

Abstract
Standard hydrogen bonds are of great importance for protein structure and function. Ionic hydrogen bonds often are significantly stronger than standard hydrogen bonds and exhibit unique properties, but their role in proteins is not well understood. We report that hydrogen/deuterium exchange causes a redshift in the visible absorbance spectrum of photoactive yellow protein (PYP). We expand the range of interpretable isotope effects by assigning this spectral isotope effect (SIE) to a functionally important hydrogen bond at the active site of PYP. The inverted sign and extent of this SIE is explained by the ionic nature and strength of this hydrogen bond. These results show the relevance of ionic hydrogen bonding for protein active sites, and reveal that the inverted SIE is a novel, to our knowledge, tool to probe ionic hydrogen bonds. Our results support a classification of hydrogen bonds that distinguishes the properties of ionic hydrogen bonds from those of both standard and low barrier hydrogen bonds, and show how this classification helps resolve a recent debate regarding active site hydrogen bonding in PYP.
Introduction
Since the classic work by Pauling, Corey, and Branson in 1951 (1), the central importance of hydrogen bonding for protein structure and function has been widely recognized (2). Backbone hydrogen bonding results in the formation of α-helices and β-sheets, whereas side-chain hydrogen bonds provide specific interactions in the native state of proteins. Functionally important hydrogen bonds are ubiquitous at protein active sites (3) and are key elements for enzymatic function (4). Biochemically relevant variations on the hydrogen bonding theme, such as low-barrier hydrogen bonds (LBHB) (5), and C-H⋅⋅⋅O hydrogen bonds (6), have attracted intense attention. However, many key questions remain unresolved, as was summarized by Perrin and Nielson in their influential review: “For all the importance of hydrogen bonding in chemistry and biology, it is remarkable that we understand it so poorly” (7). Here, we study how the unique properties of ionic hydrogen bonds, in which the hydrogen bond donor-acceptor pair carries a formal charge, affect the properties of proteins. Our results support a hydrogen bonding classification that distinguishes between ionic hydrogen bonds and LBHBs.
We examine ionic hydrogen bonding in photoactive yellow protein (PYP), a small protein that serves as a model system for protein-ligand interactions at active sites (8,9). PYP (10) exhibits a light-triggered photocycle based on its pCA chromophore (11). The PYP from the photosynthetic bacterium Halorhodospira halophila (Hh PYP) is a highly studied (8,9) member of a growing family of bacterial blue-light receptors (12). The crystal structure of Hh PYP consists of a central antiparallel 6-stranded β-sheet flanked by five α-helices (13). The structure of the pCA binding pocket indicates Tyr-42 and Glu-46 as key residues interacting with the phenolic oxygen of the pCA chromophore (Fig. 1, A and B).
Figure 1.
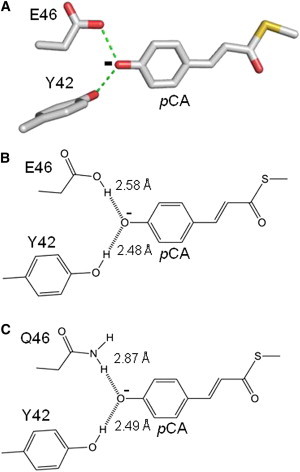
Anionic hydrogen bonding of pCA at the active site of PYP. A three-dimensional depiction of hydrogen bonding of the anionic pCA at the active site of wtPYP based on its crystal structure (PDB ID 1NWZ) (A). A two-dimensional depiction of hydrogen bonding of the pCA with residues 42 and 46, including hydrogen bonding lengths based on the crystal structures of wtPYP (B) and E46Q PYP (C) (PDB IDs: 1NWZ and 1OTA). To see this figure in color, go online.
