allele is missing sufficient information to recombine with sam1-∆Bgl II-HOcs. Black bars indicate the positions of the mutations. (B) Rates of ectopic gene conversion in wild-type and single mutant strains. Rates were determined from a minimum of 10 independent cultures as described in the Methods. Fold decreases (−) and increases (+) from wild-type are indicated in boxes. (C) Rates of ectopic gene conversion in rad27 rad59 double mutant strains. (D) Rates of ectopic gene conversion in rad51::LEU2 and srs2::TRP1 single mutant, and rad51::LEU2 rad59-Y92A and srs2::TRP1 rad59-Y92A double mutant strains.
The robust stimulatory effect of the loss of the RAD27 gene on ectopic gene conversion suggested that it could be used for examining the relationship between HR, and growth in the viable rad27 rad59 double mutants. As observed previously [40], the rad59::LEU2 mutation conferred a statistically significant 2.7-fold reduction in the rate of ectopic gene conversion (Figure 3B; Additional file 1: Table S2), confirming that RAD59 plays a role in spontaneous HR between unlinked repeats. While neither the rad59-K174A nor rad59-F180A mutations had significant effects on their own, they led to significant, 3.1- and 9.3-fold reductions in the stimulatory effect of the rad27::LEU2 allele in the rad27::LEU2 rad59-K174A and rad27::LEU2 rad59-F180A double mutants (Figure 3C; Additional file 1: Table S2), suggesting that they confer defects in the utilization of replication lesions by HR.
In contrast to the rad59-K174A and rad59-F180A mutations, the rad59-Y92A mutation caused an 86-fold increased rate of spontaneous ectopic gene conversion (Figure 3B; Additional file 1: Table S2), and, when combined with the rad27::LEU2 mutation, stimulated the rate of ectopic gene conversion by a statistically significant 7.7-fold over that observed in the rad27::LEU2 single mutant (Figure 3B and C; Additional file 1: Table S2). The synergistically increased rate of ectopic gene conversion in the rad27::LEU2 rad59-Y92A double mutant is consistent with rad59-Y92A stimulating HR by a mechanism distinct from the accumulation of replication lesions that results from loss of RAD27.
The hyper-rec effects of the rad59-Y92A and srs2::TRP1 alleles are genetically equivalent
Previous work indicating that rad59-Y92A decreases spontaneous RAD51-independent HR between directly repeated sequences [27] suggests that the stimulation of ectopic gene conversion is not due to accumulation of recombinogenic lesions. Ectopic gene conversion requires Rad51 to work after lesion formation to catalyze the strand invasion that begins the interaction between unlinked sequences that will repair the lesion [40,42]. If stimulation of HR by rad59-Y92A is the result of changes subsequent to Rad51-DNA filament formation, loss of RAD51 should abolish the stimulatory effect. The rate of ectopic gene conversion in the rad51::LEU2 rad59-Y92A double mutant was reduced 50-fold from wild-type, which was nearly identical to the rate in rad51::LEU2 single mutant cells (Figure 3D; Additional file 1: Table S2). Therefore, stimulation by rad59-Y92A requires formation of Rad51-DNA filaments.
Like the rad59-Y92A mutation, a null allele of the SRS2 gene, which encodes a DNA helicase [43] that facilitates the disassembly of Rad51-DNA filaments [36,37], has been shown to stimulate spontaneous gene conversion between non-allelic sequences [44,45]. Consistent with this, we observed a 31-fold increased rate of spontaneous ectopic gene conversion in an srs2::TRP1 mutant (Figure 3D; Additional file 1: Table S2). As the effects of srs2::TRP1 and rad59-Y92A were similar we examined ectopic gene conversion in the srs2 rad59-Y92A double mutant and observed a 38-fold increase over wild-type that was not significantly different from the rates in the srs2::TRP1 or rad59-Y92A single mutants (Figure 3B and 3D; Additional file 1: Table S2). This indicates that rad59-Y92A and srs2::TRP1 are mutually epistatic.
rad59-Y92A is a dominant mutation that stimulates RAD51-dependent HR between homologs
RAD51-dependent HR between sister-chromatids and homologous chromosomes are thought to be the primary mechanisms for rescuing lesions, and supporting viability in rad27 mutant cells [18-20]. One form of inter-homolog HR that requires RAD51 is recombination between mutant alleles at the same locus, referred to as heteroallelic recombination [46]. Accordingly, the rate of spontaneous recombination between heteroalleles of the SAM2 gene was reduced 10.5-fold in the rad51::LEU2/rad51::LEU2 homozygote (Figure 4B; Additional file 1: Table S2). Consistent with its effect on ectopic gene conversion, loss of RAD27 increased the rate of heteroallelic recombination 2,400-fold, confirming that accumulation of replication lesions robustly stimulates heteroallelic recombination [18].
Figure 4.
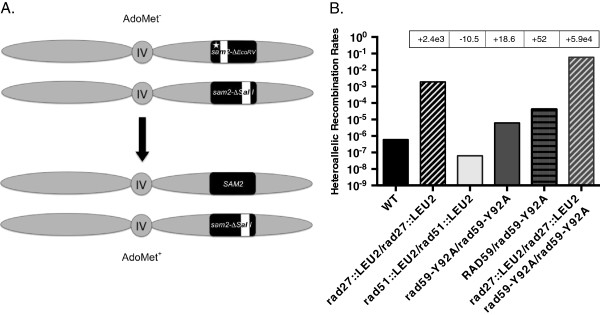
The rad59-Y92A mutation has a dominant, hyper-rec effect on hetero-allelic recombination in diploid strains. (A) The spontaneous hetero-allelic recombination system: Diploid strains possessing a sam2-∆EcoR V-HOcs allele at the SAM2 locus on one copy of chromosome IV, a sam2-∆SalI allele on the other copy, and a sam1::LEU2 allele at the SAM1 locus on both copies of chromosome XII (not pictured) were grown to saturation in YPD medium supplemented with AdoMet, and plated onto medium lacking AdoMet to select for cells in which a recombination event generates a functional SAM2 gene and an AdoMet prototrophic cell. Either reciprocal or non-reciprocal recombination events between sam2-∆EcoR V-HOcs and sam2-∆Sal I can generate AdoMet+ recombinants. The sam1::LEU2 allele is missing sufficient information to recombine with the sam2 alleles. The white bars indicate the positions of the sam2 mutations. (B) Rates of heteroallelic recombination in wild-type, heterozygous and homozygous mutant strains. Rates were determined from a minimum of 10 independent cultures as described in the Methods. Fold decreases (−) and increases (+) from wild-type indicated in boxes.
