

Abstract
Congenital poikiloderma is characterized by a combination of mottled pigmentation, telangiectasia, and epidermal atrophy in the first few months of life. We have previously described a South African European-descent family affected by a rare autosomal-dominant form of hereditary fibrosing poikiloderma accompanied by tendon contracture, myopathy, and pulmonary fibrosis. Here, we report the identification of causative mutations in FAM111B by whole-exome sequencing. In total, three FAM111B missense mutations were identified in five kindreds of different ethnic backgrounds. The mutation segregated with the disease in one large pedigree, and mutations were de novo in two other pedigrees. All three mutations were absent from public databases and were not observed on Sanger sequencing of 388 ethnically matched control subjects. The three single-nucleotide mutations code for amino acid changes that are clustered within a putative trypsin-like cysteine/serine peptidase domain of FAM111B. These findings provide evidence of the involvement of FAM111B in congenital poikiloderma and multisystem fibrosis.
Main Text
Poikiloderma is characterized by mottled pigmentation, telangiectasia, and epidermal atrophy (Figure 1). The eponymous Weary form of hereditary sclerosing poikiloderma (MIM 173700), Rothmund-Thomson syndrome (RTS [MIM 268400]), and Werner syndrome (WRN [MIM 277700]) are well-documented types of inherited poikiloderma.1–3 We previously described a distinct autosomal-dominant form of hereditary fibrosing poikiloderma (HFP) in a South African family.4 In this two-generation family, five members were affected by this syndrome, comprising poikiloderma, tendon contractures, and progressive pulmonary fibrosis. Clinical manifestations were poikiloderma from early childhood and telangiectasia and pigmentary anomalies especially on the face and sun-exposed areas. Tendon contractures especially involved the ankles and feet and caused gait disturbance. Pulmonary involvement was noted during the second decade of life, and progressive dyspnea and restrictive impairment of lung function were linked to pulmonary fibrosis.
Figure 1.
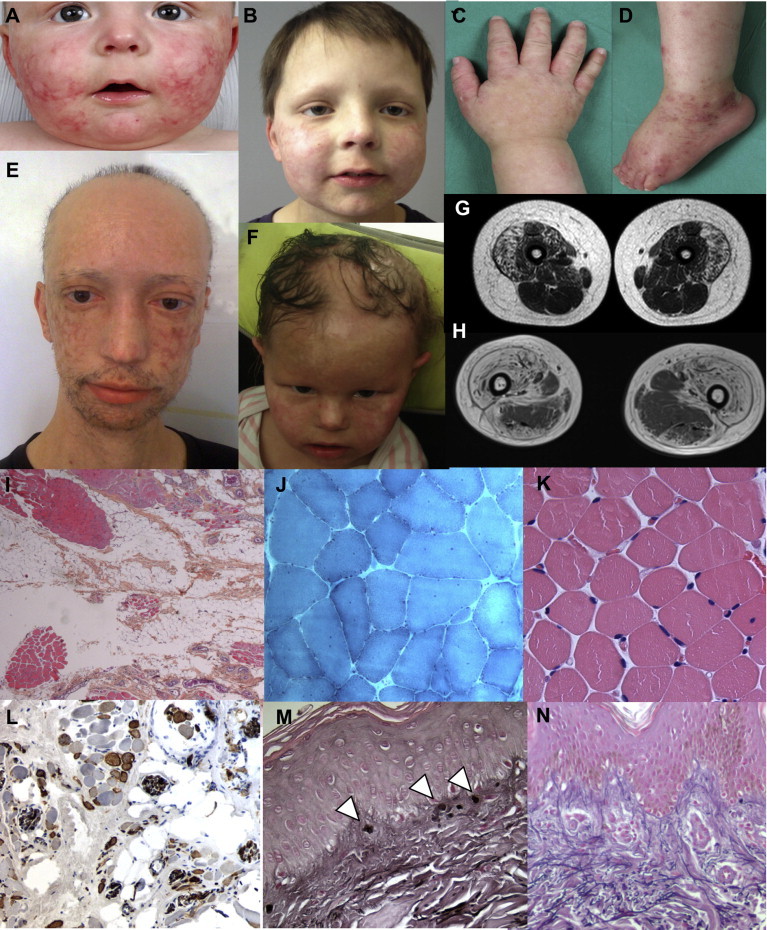
Clinical, Imaging, and Pathological Data of Affected Individuals with FAM111B Mutations
(A and B) Facial skin lesions of poikiloderma with hypopigmentation, telangiectasiae, epidermal atrophy, and alopecia of the scalp, eyebrows, and eyelashes in individual F1-II2 at the age of 12 months (A) and improvement of the lesions at 7 years (B).