The active site of PYP exhibits a number of unusual properties. The protonation states of the pCA chromophore and Glu-46 in PYP are opposite compared to the situation in solution. This causes the pCA to be present in its deprotonated form (14,15) and allows for biologically important proton transfer events during the PYP photocycle (14,15). In addition, the absorbance maximum (λmax) of the pCA in PYP is strongly shifted from its value in solution, providing an example of the phenomenon of biological spectral tuning that is also at the basis of human color vision. Studies comparing the λmax of pCA derivatives in water with the λmax of PYP have yielded a view in which three major factors contribute to the spectral tuning effect PYP (16). First, the formation of the thioester bond between the pCA and Cys-69 results in a redshift from 284 to 335 nm. Second, the deprotonation of the phenolic oxygen of the pCA causes a further shift to 400 nm. Finally, specific protein-chromophore interactions cause the final redshift to 446 nm in the case of Hh PYP (10) and 432 nm for Salinibacter ruber (Sr PYP) (17).
An alternative approach to understand spectral tuning in proteins is to compare the λmax of pCA derivatives in vacuum with that observed of pCA in the natively folded protein. Experimental studies on the ultraviolet/visible (UV/vis) absorbance spectra of pCA derivatives in vacuum have yielded two unexpected results (18,19). First, the experimentally observed λmax of free pCA derivatives in vacuum is in the 430–460 nm range, implying that although placing these compounds in water causes a very strong blueshift (to ∼300 nm), placing them in the PYP chromophore binding pocket does not cause a significant shift in λmax. Second, deprotonation of the phenolic oxygen was found to have a negligible effect on λmax in vacuum, in contrast to the strong redshift that accompanies this deprotonation event in solution. Multiple computational studies on pCA derivatives in vacuum and in the protein binding pocket formed by PYP have been reported, for example see (18,19).
The hydrogen bond between Glu-46 and the pCA has been shown to be important in determining the λmax of Hh PYP. Weakening of this hydrogen bond in the E46Q mutant (Fig. 1 C) (20–22) results in a redshift of the λmax to 460 nm (23,24), and its deletion in the E46I, V, and L mutants causes a further redshift to 478 nm (25). High-resolution crystal structures (13,20) and NMR spectroscopy (21) have revealed a length of 2.6 Å for the Glu-46-pCA hydrogen bond. In the E46Q this hydrogen bond is lengthened by 0.3 Å (20,22). The proposed mechanism by which the strength of the hydrogen bond between residue 46 and the pCA affects the λmax of PYP is a classic effect based on modulating the degree of conjugation of the pCA: a reduction in hydrogen bonding strength decreases the degree of electron localization at this hydrogen bond and therefore increases the degree of electron delocalization over the pCA (23,25). We have provided evidence that the changes in conjugation of the pCA caused by changes in hydrogen bonding between residue 46 and the pCA affect the λmax of PYP not by altering the energy gap between the ground and electronically excited state, but by changing the width of the excited state energy potential as a result of the change in bond orders in the pCA (26). Thus, a precise description of the relation between hydrogen bond strength and λmax is complex. It involves the identification of vibrational modes coupled to the optical transition, the determination of how these vibrational modes are affected by changes in electron delocalization caused by hydrogen bonding, and an evaluation of the Franck-Condon factors associated with these vibrational modes. Here, we use the known empirical dependence of the λmax of PYP on the strength of the hydrogen bond between residues 46 and the pCA.
Materials and Methods
Mutagenesis and protein purification
The pyp gene from S. ruber was obtained by direct polymerase chain reaction (PCR) amplification using lyophilized cell material and primers that introduced NcoI and BamH1 restriction sites. The resulting PCR product was cloned into the pET-16b vector (Novagen, Billerica, MA), transformed to Escherichia coli XL1, and confirmed by DNA sequence analysis. The recombinant plasmid DNA was transformed into E. coli BL21 (DE3). The E46Q mutant of Sr PYP E46Q mutation was obtained by PCR using the corresponding mutagenic primers.