Similar to its effect on ectopic gene conversion, we observed that rad59-Y92A increased the rate of heteroallelic recombination by 19-fold (Figure 4B; Additional file 1: Table S2). Interestingly, the effect of rad59-Y92A was dominant with respect to RAD59, as the rate in the RAD59/rad59-Y92A heterozygote was not significantly different from that in the rad59-Y92A/rad59-Y92A homozygote. Like with ectopic gene conversion, combining the rad27::LEU2 and rad59-Y92A alleles in the rad27/rad27 rad59-Y92A/rad59-Y92A double homozygote had a synergistic effect on heteroallelic recombination, increasing the rate 25-fold over that observed in the rad27::LEU2/rad27::LEU2 homozygote. This astonishing, 59,000-fold increased rate of heteroallelic recombination corresponds to a median frequency of recombination where 85% of the surviving colonies are recombinants.
The rad59 alleles do not affect a variety of genome destabilizing processes stimulated by the accumulation of replication lesions
Loss of RAD27 stimulates a variety of mutagenic and clastogenic events [8,16,18,47,48]. The rate of spontaneous mutation at the CAN1 locus is greatly increased in rad27::LEU2 mutant cells [8,18,49]. Characterization of these mutations revealed that the majority are short duplications flanked by short, directly repeated sequences that may be created by multiple HR mechanisms [18]. Our data confirm the previous analyses as we observed a 50-fold increased rate of spontaneous mutation at the CAN1 locus in a rad27::LEU2 mutant (Table 2; Additional file 1: Table S2). In contrast, the rad59::LEU2, rad59-Y92A, rad59-K174A, and rad59-F180A alleles did not have significant effects on the rate of CAN1 mutation, nor did the missense alleles have significant effects when combined with the rad27::LEU2 allele.
Table 2.
Rates of mutation and unequal sister chromatid recombination in wild-type and mutant haploid strains
| Genotype | Mutation rate (10-7) | USCR rate (10-6) |
|---|---|---|
| Wild-type |
4.0 (3.8, 7.4) [1] |
1.0 (0.8, 1.2) [1] |
|
rad51::LEU2 |
n.d. |
1.4 (1.0, 1.8) [+1.4] |
|
rad59::LEU2 |
7.5 (6.6, 8.6) [+1.9] |
0.82 (0.43, 1.4) [-1.3] |
|
rad59-Y92A |
4.4 (3.9, 5.3) [+1.1] |
1.3 (1.1, 1.8) [+1.3] |
|
rad59-K174A |
3.2 (1.8, 5.5) [-1.3] |
1.1 (0.85, 2.1) [+1.1] |
|
rad59-F180A |
4.8 (4, 6.9) [+1.2] |
0.61 (0.47, 0.95) [-1.6] |
|
rad27::LEU2 |
200 (90, 590) [+50] |
47 (39, 100) [+47] |
|
rad27::LEU2 rad59-Y92A |
220 (60, 510) [+55] |
39 (25, 99) [+39] |
|
rad27::LEU2 rad59-K174A |
130 (110, 190) [+32.5] |
38 (33, 53) [+38] |
| rad27::LEU2 rad59-F180A | 190 (110, 500) [+47.5] | 60 (49, 120) [+60] |
Rates of CAN1 mutation or USCR were determined from at least 10 independent cultures as described in the Methods. The 95% confidence intervals are in parentheses. Fold decreases (−) and increases (+) from wild-type are in brackets. n.d. – not determined.
Loss of RAD27 has been previously observed to strongly stimulate unequal sister chromatid recombination (USCR) (Additional file 1: Figure S2) [8,50]. We observed a 47-fold increased rate of USCR in rad27::LEU2 cells (Table 2; Additional file 1: Table S2), confirming the previous results, while loss of RAD51 had no significant effect. The rad59::LEU2, rad59-Y92A, rad59-K174A, and rad59-F180A alleles did not have significant effects on the rate of USCR, nor did the missense mutations have effects in combination with rad27::LEU2, suggesting that RAD59 does not influence this mechanism of genome rearrangement.
Disrupting lagging strand synthesis by imposing a defect in the processivity of Pol δ, or loss of RAD27, was shown previously to substantially increase rates of loss of heterozygosity (LOH) by chromosome loss, and HR between homologs [2,8,10,11,18]. In the present analysis, LOH was examined in diploid strains by simultaneously monitoring changes in the genetic state at three loci on chromosome V (HXT13, CAN1 and HOM3) in order to separately determine rates of chromosome loss (reduction to hemizygosity at all three loci), terminal LOH (homozygosity at HXT13 and CAN1), and interstitial LOH (homozygosity at CAN1) (Additional file 1: Figure S3; Table 3; Additional file 1: Table S2). Rates of all three events increased substantially in rad27::LEU2/rad27::LEU2 homozygotes; chromosome loss increased 12-fold, terminal LOH increased 37-fold, and interstitial LOH increased 11-fold, strongly suggesting that replication lesions stimulate HR between homologs when they are repaired, and chromosome loss when they are not. Interestingly, we observed an 18-fold increase in the rate of chromosome loss in rad51::LEU2/rad51::LEU2 homozygotes, consistent with a requirement for RAD51 in the rescue of broken chromosomes. In contrast, loss of RAD51 did not have significant effects on interstitial LOH or terminal LOH, indicating that these inter-chromosomal HR events do not require Rad51.
Table 3.