(C and D) Eczema-like dermatosis of the hands (C) and the feet (D) in individual F1-II2 were associated with chronic lymphoedema.
(E and F) Facial lesions in individuals F2-II4 (E) and F4-II1 (F).
(G and H) Muscle MRI (T1-weighted sequence) shows a diffuse bright appearance of the anterior compartment of the thighs, particularly in the vastus lateralis muscles in individual F1-II2 at 7 years (G) and at a more severe stage with a relative sparing of the posterior compartment in the 30-year-old F2-II4 (H).
(I–N) Muscle and skin microscopy. (I) Fatty tissue and fragmented muscle fascicles; heterogeneous dystrophic pattern (individual F1-II2; Hematoxylin and eosin staining [H&E]; ×20 magnification). (J) Nonspecific myopathic changes with variation in fiber size, internal nuclei, and splitting (individual F2-II4; Gomori trichrome; ×150 magnification). (K) Age-matched normal muscle fascicles (H&E; ×150 magnification). (L) N-CAM-positive myocytes in dystrophic fascicles (individual F4-II1; immunolabeling with a DAB system (Dako); ×100 magnification). (M) Elastic dystrophy (arrowheads) with conspicuous superficial dermal changes on skin biopsy (individual F1-II2; Weigert staining; ×150 magnification). (N) Age-matched normal skin biopsy (Weigert staining; ×150 magnification).
This study was approved by the institutional review boards of the Hospital of Nantes and the University of Cape Town, and written informed consent was obtained from each adult participant and the parents of the participating children. We identified four additional cases that validated HFP with tendon contracture, myopathy, and pulmonary fibrosis as a distinct entity. Affected persons were of French, Algerian, Italian, and Moroccan origins. The first affected person (individual II2 in family 1 [F1-II2]), a French boy born to nonconsanguineous parents, presented with a photodistributed poikiloderma at 1 month of age (Figures 1A and 1B). He also had eczematous lesions of the hands and feet, alopecia (involving the scalp, eyebrows, and eyelashes), and heat intolerance causing hypohidrosis (Figures 1C and 1D). Nails, teeth, and developmental milestones were normal. He developed lymphoedema of all limbs, complicated by cellulitis at 6 years of age, which was accompanied by rapidly progressive muscle weakness and tendon contractures of both feet, necessitating Achilles tendon lengthening at 7 years of age. Blood tests (including full blood count, autoimmune screen, and serum creatine kinase) were normal except for eosinophilia. Electromyography (EMG) showed a myogenic pattern of the lower limbs, and MRI of skeletal muscle revealed a diffuse fatty infiltration, especially affecting the anterior thigh and posterior leg muscles (Figure 1G). The vastus lateralis muscle biopsy confirmed an extensive fatty infiltration in which residual muscle tissue was composed of fragmented muscle fascicles with either normal or dystrophic myocytes (Figure 1I). Neither neurogenic features nor necrosis was identified by histochemical studies and immunolabeling. N-CAM (CD56)-positive fibers were, however, observed in most dystrophic areas. Focal nerve proliferation was seen. Microscopic examination of skin biopsies revealed epidermal atrophy and conspicuous alterations of the elastic network in the superficial and deep dermal layers. Enlarged and fragmented elastic fibers and the formation of elastic globules in the papillary dermis were accompanied by a diffuse collagen sclerosis (Figure 1L).