Hh PYP was obtained as described in (27). Sr PYP apoprotein was overexpressed in E. coli BL21 (DE3) following a similar procedure. Cells were harvested by centrifugation (2700 × g, 20 min) and lysed in 8 M urea. Cell debris was removed by centrifugation (17,200 × g, 20 min). The supernatant was diluted twofold in Tris-HCl buffer (pH7.5), and the anhydride of p-coumaric acid was added. The resulting reconstituted protein was dialyzed against 10 mM Tris buffer overnight at 4°C and applied to a DEAE Sepharose fast flow (GE Healthcare, Pittsburgh, PA) chromatography column. Elution was performed using a NaCl gradient in Tris buffer; Sr PYP eluted at 300 mM NaCl-Tris buffer (pH7.5). DNase I was added to the pooled yellow Sr PYP fractions. After overnight dialysis at 4°C the chromatography was repeated until an optical purity index (Abs276/Abs432) of 1.0 was achieved. Hh PYP and Sr PYP were then exchanged to 50 mM potassium phosphate buffer (pH 7.0) and concentrated using Centricon and Microcon (Millipore, Billerica, MA) membrane filters (Amicon; YM-10, 10,000 MWCO). Protein samples in D2O were obtained using the same procedure by washing the protein sample in a potassium phosphate buffer in D2O (pH∗ 7.0) and concentration with a Microcon centrifuge filter.
Absorbance spectroscopy
UV/vis absorbance spectra were measured using a Cary 300 UV-Visible spectrophotometer (Varian, Agilent, Santa Clara, CA) in double-beam mode using a spectral bandwidth of 2.0 nm, a data interval 1.0 nm, and a scanning rate of 150 nm/min. The wavelength accuracy of this spectrophotometer is ±0.04 nm near the visible absorbance peak of PYP.
Results and Discussion
Prediction and experimental detection of a spectral isotope effect in PYP
The exchange of the hydrogen atom in a hydrogen bond to deuterium modestly alters the strength of the hydrogen bonding interaction. In water hydrogen bonds are weaker than deuterium bonds by ∼0.6 kJ/mol, which corresponds to a 5% weakening (28,29). In contrast, both experimental gas phase studies on small molecules and computational studies have revealed that ionic hydrogen bonds are characterized by the opposite effect, with a deuterium bond that is ∼5% weaker (29,30). The effect of hydrogen/deuterium (H/D) exchange on hydrogen bonding strength is based on subtle changes in intramolecular donor-acceptor vibrational modes across the hydrogen bond, which results in opposite effects upon H/D exchange for neutral versus ionic hydrogen bonds (29,30). The influence of H/D exchange on the strength of hydrogen bonding is also reflected in a change in hydrogen bonding geometry, which is the Ubbelohde effect (31). Recently, ab initio calculations revealed such an effect for the pCA-side-chain hydrogen bonds at the active site in on PYP (32).
Based on the empirical dependence of the λmax of PYP on the strength of the hydrogen bond between residue 46 and the pCA, and reported effects of H/D exchange on hydrogen bonding strength, the effect of H/D exchange of the side chain of Glu-46 on the λmax of PYP can be predicted. Because the Glu-46-pCA hydrogen bond is ionic and therefore expected to be weakened upon H/D exchange, and because a weakened Glu-46-pCA hydrogen bond results in a redshift, we expect that the λmax of PYP in D2O is slightly redshifted. We refer to changes in position of the peak of an electronic transition λmax upon H/D exchange as a spectral isotope effect (SIE).
An estimate of the size of this SIE pCA can be obtained if both the change in hydrogen bonding strength upon H/D exchange (ΔHBSH/D) and the shift in λmax caused by a change in hydrogen bonding strength (Δλmax,HBS) are known: SIE = ΔHBSH/D × Δλmax,HBS.
We estimate the dependence of the λmax of Hh PYP on the hydrogen bonding strength between residue 46 and the pCA based on the spectral shifts caused by the E46Q and E46V mutations (23–25). The E46Q mutation causes a 14 nm redshift (from 446 to 460 nm). We use the approximation that the E46Q mutation reduces the hydrogen bonding strength between the pCA and residue 46 by half (estimated at ∼25 kJ/mol, i.e., half of a typical ionic hydrogen bond, see below), corresponding to a 721 cm−1 redshift in λmax for a ∼2000 cm−1 reduction in hydrogen bond strength (see Table 1). These data yield a value for Δλmax,HBS of ∼0.6 nm redshift per 1 kJ/mol loss in hydrogen bond strength. A similar estimate is obtained when the 32 nm shift in λmax upon the complete deletion of this hydrogen bond (estimated at ∼50 kJ/mol) in E46V PYP is considered. Based on published results (29), we use a value of a 5% reduction in the strength of the ionic hydrogen bond between residue 46 and the pCA upon H/D exchange. Therefore, a 5% reduction (2.5 kJ/mol) in the strength of the Glu-46-pCA hydrogen bond caused by H/D exchange would be expected to result in a ∼1.5 nm redshift in the λmax of PYP. We experimentally tested this prediction for both the highly studied PYP from Hh PYP and the recently identified PYP from the extremely halophilic bacterium Sr PYP, which was found to exhibit significantly different spectral and kinetic properties (12,17).