Rates of loss of heterozygosity in wild-type and mutant diploid strains
| Genotype | ILOH rate (10-5) | TLOH rate (10-4) | CL rate (10-5) |
|---|---|---|---|
| Wild-type |
2.5 (2.1, 3.1) [1] |
0.92 (0.62, 1.2) [1] |
3.0 (2.5, 3.9) [1] |
|
rad51::LEU2/rad51::LEU2 |
1.2 (0.92, 2.5) [−2] |
1.3 (0.38, 2) [+1.4] |
54 (19, 64) [+18] |
|
rad59::LEU2/rad59::LEU2 |
1.8 (1.2, 2.9) [−1.4] |
1.4 (1.1, 1.9) [+1.5] |
6.2 (5.8, 10.2) [+2] |
|
rad59-Y92A/rad59-Y92A |
3.2 (2.7, 4.8) [+1.3] |
0.95 (0.83, 1.5) [1] |
2.5 (2.0, 3.6) [−1.2] |
|
rad59-K174A/rad59-K174A |
2.0 (1.3, 3.5) [−1.3] |
0.76 (0.40, 1.1) [−1.2] |
5.6 (2.9, 8.4) [+1.9] |
|
rad59-F180A/rad59-F180A |
3.8 (3.1, 5.1) [+1.5] |
0.82 (0.63, 1.7) [−1.1] |
3.0 (1.5, 7.9) [1] |
|
rad27::LEU2/rad27::LEU2 |
28 (25, 64) [+11] |
34 (24, 47) [+37] |
38 (29, 54) [+13] |
|
rad27::LEU2/rad27::LEU2 rad59-Y92A/rad59-Y92A |
28 (13, 56) [+11] |
36 (17, 50) [+39] |
29 (23, 74) [+9.7] |
|
rad27::LEU2/rad27::LEU2 rad59-K174A/rad59-K174A |
26 (22, 55) [+10] |
33 (24, 39) [+36] |
32 (18, 48) [+11] |
| rad27::LEU2/rad27::LEU2 rad59-F180A/rad59-F180A | 52 (29, 76) [+21] | 35 (22, 57) [+38] | 57 (18,124) [+19] |
Rates of interstitial LOH (ILOH), terminal LOH (TLOH), and chromosome loss (CL) from a minimum of 12 independent cultures were determined as described in the Methods. The 95% confidence intervals are in parentheses. Fold decreases (−) and increases (+) from wild-type are in brackets.
As observed above for mutation and USCR (Table 2; Additional file 1: Table S2), the rad59-Y92A, rad59-K174A, and rad59-F180A alleles had no significant effect on the rates of interstitial LOH, terminal LOH, and chromosome loss in the rad59/rad59 single mutants, or in the double mutant combinations with the rad27::LEU2 allele (Table 3; Additional file 1: Table S2). Similarly, rad59::LEU2 had no significant effect on the rates of interstitial LOH and terminal LOH, but conferred a small (two-fold), statistically significant increase in chromosome loss. These data suggest that RAD59 has little influence on these mechanisms of LOH.
Discussion
We have explored the role of RAD59 in mediating responses to DNA lesions that accumulate in rad27::LEU2 mutant cells, and found that it supports multiple, genetically separable functions. Like the rad59::LEU2 allele, we found that the rad59-K166A allele, which alters a lysine in a conserved, putative α-helical domain (Additional file 1: Figure S1B) [27,34,35], results in synthetic lethality when combined with the rad27::LEU2 allele (Figure 1). In previous experiments, we found that rad27::LEU2 mutant cells display a profusion of DSBs [8]. As both rad59::LEU2 and rad59-K166A substantially reduce association of Rad52 with DSBs [21], we speculate that a critical reduction in the association of Rad52 with the many DSBs in rad27::LEU2 rad59::LEU2 and rad27::LEU2 rad59-K166A double mutants may inhibit their rescue by HR, and results in a lethal level of chromosome loss. The rad59-F180A and rad59-K174A alleles, which change conserved residues in the same α-helical domain altered by rad59-K166A, may have incrementally less severe effects on association of Rad52 with DSBs. This may result in their serially reduced inhibition of repair of replication-induced DSBs by HR (Figure 3C; Additional file 1: Table S2) and commensurate effects on growth (Table 1; Additional file 1: Table S2) when combined with rad27. An accumulation of rad27::LEU2 rad59-F180A double mutant cells in the G2 phase of the cell cycle, as compared to rad27::LEU2 single mutant or rad27::LEU2 rad59-K174A double mutant cells is consistent with more deficient repair of replication-induced DSBs by HR (Figure 3). This further supports the notion that RAD59 promotes the survival of rad27::LEU2 mutant cells by facilitating the rescue of replication lesions by HR. Recently, RAD59 has been shown to be required for the viability of DNA ligase I-deficient mutants, verifying the requirement for this factor in accommodating to incomplete DNA replication [51].
In striking contrast to the other rad59 alleles, rad59-Y92A stimulated HR (Figure 3B; Figure 4B). This hyper-recombinogenic effect was distinct from that caused by rad27 as it was not accompanied by significant effects on doubling time (Table 1), cell cycle profile (Figure 2), mutation (Table 2), unequal sister chromatid exchange, or LOH (Table 3), suggesting that rad59-Y92A does not cause an accumulation of replication lesions. The observation that the stimulatory effect of rad59-Y92A was completely suppressed by a null allele of RAD51, and was mutually epistatic with a null allele of SRS2 (Figure 3D), suggests that rad59-Y92A may increase HR by increasing the stability of Rad51-DNA filaments, perhaps by changing its interaction with Rad51 (24). An increase in DSBs combined with an increase in the stability of Rad51 filaments at the DSBs may underlay the synergistically increased rates of HR observed in rad27 rad59-Y92A double mutants (Figures 3C and 4B). However, since Rad59 also interacts with RPA [52] and RSC [53], the increase in HR observed in rad59-Y92A mutant cells may also involve changes in additional processes.
While our results support a prominent role for RAD59-dependent HR in the repair of replication lesions in rad27::LEU2 mutants, HR mechanisms that do not depend on RAD59 were also strongly stimulated in rad27::LEU2 mutants. In particular, inter-chromosomal HR leading to interstitial and terminal LOH events was strongly stimulated by rad27, but was unaffected by the rad59 alleles (Table 3). Inter-chromosomal HR leading to LOH is thought to occur by break-induced replication (BIR) [54]. BIR has been proposed to utilize a single-ended DSB on one homolog to generate a replication fork-like intermediate with the unbroken homolog that may potentially proceed until reaching the end of the donor chromosome (Additional file 1: Figure S4A) [22]. In contrast, RAD59-dependent heteroallelic recombination is thought to utilize a double-ended DSB where both ends are rescued, either through concerted interactions with the unbroken homolog, or through the first end interacting with the homolog followed by the second end annealing with the first after gaining sequences copied from the unbroken homolog (Additional; file 1: Figure S4B). The stimulation of both mechanisms of HR between homologs suggests that loss of RAD27 leads to the accumulation of both single- and double-ended DSBs. DSBs may arise when the failure to remove flaps on the 5′ ends of Okazaki fragments leads to accumulation of nicks on newly replicated lagging strands (Figure 5). Persistence of these nicks into the subsequent cell cycle will leave discontinuities on the template for leading strand synthesis that will stall replication and form single-ended DSBs. If a second replication fork from an adjacent replicon collides with the first stalled fork, a double-ended DSB can arise. A genome-wide increase in replication-induced DSB formation, like that induced by many chemotherapeutic agents, would therefore require a robust response by the HR apparatus to prevent chromosome loss, potentially explaining the critical role of HR in determining sensitivity to these drugs in humans [55,56].