The second affected person (individual II-4 in family 2 [F2-II4]) was a 30-year-old man with no family history of poikiloderma and normal developmental milestones and was born to consanguineous Algerian parents (Figure 1E). He had a history of congenital poikiloderma (worse on the cheeks) and biceps brachii muscle contractures from early childhood. He developed contractures of the triceps surae muscles at the age 6 of years, requiring Achilles tendon lengthening at 11 years of age. By the age of 30 years, he had dysphagia and severe difficulties with mobility as a result of multiple contractures of the arms and digits, scoliosis, and impaired lung function. Serum creatine kinase was slightly increased (460 IU/l). Muscle MRI showed fat infiltration, but the tibialis anterior and the posterior compartment of the thigh were spared (Figure 1H). Muscle biopsy of the left deltoid muscle revealed extensive fatty infiltration with focal lymphocytic (CD3+) and macrophage (CD68+) infiltrates without associated necrosis or regeneration. Nonspecific myogenic lesions without any neurogenic features (i.e., normal ATPase pattern) were found on histochemistry (Figure 1J). An immunoblot analysis showed a decreased quantity of calpain.
The third affected person (individual II1 in family 3 [F3-II1]) was a girl born to nonconsanguineous Italian parents. At 13 years of age, she was referred with congenital poikiloderma and growth retardation. She presented with alopecia (involving the scalp, eyebrow, and eyelashes) and nail dysplasia. She was underweight (body mass index of 14.3 kg/m2) and had short stature (height 145 cm [−1.5 SDs]) and delayed puberty, which were attributed to rickets. She also had contractures of the biceps brachii and triceps surae muscles, diffuse muscle atrophy and weakness, and thoracolumbar scoliosis, lower-limb lymphoedema, and hepatomegaly. Blood tests revealed cholestasis, which was treated with ursodesoxycholic acid.
The fourth affected person (individual II1 in family 4 [F4-II1]) was a 6-year old girl who had no family history of poikiloderma and was the only daughter of a French mother and a Moroccan father. She presented with congenital poikiloderma of the face and extremities, diffuse alopecia, and anhidrosis (Figure 1F). Frequent falls and delayed walking were noted by 18 months of age. She developed progressive muscle weakness, contractures of the triceps muscles and wrist extensors, and lymphedema of the extremities by 4 years of age and required a wheelchair at 6 years of age. Cognitive development was normal. Electroencephalography revealed multifocal spike-and-wave discharges without clinical seizures. Serum creatine kinase was slightly increased in infancy (340 IU/l). EMG showed a myogenic profile at 22 months. On muscle biopsy, fatty infiltration was associated with heterogeneous degeneration of fragmented muscle fascicles. No necrosis or regeneration was observed, but numerous N-CAM-positive cells were seen, especially in the most dystrophic parts (Figure 1K). ATPase patterns and the mitochondrial network were normal. Focal nerve proliferation was present. Skin biopsy was in favor of RTS, but this diagnosis was ruled out because genetic testing failed to detect any RECQL4 (MIM 603780) mutation.
A whole-exome sequencing strategy was first applied to members from two of the families affected by HFP with tendon contracture, myopathy, and pulmonary fibrosis: individual F1-II2 and his two unaffected parents from French family 1 (trio design) and three affected siblings (one deceased) and their unaffected mother from the South African family (Figure 2 and Figure S1, available online). Whole-exome sequencing was carried out by Oxford Gene Technology in the French family and at the Institute of Genetic Medicine at Newcastle University in the South African family. In both families, capture was done with the SureSelect XT Human All Exon V4 kit (Agilent Technologies) and was followed by Illumina sequencing. Read files (Fastq) were generated from the sequencing platform with the manufacturer’s proprietary software. Reads were mapped to their location in build hg19 (UCSC Genome Browser) of the human reference genome with the Burrows-Wheeler Aligner package (v.0.6.1). Local realignment of the mapped reads around potential indel sites was carried out with the Genome Analysis Toolkit (GATK; v.1.6).5 Duplicate reads were marked with Picard and discarded. SNPs and indel variants were called with the GATK Unified Genotyper for each sample. SNP novelty was determined against dbSNP releases 132 and 135, 1000 Genomes (releases from 2010 to 2012), and the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project Exome Variant Server (ESP6500). Given the dominant mode of inheritance inferred from the original description,4 we focused on heterozygous de novo mutations in F1-II2. Candidate de novo mutations were determined with KGGSeq v.0.36 in the French simplex family, and these were cross-referenced to the list of heterozygous potentially damaging variants that were present in the affected siblings but absent from their unaffected mother in the South African family. FAM111B (RefSeq accession number NM_198947.3) appeared as the only candidate gene in common between the two families: c.1879A>G (p.Arg627Gly) was the unique de novo variant found in F1-II2, whereas c.1861T>G (p.Tyr621Asp) was observed in the affected South African siblings and absent from their unaffected mother. DNA from the affected father in the South African pedigree was not available for sequencing (Table 1, Figure 2, and Figure S1). The two rare variants were absent from all public genetic variant databases tested and they were validated by Sanger sequencing.