Table 1.
Experimentally observed shifts in absorbance maxima of Sr PYP and Hh PYP and their E46Q mutants caused by H/D exchange compared to tentative estimates for the strength of the hydrogen bond between the pCA and residue 46
| Protein | Absorbance maximum |
Tentative estimate of pCA-residue 46 HB strengtha |
||
|---|---|---|---|---|
| nm | cm−1 | kJ/mol | cm−1 | |
| Sr PYP | ||||
| H2O | 431.8 | 23,158 | 50 | 4,179 |
| D2O | 437.1 | 22,878 | 47.7 | 3,970 |
| Δ by H/D exch | 5.3 | 280 | 2.5 | 209 |
| Sr E46Q PYP | ||||
| H2O | 455.1 | 21,973 | 25 | 2,089 |
| D2O | 457.8 | 21,843 | 23.8 | 1,989 |
| Δ by H/D exch | 2.7 | 130 | 1.3 | 100 |
| Δ by mutation | 23.3 | 1,185 | 25 | 2,090 |
| Hh PYP | ||||
| H2O | 445.7 | 22,436 | 45 | 3,761 |
| D2O | 447.5 | 22,346 | 43 | 3,594 |
| Δ by H/D exch | 1.8 | 90 | 2 | 167 |
| Hh E46Q PYP | ||||
| H2O | 460.5 | 21,715 | 23 | 1,922 |
| D2O | 461.5 | 21,668 | 22 | 1,839 |
| Δ by H/D exch | 1.0 | 47 | 1 | 83 |
| Δ by mutation | 14.8 | 721 | 22 | 1,880 |
The depicted values are estimates based on the following assumptions: i), pCA-Glu-46 hydrogen bond in Sr PYP is 50 kJ/mol, a typical value for ionic hydrogen bonds. ii), This hydrogen bond is 10% weaker in Hh PYP. iii), The E46Q mutation weakens this hydrogen bond by 50% in both Sr PYP and Hh PYP. iv), H/D exchange reduces the strength of this hydrogen bond by 5% in both Sr PYP and Hh PYP.
We determined the effect of H/D exchange on the absorbance spectrum of PYP by measuring the UV/vis absorbance spectra of Hh PYP and Sr PYP in buffered H2O and D2O solutions (pH 7.0). This measurement revealed small but measurable shifts in the absorbance spectra of both Hh PYP and Sr PYP (Fig. 2, Table 1). The first derivatives of the absorbance spectra were used to extract the exact peak positions of the chromophore absorbance bands. This quantification of the SIE in Hh PYP and Sr PYP yielded two key observations. First, the λmax of Hh PYP at 445.7 nm exhibits a 1.8 nm redshift in D2O (Fig. 2), consistent with previous results (33). Upon incubation in D2O and the resulting H/D exchange, the λmax of Sr PYP at 431.8 nm is redshifted by 5.3 nm (Fig. 2). Thus, for both PYPs a SIE is observed. The direction of this SIE is consistent with the shift expected for an ionic hydrogen bond derived previously. We refer to this effect as an inverted SIE, because it is opposite of what would be expected for a standard hydrogen bond. Second, the inverted SIE for Sr PYP is larger by a factor three compared to that for Hh PYP.
Figure 2.