Figure 5.

Models for initiation of RAD51- and RAD59-dependent and –independent HR by defective lagging strand synthesis. 1.) Accumulation of daughter strand nicks in the absence of Rad27 nuclease causes replication fork stalling during the next S phase when the lagging strand becomes the template for leading strand synthesis and the replication fork encounters the discontinuity. 2.) The stalled fork is converted into an intact chromatid and a single-ended DSB. The single-ended DSB becomes a substrate for RAD51- and RAD59-independent HR mechanisms, such as interstitial and terminal LOH (Additional file 1: Figure S3). 3.) The replication fork from an upstream replicon converges with the previously stalled fork. 4.) Converged forks are converted into an intact chromatid and a double-ended DSB. The double-ended DSB becomes a substrate for RAD51- and RAD59-dependent HR mechanisms, such as ectopic gene conversion and heteroallelic recombination (Figures 3A and 4A).
Conclusions
RAD59 encodes one of several homologous recombination (HR) factors required for viability of budding yeast cells lacking the DNA replication factor, Rad27. This demonstrates that the HR apparatus is required to prevent the lethal effects of dysfunctional replication, but no link between a specific HR mechanism and survival has been previously established. In this analysis, we show that two rad59 alleles that diminish association of Rad52 with double-strand breaks are synthetically lethal with rad27, while two others coordinately reduce RAD51-dependent HR and growth, thus linking RAD51-dependent repair with survival. Another allele stimulates HR by stabilizing Rad51-DNA filaments. Therefore, Rad59 influences the repair of replication lesions by HR through its interactions with multiple HR factors. We speculate that the massive increase in replication failure genome-wide that results from loss of Rad27 may be similar to that caused by chemotherapeutic agents in human cells, potentially explaining why the HR apparatus is critical in determining sensitivity to these drugs.
Methods
Strains
All strains used in this study were isogenic and are listed in Additional file 1: Table S1. Standard techniques for yeast strain construction and growth were used [57]. Construction of the rad27::LEU2, rad51::LEU2, rad59::LEU2, rad59-Y92A, rad59-K166A, rad59-K174A, rad59-F180A and srs2::TRP1 alleles have been described previously [27,58-60]. The rad27::LEU2 allele can be followed in crosses by PCR, using the forward primer 5′-GCG TTG ACA GCA TAC ATT-3′, and reverse primer 5′-CGT ACA AAC CAA ATG CGG-3′. The rad59::LEU2 allele is followed by PCR using the forward primer 5′-GCC ACA GTT TGG CAA GGG-3′, and the reverse primer 5′-GGG TTT GTT GCC ATC TGC G-3′. The rad59 missense alleles were followed in crosses by allele-specific PCR [27]. Unique forward primers were used to detect rad59-Y92A (5′-GCT AAT GAA ACA TTC GGG GC-3′), rad59-K166A (5′-AAT GTT ATA ACA GGT CGA AAG C-3′), rad59-K174A (5′-AAG GGT TAC GTA GAG GAG AAG-3′), and rad59-F180A (5′-AAG AAG GCG TTA TTG AGC GC-3′). All allele-specific PCRs use the same reverse primer (5′-TAT ATA AGT ACG TGA GAT CTA TTT G-3′). Presence of the rad59-K174A allele is scored by digesting the PCR product with MseI restriction endonuclease. DNA was purified for PCR analysis using a standard method [61].
Synthetic lethality
Diploid yeast strains heterozygous for each of the rad59 alleles (rad59/RAD59) and the rad27::LEU2 allele (rad27::LEU2/RAD27) were sporulated and dissected. After 72 h, five representative tetrads from each diploid were selected. The presence of rad27 and rad59 mutant alleles in each of the colonies that arose from the spores was scored using PCR as described above.
Doubling time
At least 10, five-milliliter YPD (1% yeast extract, 2% peptone, 2% dextrose) cultures were inoculated with colonies arising from the spores of freshly dissected tetrads and grown overnight at 30°. These were sub-cultured into Klett tubes containing five milliliters of YPD medium that were incubated at 30° while shaking. Cell density was measured by monitoring culture turbidity with a Klett-Summerson colorimeter each hour over a 10 h period. Doubling times were calculated using a standard algorithm [62]. The 95% confidence intervals and Mann–Whitney values were determined using the Prism statistics package (GraphPad, La Jolla, CA).
Flow cytometry
At least five, five-milliliter YPD cultures were inoculated with colonies arising from freshly dissected tetrads and grown overnight at 30°. Overnight cultures were sub-cultured into five milliliters of YPD medium and grown to mid-log phase at 30° defined by growth curve using a Klett-Summerson colorimeter. Cells were processed for flow cytometry using the following adaptation of a published method [63]. The cell density was determined by hemacytometer count and aliquots containing 107 cells were pelleted, resuspended in 70% ice-cold ethanol, and fixed while rotating at 4° overnight. Fixed cells were pelleted, resuspended in 1 ml of citrate buffer (50 mM Na citrate, pH 7.2), and sonicated (Misonix 3000, Farmingdale, NY). Sonicated cells were pelleted, resuspended in citrate buffer and treated with 25 μl of 10 mg/ml RNase A, at 50° for one h, followed by treatment with 50 μl of 20 mg/ml Proteinase K and incubation at 50° for one h. Cells were pelleted and resuspended in 1 ml of citrate buffer, and either rotated overnight at 4°, or stained immediately by adding 16 μl of 1 mg/ml propidium iodide and rotating for 45 min at room temperature in the dark before processing by flow cytometry (Beckman Coulter CyAn ADP 9color, Miami FL). Fractions of cells in the G1, S and G2/M phases of the cell cycle were determined using FlowJo v.7.6.5 image processing software (Tree Star, Ashland, OR). The ratio of cells in G1 vs. S + G2/M were calculated for each trial and the median value for each strain used for comparing cell cycle distributions in different strains. The Mann–Whitney test was used to assess the statistical significance of differences between strains.