Figure 2.
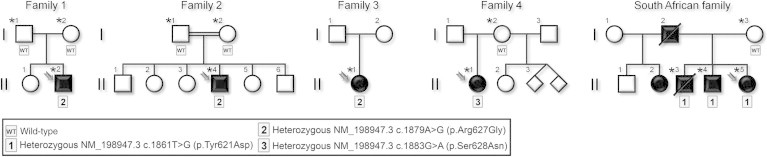
Pedigrees of the Participating HFP-Affected Families
The families are related to the four affected individuals (F1-II2, F2-II4, F3-II1, and F4-II1) described in the present report and to the three additional affected members (FSA-II3, FSAII-4, and FSA-II5) from the South African family previously reported.4 Affected individuals are represented by a solid square or circle, depending upon the gender, and index cases are indicated by arrows. Of note, the twins in family 4 have not been born yet. Individuals tested by exome and/or Sanger sequencing are marked by an asterisk, and their genotypes are noted below in boxes: WT indicates a wild-type genotype, whereas the three different mutations observed in FAM111B (RefSeq NM_198947.3: c.1861T>G [p.Tyr621Asp], c.1879A>G [p.Arg627Gly], and c.1883G>A [p.Ser628Asn]) are numbered 1–3.
Table 1.
Clinical Characteristics of the Index Cases
| Characteristics | Individual F1-II2 | Individual F2-II4 | Individual F3-II1 | Individual F4-II1 | South African Family4 | |||
|---|---|---|---|---|---|---|---|---|
| Sex | male | male | female | female | female (proband) | male (father) | male (brother) | male (brother) |
| Age at last examination | 7 years | 30 years | 14 years | 6 years | 26 years | (death at 56 years) | (death at 30 years) | 31 years |
| General | ||||||||
| Growth retardation | − | + | + | − | − | − | − | − |
| Delayed puberty | ND | + | + | ND | ND | ND | ND | ND |
| Skin | ||||||||
| Congenital poikiloderma | + | + | + | + | + | + | + | + |
| Face | + | + | + | + | + | + | + | + |
| Exposed area | + | + | + | + | + | + | + | + |
| Upper and/or lower limbs | + | + | + | + | + | + | + | + |
| Eczematiform | + | + | ND | + | − | − | − | − |
| Psoriasiform | − | − | ND | + | − | − | − | − |
| Erysipelas | + | − | + | − | − | − | − | − |
| Lymphoedema of extremities | + | − | + | + | − | − | − | − |
| Sclerosis of the digits | − | + | − | − | + | ND | ND | + |
| Hypohidrosis and/or heat intolerance | + | + | ND | + | + | + | + | + |
| Hair | ||||||||
| Hypotrichosis and/or alopecia | + | + | + | + | + | ND | ND | + |
| Scalp hair | + | + | + | + | + | + | + | + |
| Eyebrows | + | + | + | + | + | + | + | + |
| Eyelashes | + | + | + | + | − | − | − | − |
| Follicular dystrophy | − | ND | ND | ND | ND | ND | ND | ND |
| Nails | ||||||||
| Dysplasia | − | − | + | − | − | − | − | − |
| Muscle | ||||||||
| Muscle weakness (age of onset) | + (7 years) | + (11 years) | + (4 years) | + (14 months) | + (9 years) | ND | ND | ND |
| Proximal lower limbs | + | + | + | + | ND | ND | ND | ND |
| Distal lower limbs | + | + | + | ND | ND | ND | ND | ND |
| Proximal upper limbs | + | + | + | ND | ND | ND | ND | ND |
| Distal upper limbs | + | + | + | + | ND | ND | ND | ND |
| Neck extensors | − | + | ND | ND | ND | ND | ND | ND |
| Amyotrophy | + | + | + | + | ND | ND | ND | ND |
| Abolition of lower-limb tendon reflexes | ND | + | + | + | ND | ND | ND | ND |
| Tendon lengthening (age of onset) | + (7 years) | + (11 years) | + (13 years) | − (ND) | + (14 years) | − (ND) | + (5 years) | − (ND) |