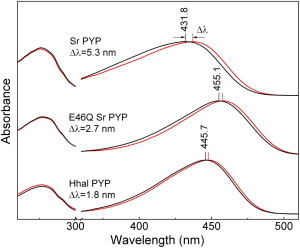
Spectral isotope effects in PYP. The UV/vis absorbance spectra of the PYPs from S. ruber (top two curves) and H. halophila (bottom two curves) and the E46Q mutant of Sr PYP (middle two curves) in H2O (black) and D2O (red). The shifts in the chromophore absorbance bands upon dissolving the proteins in D2O are indicated. To see this figure in color, go online.
Assignment and analysis of the SIE to the Glu-46-pCA hydrogen bond
As a first step toward a solid assignment of the H/D exchanging group responsible for the observed inverted SIE in PYP, we note that the ionized pCA molecule in PYP does not contain any exchangeable protons (see Fig. 1). Therefore, any shifts in λmax upon H/D exchange are caused by indirect effects through interactions between the pCA and side chain and/or backbone OH/NH groups in the protein that undergo H/D exchange. One possibility is that the observed SIE is the cumulative result of multiple very small effects caused by H/D exchange events at a number of sites. An alternative explanation is that the observed SIE is caused by an exchangeable group that directly interacts with the pCA. Below, we explicitly consider these two options.
The effects of mutations on the λmax of Hh PYP was studied through site-directed mutations at 22 residues selected based on the crystal structure of PYP (summarized in (12)), placing each of the 19 possible side chains at position 46 (25,26), and a complete alanine scan of PYP (34). The perturbations caused by these mutations in general tend to cause small spectral shifts both toward the blue and the red sides of the spectrum. Notably, by far the largest shift is caused by substitutions at position 46, implying that this residue plays an unusual role in the spectral tuning of PYP. Based on these data it would be expected that in the case that a substantial number of H/D exchange events affect the λmax of Sr PYP, these independent small effects would cause small shifts in λmax both toward the blue and toward the red, and would therefore largely cancel out (Δλmax≈0). Another consideration is that thermodynamic stability of proteins against unfolding is known to be slightly affected by H/D exchange (35). However, in systematic mutation studies of Hh PYP no correlation was found between the thermodynamic stability of a mutant and its λmax (34). This implies that the stability of PYP against unfolding is not a mechanism involved in spectral tuning in this protein, and therefore a small change in its stability caused by H/D exchange would not be expected to alter its λmax. These considerations argue against an explanation of the observed SIE based on small shifts in λmax caused by H/D exchange events at multiple different sites.
In considering a possible origin of the SIE in the H/D exchange of a group in the protein binding pocket of the pCA it should be noted that the weak interactions between the chromophore and the protein, such as hydrogen bonding and counterions, are electronic in nature, whereas H/D exchange is a nuclear perturbation that does not change electronic structure. In the case of hydrogen bonding, H/D exchange causes a ∼5% change in the strength of the interaction. Thus, the weak interaction that is affected by H/D exchange resulting in the observed SIE would need to have a substantial effect on λmax. Based on the crystal structure of PYP the side-chain Arg-52 was initially proposed to be the counter ion for the negatively charged pCA (36). However, subsequent studies on the R52A and R52Q mutants have revealed only relatively small effects on both the λmax and pKa of the pCA. Based on the small effects of the much more dramatic R52A and R52Q mutations on the absorbance spectrum of PYP, we conclude that the effect of H/D exchange of Arg-52 would be too weak to cause the observed SIE. This leaves the phenolic OH group of Tyr-42 and the COOH group of Glu-46 as candidates; mutations at both residues have relatively strong effects on the λmax of Hh PYP. In addition, the backbone NH group of Cys-69, which forms a hydrogen bond between the carbonyl oxygen of the pCA is a candidate for the H/D exchanging group contributing to the observed SIE; published NMR measurements (37) indicate that the backbone amide of Cys-69 indeed will undergo H/D exchange under the measurement conditions used here. The side chain of Glu-46 is the most likely option, because the SIE predicted based on the H/D exchange of this side chain derived previously matches the observed effect fairly well.