Spontaneous ectopic gene conversion
Spontaneous ectopic gene conversion in haploid strains was assayed as described previously [64], but using substrates described in a separate analysis [41]. All strains contained the sam1-ΔBgl II-HOcs allele at the SAM1 locus on chromosome XII, the sam1-ΔSal I allele adjacent to the HIS3 locus on chromosome XV, and a HIS3 gene replacing the SAM2 coding sequence at the SAM2 locus (sam2::HIS3) on chromosome IV. The sam1-∆Bgl II-HOcs allele has a 117 bp fragment of the MAT locus disrupting the Bgl II site in the SAM1 coding sequence, while the sam1-ΔSal I allele has a 4 bp insertion at the Sal I site [41]. The sam1-ΔSal I allele lacks a promoter, preventing conversion events at this locus from generating AdoMet+ recombinants. The sam1-∆Bgl II-HOcs and sam1-ΔSal I alleles are also in opposite orientations relative to their centromeres, preventing the isolation of single crossover recombinants.
At least ten freshly dissected haploid segregants of each strain were inoculated into five-milliliter YPD cultures supplemented with 100 μg/ml of S-adenosylmethionine (AdoMet) and grown to saturation at 30°. Appropriate dilutions of each culture were plated onto YPD + AdoMet plates to determine the number of viable cells, and onto YPD plates lacking AdoMet to determine the number of AdoMet prototrophic recombinants.
All rates were determined by the method of the median [65]. Rates and 95% confidence intervals were calculated as described previously [66].
Spontaneous hetero-allelic recombination
Rates of spontaneous hetero-allelic recombination were determined as for ectopic gene conversion except that different substrates were used in diploid cells. All strains contained the sam2-ΔEcoR V-HOcs allele at the SAM2 locus on one copy of chromosome IV, the sam2-ΔSal I allele on the other, and a LEU2 marker replacing the SAM1 coding sequence at the SAM1 locus on both copies of chromosome XII. The sam2-ΔEcoR V-HOcs allele has a 117 bp fragment of the MAT locus disrupting the EcoR V site, while the sam2-ΔSal I allele has a 4 bp insertion disrupting the Sal I site [41].
Mutation rate
Rates of mutation at the CAN1 locus were examined using a previously published assay [8,10,18]. At least ten freshly dissected segregants were used to inoculate one-milliliter YPD cultures that were grown to saturation at 30°. Appropriate dilutions were plated onto YPD to determine viability and synthetic medium lacking arginine but containing 60 μg/ml of canavanine to select for mutants.
Unequal sister chromatid recombination (USCR)
Rates of USCR were determined using a previously published assay [8,10,67]. At least ten freshly dissected segregants containing the USCE construct at the TRP1 locus on chromosome IV and the his3∆200 allele at the HIS3 locus on chromosome XV, were struck out to single colonies on YPD. After three days of growth at 30°, single colonies were used to inoculate one-milliliter YPD cultures, and grown to saturation at 30°. Appropriate dilutions were plated onto YPD to assess viability and onto medium lacking histidine to determine the number of histidine prototrophic recombinants.
Loss of heterozygosity (LOH)
Rates of spontaneous LOH by three different mechanisms were assessed using a previously published assay [8]. Freshly dissected haploid segregants containing either the hxt13::URA3, CAN1, and HOM3 alleles, or the HXT13, can1-100, and hom3-10 alleles on chromosome V were crossed and the resulting diploids struck out to single colonies on YPD. At least 12 independent colonies were inoculated into one-milliliter YPD liquid cultures and grown to saturation at 30°. Appropriate dilutions were plated onto YPD for viability and synthetic medium lacking arginine, but containing 60 μg/ml of canavanine to select for clones resistant to canavanine. After three days of growth at 30° canavanine-resistant (CanR) colonies were replica plated onto synthetic medium lacking either uracil or threonine to assay for the presence of the hxt13::URA3 (Ura+) and HOM3 (Thr+) alleles, respectively. Rates of interstitial LOH, terminal LOH, and CL were determined from the number of Ura+CanRThr+, Ura-CanRThr+, and Ura-CanRThr- recombinant colonies, respectively.
Modeling the Rad59 protein
The crystal structure of the N-terminus of human Rad52 [34] was obtained from the RSCB Protein Data Bank (http://www.rcsb.org/pdb/). This structure was imaged using the molecular modeling program, SYBYL, and the amino acids corresponding to those mutated in the rad59 missense alleles were identified, and highlighted.
Availability of supporting data
The data sets supporting the results of this article are included within the article and in Additional file 1.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
LL carried out the synthetic lethality experiments, LOH genetic studies, flow cytometric analysis, sequence alignment, designed the figures and tables, and drafted the manuscript. GM performed the growth, mutation and USCR rate studies. SO assisted with the synthetic lethality and LOH experiments. BF contributed to the LOH experiments. AB conceived of the study, designed and carried out the ectopic gene conversion and hetero-allelic recombination analyses, and helped draft the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Saccharomyces cerevisiae strains used in this study. Table S2. Summary of quantitative data. Figure S1. A. Multiple amino acid sequence alignment of ScRad59 with ScRad52 and HsRad52. B. Molecular modeling of the proteins encoded by the rad59 missense alleles demonstrates that Rad59-Y92A is in a different structural motif. Figure S2. The unequal sister chromatid recombination (USCR) assay for measuring spontaneous homologous recombination between sister chromatids in haploid yeast. Figure S3. The loss of heterozygosity assay for measuring spontaneous Rad51-independent homologous recombination. Figure S4. LOH is the recombination product of a single-ended DSB, whereas HAR results from repair of a double-ended DSB. A) LOH results from the repair of a single-ended DSB by HR. B) HAR results from the repair of a double-ended DSB by HR.
Contributor Information
Lauren C Liddell, Email: lliddell@coh.org.
Glenn M Manthey, Email: gmanthey@coh.org.
Shannon N Owens, Email: owenss@oxy.edu.
Becky XH Fu, Email: xuhua@stanford.edu.
Adam M Bailis, Email: abailis@coh.org.