| Joints | ||||||||
| Distal lower-limb contractures and/or pes varus (age of onset) | + (6 years) | + (2–3 years) | + | + (2 years) | + | ND | ND | − |
| Upper-limb contractures | − | + | + | + | − | ND | ND | − |
| Spine | ||||||||
| Scoliosis | − | + | + | − | − | − | − | − |
| Lungs | ||||||||
| Restrictive syndrome | + | + | ND | ND | + | + | + | ND |
| Liver | ||||||||
| Hepatomegaly | − | ND | + | − | − | − | − | − |
| Eyes | ||||||||
| Cataract | − | − | + | − | − | − | − | − |
| Blood Test | ||||||||
| Serum creatine kinase (IU/l) | N | 460 | 500 | 340 | ND | ND | ND | ND |
| Muscle Exploration | ||||||||
| EMG: myogenic profile | + | ND | + | + | ND | ND | ND | ND |
| MRI | adiposis | adiposis | ND | ND | ND | ND | ND | ND |
| Pathology | ||||||||
| Peripheral skeletal muscle | adiposis | dystrophy, fibrosis, adiposis | dystrophy, fibrosis, adiposis | fibrosis, atrophy | ND | ND | fatty infiltration | ND |
| Skin | sclerodermiform aspect, fibrosis, elastic tissue degeneration | ND | ND | RTS-like | ND | ND | sclerodermiform aspect, fibrosis, elastic tissue degeneration | ND |
| Visceral organs | ND | ND | ND | ND | ND | pulmonary fibrosis | pulmonary, esophageal, and mediastinal lymph node fibrosis; pancreas fatty infiltration | ND |
| Vasculature | ND | ND | ND | ND | ND | ND | elastic degeneration, medial calcification | ND |
| Mutational Analysis | ||||||||
| RECQL4 | − | − | − | − | ND | − | − | ND |
| Other candidate genes | − | CAPN3, LMNA, CAV3 (no mutation) | − | SMN1 (no mutation) | − | − | − | − |
| FAM111B | c.1879A>G (p.Arg627Gly) | c.1879A>G (p.Arg627Gly) | c.1879A>G (p.Arg627Gly) | c.1883G>A (p.Ser628Asn) | c.1861T>G (p.Tyr621Asp) | ND | c.1861T>G (p.Tyr621Asp) | c.1861T>G (p.Tyr621Asp) |
The following abbreviations are used: EMG, electromyography; N, normal; ND, not documented; and RTS, Rothmund-Thomson syndrome.
Direct mutation analysis of FAM111B was performed by Sanger sequencing in the three additional simplex individuals (F2-II4, F3-II1, and F4-II1 in Figure 2). Although they were unrelated and of different ancestries, individuals F2-II4 and F3-II1 had the same FAM111B variant as individual F1-II2; the de novo occurrence of the mutation was confirmed in individual F2-II4, but this could not be tested in individual F3-II1 because no parental samples were available. Individual F4-II1 was found to have the variant c.1883G>A (p.Ser628Asn), located one codon downstream of the de novo mutation (Figure S1) and absent from public genetic variant databases; only the mother of individual F4-II1 could be tested, and she did not have the mutation. All three putative causative variants were absent on Sanger sequencing of 388 healthy controls, including 96 Algerian, 127 Moroccan (half Berber and half Arab), and 165 South African (93 of white European origin and 72 of mixed ancestry) individuals. All three variants were predicted to be deleterious by bioinformatic programs PolyPhen-2 and SIFT and affect amino acids conserved across mammalian species or primates (Figure S1 and Table S1).