To experimentally test the possibility that the significant SIE in Sr PYP is due to the hydrogen bonds of the pCA with Tyr-42 or Glu-46, and to dissect the contributions of these two interactions to the SIE, we prepared the Y42F and E46Q mutants of Sr PYP. These mutants strongly diminish their hydrogen bonding to the pCA and would therefore also reduce the SIE associated with these hydrogen bonds. Y42F Sr PYP exhibited a very low expression yield and significant instability, precluding its purification for spectroscopic studies. For the E46Q mutant of Sr PYP we detected a 2.7 nm redshift in its λmax at 455.1 nm upon incubation in D2O (Fig. 2). The ∼twofold reduction in SIE of E46Q Sr PYP compared to wild-type Sr PYP establishes that the strong hydrogen bonding interaction between Glu-46 and the pCA is directly involved in this effect. The E46Q mutant is a particularly attractive variant to study the SIE in PYP, because it is known to cause only local perturbations at the pCA binding pocket (20,22). Further evidence for this assignment is provided by the similarities in the behavior of the SIE in Sr PYP and Hh PYP, which share only 28% amino acid sequence identity but retain functionally important active site residues: for both PYPs i), the λmax in D2O is shifted toward the red and ii), the SIE is substantially reduced SIE by the E46Q mutation (Fig. 2 and Table 1).
Structural and spectroscopic studies provide a framework for the interpretation of these spectroscopic data, and indicate two factors that are important for understanding the inverted SIE of PYP. The first factor is that the hydrogen bonds of Tyr-42 and Glu-46 with the pCA are short. X-ray crystallographic studies at 0.82 and 1.00 Å resolution have revealed that the hydrogen bonds of side chains of Glu-46 and Tyr-42 to the pCA chromophore are 2.6 and 2.5 Å, respectively (13,20) (Fig. 1 B), and thus are significantly shorter than standard hydrogen bonds. Neutron diffraction studies confirmed the presence of these short hydrogen bonds (38,39). Independent information on the presence of these strong short hydrogen bonds with the pCA was obtained by both Fourier transform infrared spectroscopy of the C=O stretching mode of the side chain of Glu-46 in Hh PYP (14,15) and by NMR spectroscopy of the far-downfield proton NMR signals of Tyr-42 and Glu-46 (21). The second consideration is that the side chains of Tyr-42 and Glu-46 are neutral, whereas the pCA in PYP is ionized (40). Thus, the negatively charged phenolic oxygen of the pCA interacts with the neutral side chains of Tyr-42 and Glu-46 via a forked ionic hydrogen bond (13,20,39). The short length and ionic nature of the Glu-46-pCA hydrogen bond are in excellent agreement with the assignment of a SIE to this interaction.
The role of the hydrogen bond between the backbone NH group of Cys-69, and the carbonyl oxygen of the pCA in spectral tuning of PYP is difficult to directly study, because the backbone atom of Cys-69 involved cannot be altered through site-directed mutagenesis. Okamoto et al. (41,42) approached this problem through the use of model compounds that combine a pCA moiety with compounds either capable or incapable of forming a 7-membered ring through intramolecular hydrogen bonding. Measurements of these deprotonated chromophore model compounds in an aprotic solvent (tetrahydrofuran) revealed that disruption of this hydrogen bond shifts the absorbance maximum of the model compound from ∼430 nm to ∼415 nm (see Fig. 5, a and b in (41)). These results indicate that the Cys-69-chromophore hydrogen bond in PYP contributes to its relatively redshifted absorbance maximum at 446 nm in the native protein. This finding implies that an increase in hydrogen bond strength between the pCA and the backbone amide group of Cys-69 results in a redshift in the λmax of PYP. Since this hydrogen bond has a standard length near 2.8 Å and because most of the negative charge of the deprotonated chromophore in PYP is centered near the phenolic oxygen, the pCA-Cys-69 hydrogen bond is of the standard type. Therefore, its H/D exchange will result in a small increase in its strength, which would be expected to cause a small redshift of the λmax of PYP upon H/D exchange. Because this shift is in the direction that we observed experimentally, we need to consider the contribution of this effect to the SIE in PYP.