Acknowledgements
We thank M. Boldin, M. Kalkum, R.-J. Lin, T. O’Connor, and J. Stark for stimulating discussions, and N. Pannunzio for comments on the manuscript. We would like to acknowledge the City of Hope Biostatistics and Bioinformatics, and Flow Cytometry Core Facilities for their assistance. This work was supported by a Morgan and Helen Chu graduate student fellowship to L.C.L, a summer undergraduate research fellowship from the Howard Hughes Medical Institute to S.N.O, a summer student fellowship from the Eugene and Ruth Roberts Summer Academy to B.X.H.F., and funds from the Beckman Research Institute of the City of Hope.
References
- Gordenin DA, Malkova AL, Peterzen A, Kulikov VN, Pavlov YI, Perkins E, Resnick MA. Transposon Tn5 excision in yeast: Influence of DNA polymerases α, δ, and ϵ and repair genes. Proc Natl Acad Sci U S A. 1992;89:3785–3789. doi: 10.1073/pnas.89.9.3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallen EA, Cross FR. Mutations in RAD27 define a potential link between G1 cyclins and DNA replication. Mol Cell Biol. 1995;15(8):4291–4302. doi: 10.1128/mcb.15.8.4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruskin B, Fink G. Mutations in POL1 increase the mitotic instability of tandem inverted repeats in Saccharomyces cerevisiae. Genetics. 1993;133:43–56. doi: 10.1093/genetics/134.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff DX, Boerger AL, Bertrand P, Filosi N, Gaida GM, Kane MF, Kolodner RD. Identification and characterization of Saccharomyces cerevisiae EXO1, a gene encoding an exonuclease that interacts with MSH2. Proc Natl Acad Sci U S A. 1997;94:7487–7492. doi: 10.1073/pnas.94.14.7487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H, Rothstein R. Holliday junctions accumulate in replication mutants via a RecA homolog-independent mechanism. Cell. 1997;90:87–96. doi: 10.1016/S0092-8674(00)80316-5. [DOI] [PubMed] [Google Scholar]
- Chen C, Kolodner R. Gross chromosomal rearrangements in Saccharomyces cerevisiae replication and recombination defective mutants. Nat Genet. 1999;23:81–85. doi: 10.1038/12687. [DOI] [PubMed] [Google Scholar]
- Pavlov YI, Shcherbakova PV, Kunkel TA. In vivo consequences of putative active site mutations in yeast DNA Polymerases a, ϵ, δ, and ζ. Genetics. 2001;159:47–64. doi: 10.1093/genetics/159.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro MS, Bi L, Bailis AM. A mutant allele of the transcription factor IIH helicase gene, RAD3, promotes loss of heterozygosity in response to a DNA replication defect in Saccharomyces cerevisiae. Genetics. 2007;176:1391–1402. doi: 10.1534/genetics.107.073056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesan RN, Treuting PM, Fuller ED, Goldsby RE, Norwood TH, Gooley TA, Ladiges WC, Preston BD, Loeb LA. Mutation at the polymerase active site of mouse DNA polymerase δ increases genomic instability and accerlerates tumorigenesis. Mol Cell Biol. 2007;27(21):7669–7682. doi: 10.1128/MCB.00002-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mito E, Mokhnatkin JV, Steele MC, Buettner VL, Sommer SS, Manthey GM, Bailis AM. Mutagenic and recombinagenic responses to defective DNA polymerase δ are facilitated by the Rev1 protein in pol3-t mutants of Saccharomyces cerevisiae. Genetics. 2008;179:1795–1806. doi: 10.1534/genetics.108.089821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli A, Cervelli T, Schiestl RH. Characterization of the hyperrecombination phenotype of the pol3-t mutation of Saccharomyces cerevisiae. Genetics. 2003;164:65–79. doi: 10.1093/genetics/164.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington JJ, Lieber MR. The characterization of a mammalian DNA structure-specific endonuclease. EMBO J. 1994;13(5):1235–1246. doi: 10.1002/j.1460-2075.1994.tb06373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Kao HI, Bambara RA. Flap endonuclease 1: A central component of DNA metabolism. Annu Rev Biochem. 2004;73:589–615. doi: 10.1146/annurev.biochem.73.012803.092453. [DOI] [PubMed] [Google Scholar]
- Wu X, Wang Z. Relationships between yeast Rad27 and Apn1 in response to apurinic/apyrimidinic (AP) sites in DNA. Nucleic Acids Res. 1999;27(4):956–962. doi: 10.1093/nar/27.4.956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng HM, Tomkinson AE. Processing and joining of DNA ends coordinated by interactions among Dnl4/Lif1, Pol4, and FEN-1. J Biol Chem. 2004;279(46):47580–47588. doi: 10.1074/jbc.M404492200. [DOI] [PubMed] [Google Scholar]
- Parenteau J, Wellinger RJ. Accumulation of single-stranded DNA and destabilization of telomeric repeats in yeast mutant strains carrying a deletion of RAD27. Mol Cell Biol. 1999;19(6):4143–4152. doi: 10.1128/mcb.19.6.4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowosielska A. Bacterial DNA repair genes and their eukaryotic homologues: 5. The role of recombination in DNA repair and genome stability. Acta Biochimica Polonica. 2007;54(3):483–494. [PubMed] [Google Scholar]
- Tishkoff DX, Filosi N, Gaida GM, Kolodner RD. A novel mutation avoidance mechanism dependent on S. cerevisiae RAD27 is distinct from DNA mismatch repair. Cell. 1997;88:253–263. doi: 10.1016/S0092-8674(00)81846-2. [DOI] [PubMed] [Google Scholar]
- Symington LS. Homologous recombination is required for the viability of rad27 mutants. Nucleic Acids Res. 1998;26(24):5589–5595. doi: 10.1093/nar/26.24.5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debrauwere H, Loeillet S, Lin W, Lopes J, Nicolas A. Links between replication and recombination in Saccharomyces cerevisiae: a hypersensitive requirement for homologous recombination in the absence of Rad27 activity. Proc Natl Acad Sci U S A. 2001;98(15):8263–8269. doi: 10.1073/pnas.121075598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannunzio NR, Manthey GM, Liddell LC, Fu BX, Roberts CM, Bailis AM. Rad59 regulates association of Rad52 with DNA double-strand breaks. Microbiology Open. 2012;1(3):285–297. doi: 10.1002/mbo3.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paques F, Haber JE. Multiple pathwyas of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Micro Mol Biol Rev. 1999;63(2):349–404. doi: 10.1128/mmbr.63.2.349-404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh BO, Symington LS. Recombination proteins in yeast. Annu Rev Genet. 2004;38:233–271. doi: 10.1146/annurev.genet.38.072902.091500. [DOI] [PubMed] [Google Scholar]