Together, the identification of (1) three rare heterozygous mutations in affected members of five unrelated families with differing ethnic backgrounds and a concordant phenotype and (2) a de novo occurrence in two families provide compelling genetic evidence that mutations in FAM111B cause HFP with tendon contracture, myopathy, and pulmonary fibrosis. Given that several putative loss-of-function variants (stop-gain and frameshift) are present in the NHLBI Exome Variant Server, i.e., in individuals very unlikely to be affected by HFP, we hypothesize that the FAM111B mutations are either gain-of-function or dominant-negative variants.
Very little is known about FAM111B, which is the second and last member of the “family with sequence similarity 111” gene family (Figure S2). It encodes a protein predicted to contain a trypsin-like cysteine/serine peptidase domain split into two parts by a loop that harbors the three alterations identified in the affected individuals in the present study (Figure S3). The cluster of alterations in this small region suggests that it has an important role in the functional activity of FAM111B. This domain is 45% homologous to the one predicted in FAM111A (Figure S4), which contains causative mutations for Kenny-Caffey syndrome (KCS [MIM 127000]) and osteocraniostenosis (OCS [MIM 602361]), two clinical entities that are very different from HFP.7 Predictions of the two- and three-dimensional protein structures of FAM111B suggest that the amino acid residue 621 (p.Tyr621Asp) is located in a β sheet, whereas residues 627 (p.Arg627Gly) and 628 (p.Ser628Asn) lie either in a β sheet or in a random coil (Figures S5–S7). Physical interactions between FAM111B and polyubiquitin-C (UBC)8,9 and ATP13A210 have been detected experimentally. FAM111B was detected in skeletal striated muscle, but not in fibroblasts, by immunoblot analysis of tissue samples from individual F1-II2 (Figure S8). FAM111B mRNA expression has been detected in many tissues, including skin, skeletal muscle, and trachea. (Figures S9 and S10). We found that in normal fibroblasts, protein levels were undetectable and mRNA expression was low.
The phenotype of our cases differs from other types of hereditary poikiloderma, such as RTS, WRN, and Kindler syndrome (MIM 173650).1,2,11,12 Of note, skin lesions improved with time (Figure 1), whereas extracutaneous symptoms became more prominent. Furthermore, tendon contracture, progressive muscle weakness, and pulmonary fibrosis have not been described in other types of hereditary poikiloderma. Nevertheless, Meier has reported an adult with putative RTS associated with progressive muscle atrophy, multiple organ fibrosis, and pulmonary cachexia.13 We postulate that simplex cases of congenital poikiloderma with muscular atrophy, organ fibrosis, or pulmonary involvement and no RECQL4 mutations might represent HFP related to FAM111B. The phenotype described here also differs from Clericuzio-type poikiloderma with neutropenia (MIM 604173), due to mutations in C16orf57 (MIM 61327), which encodes the highly conserved USB1, given that there was no associated neutropenia.6,14,15 The Weary form of hereditary poikiloderma is characterized by cutaneous fibrous bands, subcutaneous calcification, and/or valvular heart disease, which were not observed in our series.3,16
Histological examination of skeletal muscle in our cases revealed a partial loss of muscle tissue associated with extensive fibrofatty tissue infiltration regardless of age. There was no indication of denervation, necrosis, or inflammation (except for one isolated focus of inflammatory cells in individual F2-II4). No noticeable labeling was observed with many different antibodies, except for N-CAM, which was present in individuals F1-II2 and F4-II1. The fatty tissue might be considered either as a passive phenomenon due to muscle degeneration or as an active process invading normal muscle tissue and leading to a secondary myopathic destruction. The skin pathology is remarkable for collagen sclerosis, elastic degeneration, and the absence of fatty infiltration, in contrast to the observation made in muscle tissue.
Our results led us to suspect that HFP involves genotype-phenotype correlations similar to those seen in OCS and KCS, due to FAM111A (MIM 615292) mutations.7 Mutations in FAM111B codons 627 and 628 indeed appear to be associated with early disease onset (in affected members from families 1–4); however, the symptoms caused by the mutation in codon 621 occurred later in the South African family, which enabled the familial transmission of the anomaly. We postulate that mutations in other regions of FAM111B might lead to phenotypes different from HFP, as observed, for instance, for Hajdu-Cheney (MIM 102500) and Alagille (MIM 610205) syndromes, which are caused by mutations located in different coding regions of the same gene, NOTCH2 (MIM 600275).17,18 As a matter of fact, in the South African family, the postmortem study of one affected member revealed a diffuse fatty infiltration and fibrosis of organs such as the lungs, esophagus, and pancreas. The lung-function impairment in individuals F1-II2 and F2-II4 and the presence of hepatomegaly in individual F3-II1 probably have the same origin, supporting that FAM111B has a multisystemic involvement that could lead to other phenotypes not yet related to this gene. Nevertheless, these tentative observations remain to be confirmed in a larger series of affected people.
In conclusion, we have demonstrated that mutations in FAM111B cause HFP with tendon contracture, myopathy, and pulmonary fibrosis. Our findings identify FAM111B as a critical gene in the development of fibrosis, which is a key pathological process in many human diseases.
Acknowledgments
We thank the families who participated in this study. B.M.M. is funded in part through grants from the South African Medical Research Council, the National Research Foundation of South Africa, and the Lily and Ernst Hausmann Research Trust. B.K. is funded by a British Heart Foundation Personal Chair. We are very grateful to all the French collaborators who also contributed to this work: Claire Bénéteau, Marc Tardieu, Isabelle Desguerre, Julie Perrier, Jean-Yves Mahé, Emmanuelle Fleurence, Jean-Benoit Courcet, Jérôme Franques, France Leturcq, Sylvie Fraitag, Olivier Pichon, Jocelyne Havard, Laëtitia de Peufeilhoux, Catherine Bodemer, and Solange Duriez, as well as Flora Bréheret, Réjane Troudet, and Céline Chevrier. We thank Nasr-eddine Benchick, who provided us with control DNAs from Algeria. We acknowledge the bioinformatics core facility of Nantes (BiRD, Biogenouest) and Oxford Gene Technology for their expert service.
Contributor Information
Stéphane Bézieau, Email: stephane.bezieau@chu-nantes.fr.
Bongani M. Mayosi, Email: bongani.mayosi@uct.ac.za.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project, http://browser.1000genomes.org/
Burrows-Wheeler Aligner, http://bio-bwa.sourceforge.net/
Ensembl, http://www.ensembl.org/index.html/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
GeneCards, http://www.genecards.org/
Human Protein Atlas, http://www.proteinatlas.org/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Picard, http://picard.sourceforge.net/
References
- 1.Epstein C.J., Martin G.M., Schultz A.L., Motulsky A.G. Werner’s syndrome a review of its symptomatology, natural history, pathologic features, genetics and relationship to the natural aging process. Medicine (Baltimore) 1966;45:177–221. doi: 10.1097/00005792-196605000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Larizza L., Roversi G., Volpi L. Rothmund-Thomson syndrome. Orphanet J. Rare Dis. 2010;5:2. doi: 10.1186/1750-1172-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weary P.E., Hsu Y.T., Richardson D.R., Caravati C.M., Wood B.T. Hereditary sclerosing poikiloderma. Report of two families with an unusual and distinctive genodermatosis. Arch. Dermatol. 1969;100:413–422. doi: 10.1001/archderm.100.4.413. [DOI] [PubMed] [Google Scholar]