- Wu Y, Kantake N, Sugiyama T, Kowalczykowski SC. Rad51 protein controls Rad52-mediated DNA annealing. J Biol Chem. 2008;283(21):14883–14892. doi: 10.1074/jbc.M801097200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis AP, Symington LS. The yeast recombinational repair protein Rad59 interacts with Rad52 and stimulates single-strand annealing. Genetics. 2001;159:515–525. doi: 10.1093/genetics/159.2.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannunzio NR, Manthey GM, Bailis AM. RAD59 is required for efficient repair of simultaneous double-strand breaks resulting in translocations in Saccharomyces cerevisiae. DNA Repair (Amst) 2008;7(5):788–800. doi: 10.1016/j.dnarep.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannunzio NR, Manthey GM, Bailis AM. Rad59 and Rad1 cooperate in translocation formation by single-strand annealing in Saccharomyces cerevisiae. Curr Genet. 2010;56(1):87–100. doi: 10.1007/s00294-009-0282-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara N, Ira G, Haber JE. DNA length dependence of the single-strand annealing pathway and the role of Saccharomyces cerevisiae RAD59 in double-strand break repair. Mol Cell Biol. 2000;20(14):5300–5309. doi: 10.1128/MCB.20.14.5300-5309.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y, Symington LS. A Rad52 homolog is required for RAD51-independent mitotic recombination in Saccharomyces cerevisiae. Genes Dev. 1996;10(16):2025–2037. doi: 10.1101/gad.10.16.2025. [DOI] [PubMed] [Google Scholar]
- Cortes-Ledesma F, Tous C, Aguilera A. Different genetic requirements for repair of replication-born double-strand breaks by sister-chromatid recombination and break-induced replication. Nucleic Acids Res. 2007;35(19):6560–6570. doi: 10.1093/nar/gkm488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mott C, Symington LS. RAD51-independent inverted-repeat recombination by a strand-annealing mechanism. DNA Repair (Amst) 2011;10(4):408–415. doi: 10.1016/j.dnarep.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes-Ledesma F, Malagon F, Aguilera A. A novel yeast mutation, rad52-L89F, causes a specific defect in Rad51-independent recombination that correlates with a reduced ability of Rad52-L89F to interact with Rad59. Genetics. 2004;168:553–557. doi: 10.1534/genetics.104.030551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q, During L, de Mayolo AA, Lettier G, Lisby M, Erdeniz N, Mortensen UH, Rothstein R. Rad52 and Rad59 exhibit both overlapping and distinct functions. DNA Repair (Amst) 2007;6(1):27–37. doi: 10.1016/j.dnarep.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Kagawa W, Kurumizaka H, Ishitani R, Fukai S, Nureki O, Shibata T, Yokoyama S. Crystal structure of the homologous-pairing domain from the human Rad52 recombinase in the undecameric form. Mol Cell. 2002;10:359–371. doi: 10.1016/S1097-2765(02)00587-7. [DOI] [PubMed] [Google Scholar]
- Lloyd JA, McGrew DA, Knight KL. Identification of residues important for DNA binding in the full-length human Rad52 protein. J Mol Biol. 2005;345(2):239–249. doi: 10.1016/j.jmb.2004.10.065. [DOI] [PubMed] [Google Scholar]
- Veaute X, Jeusset J, Soustelle C, Kowalczykowski SC, Fabre F. The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature. 2003;423:309–312. doi: 10.1038/nature01585. [DOI] [PubMed] [Google Scholar]
- Antony E, Tomko EJ, Xiao Q, Krejci L, Lohman TM, Ellenberger T. Srs2 disassembles Rad51 filaments by a protein-protein interaction triggering ATP turnover and dissociation of Rad51 from DNA. Mol Cell. 2009;35(1):105–115. doi: 10.1016/j.molcel.2009.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung P. Catalysis of ATP-dependent homologous DNA pairing and strand exchange by yeast RAD51 protein. Science. 1994;265(5176):1241–1243. doi: 10.1126/science.8066464. [DOI] [PubMed] [Google Scholar]
- Bai Y, Davis AP, Symington LS. A novel allele of RAD52 that causes severe DNA repair and recombination deficiencies only in the absence of RAD51 or RAD59. Genetics. 1999;153:1117–1130. doi: 10.1093/genetics/153.3.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonovich Z, Liefshitz B, Steinlauf R, Kupiec M. Characterization of the role played by the RAD59 gene of Saccharoymces cerevisiae in ectopic recombination. Curr Genet. 1999;36:13–20. doi: 10.1007/s002940050467. [DOI] [PubMed] [Google Scholar]
- Bailis AM, Maines S, Negritto MT. The essential helicase gene RAD3 suppresses short-sequence recombination in Saccharomyces cerevisiae. Mol Cell Biol. 1995;15(5):3998–4008. doi: 10.1128/mcb.15.8.3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liefshitz B, Parket A, Maya R, Kupiec M. The role of DNA repair genes in recombination between repeated sequences in yeast. Genetics. 1995;140:1199–1211. doi: 10.1093/genetics/140.4.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong L, Klein HL. Purification and characterization of the SRS2 DNA helicase of the yeast Saccharomyces cerevisiae. J Biol Chem. 1993;268(2):1252–1259. [PubMed] [Google Scholar]
- Rong L, Palladino F, Aguilera A, Klein HL. The hyper-gene conversion hpr5-1 mutation of Saccharomyces cerervisiae is an allele of the SRS2/RADH gene. Genetics. 1991;127:75–85. doi: 10.1093/genetics/127.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palladino F, Klein HL. Analysis of mitotic and meiotic defects in Saccharomyces cerevisiae SRS2 DNA helicase mutants. Genetics. 1992;132(1):23–37. doi: 10.1093/genetics/132.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison DP, Hastings PJ. Characterization of the mutator mutation mut5-1. Mol Gen Genet. 1979;175(1):57–65. doi: 10.1007/BF00267856. [DOI] [PubMed] [Google Scholar]