- 4.Khumalo N.P., Pillay K., Beighton P., Wainwright H., Walker B., Saxe N., Mayosi B.M., Bateman E.D. Poikiloderma, tendon contracture and pulmonary fibrosis: a new autosomal dominant syndrome? Br. J. Dermatol. 2006;155:1057–1061. doi: 10.1111/j.1365-2133.2006.07473.x. [DOI] [PubMed] [Google Scholar]
- 5.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Piard J., Holder-Espinasse M., Aral B., Gigot N., Rio M., Tardieu M., Puzenat E., Goldenberg A., Toutain A., Franques J. Systematic search for neutropenia should be part of the first screening in patients with poikiloderma. Eur. J. Med. Genet. 2012;55:8–11. doi: 10.1016/j.ejmg.2011.07.004. [DOI] [PubMed] [Google Scholar]
- 7.Unger S., Górna M.W., Le Béchec A., Do Vale-Pereira S., Bedeschi M.F., Geiberger S., Grigelioniene G., Horemuzova E., Lalatta F., Lausch E. FAM111A Mutations Result in Hypoparathyroidism and Impaired Skeletal Development. Am. J. Hum. Genet. 2013 doi: 10.1016/j.ajhg.2013.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Emanuele M.J., Elia A.E., Xu Q., Thoma C.R., Izhar L., Leng Y., Guo A., Chen Y.N., Rush J., Hsu P.W. Global identification of modular cullin-RING ligase substrates. Cell. 2011;147:459–474. doi: 10.1016/j.cell.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Povlsen L.K., Beli P., Wagner S.A., Poulsen S.L., Sylvestersen K.B., Poulsen J.W., Nielsen M.L., Bekker-Jensen S., Mailand N., Choudhary C. Systems-wide analysis of ubiquitylation dynamics reveals a key role for PAF15 ubiquitylation in DNA-damage bypass. Nat. Cell Biol. 2012;14:1089–1098. doi: 10.1038/ncb2579. [DOI] [PubMed] [Google Scholar]
- 10.Usenovic M., Knight A.L., Ray A., Wong V., Brown K.R., Caldwell G.A., Caldwell K.A., Stagljar I., Krainc D. Identification of novel ATP13A2 interactors and their role in α-synuclein misfolding and toxicity. Hum. Mol. Genet. 2012;21:3785–3794. doi: 10.1093/hmg/dds206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kindler T. Congenital poikiloderma with traumatic bulla formation and progressive cutaneous atrophy. Br. J. Dermatol. 1954;66:104–111. doi: 10.1111/j.1365-2133.1954.tb12598.x. [DOI] [PubMed] [Google Scholar]
- 12.Wang L.L., Levy M.L., Lewis R.A., Chintagumpala M.M., Lev D., Rogers M., Plon S.E. Clinical manifestations in a cohort of 41 Rothmund-Thomson syndrome patients. Am. J. Med. Genet. 2001;102:11–17. doi: 10.1002/1096-8628(20010722)102:1<11::aid-ajmg1413>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 13.Meier M., Schwarz A. Rothmund-Thomson syndrome—a single case report with systemic muscular atrophy, multiple organ fibrosis and pulmonary cachexia. Rheumatology (Oxford) 2012;51:2109–2111. doi: 10.1093/rheumatology/kes143. [DOI] [PubMed] [Google Scholar]
- 14.Arnold A.W., Itin P.H., Pigors M., Kohlhase J., Bruckner-Tuderman L., Has C. Poikiloderma with neutropenia: a novel C16orf57 mutation and clinical diagnostic criteria. Br. J. Dermatol. 2010;163:866–869. doi: 10.1111/j.1365-2133.2010.09929.x. [DOI] [PubMed] [Google Scholar]
- 15.Hilcenko C., Simpson P.J., Finch A.J., Bowler F.R., Churcher M.J., Jin L., Packman L.C., Shlien A., Campbell P., Kirwan M. Aberrant 3′ oligoadenylation of spliceosomal U6 small nuclear RNA in poikiloderma with neutropenia. Blood. 2013;121:1028–1038. doi: 10.1182/blood-2012-10-461491. [DOI] [PubMed] [Google Scholar]
- 16.Grau Salvat C., Pont V., Cors J.R., Aliaga A. Hereditary sclerosing poikiloderma of Weary: report of a new case. Br. J. Dermatol. 1999;140:366–368. doi: 10.1046/j.1365-2133.1999.02684.x. [DOI] [PubMed] [Google Scholar]
- 17.McDaniell R., Warthen D.M., Sanchez-Lara P.A., Pai A., Krantz I.D., Piccoli D.A., Spinner N.B. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway. Am. J. Hum. Genet. 2006;79:169–173. doi: 10.1086/505332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simpson M.A., Irving M.D., Asilmaz E., Gray M.J., Dafou D., Elmslie F.V., Mansour S., Holder S.E., Brain C.E., Burton B.K. Mutations in NOTCH2 cause Hajdu-Cheney syndrome, a disorder of severe and progressive bone loss. Nat. Genet. 2011;43:303–305. doi: 10.1038/ng.779. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.