- Lopes J, Ribeyre C, Nicolas A. Complex minisatellite rearrangements generated in the total or partial absence of Rad27/hFEN1 activity occur in a single generation and are Rad51 and Rad52 dependent. Mol Cell Biol. 2006;26(17):6675–6689. doi: 10.1128/MCB.00649-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freudenreich CH, Kantrow SM, Zakian VA. Expansion and length-dependent fragility of CTG repeats in yeast. Science. 1998;279(853):853–856. doi: 10.1126/science.279.5352.853. [DOI] [PubMed] [Google Scholar]
- Johnson RE, Kovvali GK, Prakash L, Prakash S. Role of yeast Rth1 nuclease and its homologs in mutation avoidance, DNA repair, and DNA replication. Curr Genet. 1998;34:21–29. doi: 10.1007/s002940050362. [DOI] [PubMed] [Google Scholar]
- Fasullo MT, Davis RW. Direction of chromosome rearrangements in Saccaromyces cerevisiae by use of his3 recombinational substrates. Mol Cell Biol. 1988;8(10):4370–4380. doi: 10.1128/mcb.8.10.4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HD, Becker J, Thu YM, Costanzo M, Koch EN, Smith S, Myers CL, Boone C, Bielinsky AK. Unligated Okazaki fragments induce PCNA ubiquitnation and a requirement for Rad59-dependent replication fork progression. PLoS One. 2013;8(6):e66379. doi: 10.1371/journal.pone.0066379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis AP, Symington LS. The Rad52-Rad59 complex interacts with Rad51 and replication protein A. DNA Repair (Amst) 2003;2:1127–1134. doi: 10.1016/S1568-7864(03)00121-6. [DOI] [PubMed] [Google Scholar]
- Oum J-H, Seong C, Kwon Y, Ji J-H, Sid A, Ramakrishnan S, Ira G, Malkova A, Sung P, Lee SE, Shim EY. RSC facilitates Rad59-dependent homologous recombination between sister chromatids by promoting cohesin loading at DNA double-strand breaks. Mol Cell Biol. 2011;31(19):3924–3937. doi: 10.1128/MCB.01269-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl TJ, Nickoloff JA. Rad51-independent interchromosomal double-strand break repair by gene conversion requires Rad52 but not Rad55, Rad57, or Dmc1. Mol Cell Biol. 2008;28(3):897–906. doi: 10.1128/MCB.00524-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolova T, Ensminger M, Lobrich M, Kaina B. Homologous recombination protects mammalian cells from replication-associated DNA double-strand breaks arising in response to methyl methanesulfonate. DNA Repair (Amst) 2010;9(10):1050–1063. doi: 10.1016/j.dnarep.2010.07.005. [DOI] [PubMed] [Google Scholar]
- Nikolova T, Hennekes F, Bhatti A, Kaina B. Chloroethylnitrosourea-induced cell death and genotoxicity: cell cycle dependence and the role of DNA double-strand breaks. HR and NHEJ. Cell Cycle. 2012;11(14):2606–2619. doi: 10.4161/cc.20862. [DOI] [PubMed] [Google Scholar]
- Sherman F, Fink F, Hicks J. Methods in Yeast Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1986. [Google Scholar]
- Schild D, Konforti B, Perez C, Gish W, Mortimer RK. Isolation and characterization of yeast DNA repair genes. I. Cloning of the RAD52 gene. Curr Genet. 1983;7:85–92. doi: 10.1007/BF00365631. [DOI] [PubMed] [Google Scholar]
- Schild D, Calderon IL, Contopoulo R, Mortimer RK. Cloning of yeast recombination repair genes and evidence that several are nonessential genes. New York: Alan R. Liss; 1983. [Google Scholar]
- Frank G, Qiu J, Somsouk M, Weng Y, Somsouk L, Nolan JP, Shen B. Partial functional deficiency of E160D flap endonuclease-1 mutant in vitro and in vivo is due to defective cleavage of DNA substrates. J Biol Chem. 1998;273(49):33064–33072. doi: 10.1074/jbc.273.49.33064. [DOI] [PubMed] [Google Scholar]
- Hoffman CS, Winston F. A ten-minute DNA preparation from yeast efficiently releases autonomous plasmids for transformation of Escherichia coli. Gene. 1987;57(2–3):267–272. doi: 10.1016/0378-1119(87)90131-4. [DOI] [PubMed] [Google Scholar]
- Singleton P. Bateria in Biology, Biotechnology, and Medicine. New York: John Wiley & Sons; 1995. [Google Scholar]
- Nash N, Tokiwa G, Anand S, Erickson K, Futcher AB. The WHI1+ gene of Saccharomyces cerevisiae tethers cell division to cell size and is a cyclin homolog. EMBO J. 1988;7(13):4335–4346. doi: 10.1002/j.1460-2075.1988.tb03332.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailis AM, Rothstein R. A defect in mismatch repair in Saccharomyces cerevisiae stimulates ectopic recombination between homeologous genes by an excision repair dependent process. Genetics. 1990;126:535–547. doi: 10.1093/genetics/126.3.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lea DE, Coulson CA. The distribution of the numbers of mutants in bacterial populations. J Genet. 1949;49:264–285. doi: 10.1007/BF02986080. [DOI] [PubMed] [Google Scholar]
- Spell RM, Jinks-Robertson S. Determination of mitotic recombination rates by fluctuation analysis in Saccaromyces cerevisiae. Methods Mol Biol. 2004;262:3–12. doi: 10.1385/1-59259-761-0:003. [DOI] [PubMed] [Google Scholar]
- Fasullo MT, Davis RW. Recombinational substrates designed to study recombination between unique and repetitive sequence in vivo. Proc Natl Acad Sci U S A. 1987;84:6215–6219. doi: 10.1073/pnas.84.17.6215. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Saccharomyces cerevisiae strains used in this study. Table S2. Summary of quantitative data. Figure S1. A. Multiple amino acid sequence alignment of ScRad59 with ScRad52 and HsRad52. B. Molecular modeling of the proteins encoded by the rad59 missense alleles demonstrates that Rad59-Y92A is in a different structural motif. Figure S2. The unequal sister chromatid recombination (USCR) assay for measuring spontaneous homologous recombination between sister chromatids in haploid yeast. Figure S3. The loss of heterozygosity assay for measuring spontaneous Rad51-independent homologous recombination. Figure S4. LOH is the recombination product of a single-ended DSB, whereas HAR results from repair of a double-ended DSB. A) LOH results from the repair of a single-ended DSB by HR. B) HAR results from the repair of a double-ended DSB by HR.