Based on the assumption that results for the previous model compounds are directly applicable to PYP, the disruption of the Cys-69-pCA hydrogen bond causes a 840 cm−1 blueshift (from 430 to 415 nm), whereas disruption of the Glu-46-pCA hydrogen bond (in E46I PYP) causes a 1501 cm−1 redshift (from 446 to 478 nm). This consideration indicates that the effect of the Glu-46-pCA hydrogen bond on the λmax of PYP is substantially stronger than that of the Cys-69-pCA hydrogen bond. In the case of the E46Q mutant of Hhal PYP the hydrogen bond between residue 46 is lengthened by 11% (from 2.58 to 2.87 Å), whereas the Cys-69-pCA is lengthened by 3% (from 2.72 to 2.80 Å). This analysis supports the interpretation that the reduced SIE in E46Q PYP is largely caused by a weakening of the hydrogen bond between the pCA and residue 46. We conclude that a possible contribution of the Cys-69-pCA hydrogen bond to the observed SIE in PYP does not affect our assignment of a large part of this effect to the Glu-46-pCA hydrogen bond.
The observation that the inverted SIE for Sr PYP is significantly larger than that for Hh PYP leads to the proposal that the Glu-46-pCA hydrogen bond in Sr PYP is stronger than that in Hh PYP. This would be unexpected, because in Hh PYP this hydrogen bond already is quite strong and short (13,20,21,39). Two considerations provide direct support for this interpretation. First, the λmax of Sr PYP is significantly blueshifted compared to that of Hh PYP. An increase in the strength of the Glu-46-pCA hydrogen bond would be expected to cause such a blueshift. Second, the E46Q mutation causes a strong 1185 cm−1 redshift in the λmax of Sr PYP at 457.8 nm (Fig. 2) compared to the smaller 721 cm−1 redshift caused by the same mutation in Hh PYP (23,24). These results support the notion that the extent of the SIE in PYP reflects the strength of the ionic hydrogen bond between Glu-46 and the pCA. In Fig. 3 we summarize the spectroscopic readout of the strength of the ionic hydrogen bond between residue 46 and the pCA at the PYP active site through an inverted SIE.
Figure 3.
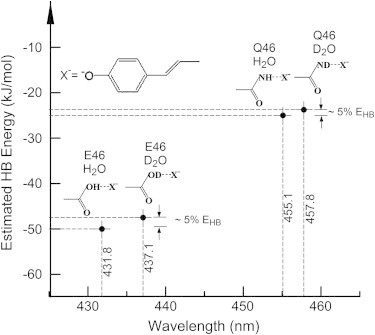
Effects of H/D exchange on active site hydrogen bonding strength and absorbance maximum in PYP. The estimated weakening of the hydrogen bond between residue 46 and the chromophore upon H/D exchange, and the resulting experimentally detected redshift in absorbance maximum are depicted for the PYP from Salinibacter ruber and its E46Q mutant.
Classification of hydrogen bonds with qualitatively different properties
Currently the most useful classification of hydrogen bonding interactions in proteins is debated. Some authors have proposed a classification based on hydrogen bonding strength, with strong (60–170 kJ/mol), moderate (15–60 kJ/mol), and weak (<15 kJ/mol) hydrogen bonds (43). Others have emphasized the shape of the hydrogen bonding potential as double-well, single-well, and LBHB (44). Yet another classification, introduced in studies on the role of C-H⋅⋅⋅O hydrogen bonds, focuses on the atom type to which the hydrogen atom forming the hydrogen bond is connected (6). Particularly the role that ionic hydrogen bonds (7,45), in which a net charge is present on the hydrogen bond donor-acceptor pair, play in proteins remains unclear.
Based on the results reported here in combination with analysis of the wealth of biophysical data available on PYP (see (8,9)) and drawing on the large body of information available on hydrogen bonding (7,43,45), we propose that it is helpful to distinguish three qualitatively different types of hydrogen bonds involving N and O atoms (Fig. 4). Based on published literature (7,43,45), these hydrogen bonds can be distinguished by their behavior along two key reaction coordinates. First, the distance between the heteroatoms in the hydrogen bond donor-acceptor pair defines the length and strength of the hydrogen bond (Fig. 4 A). Second, the position of the proton between these two heteroatoms describes proton transfer along the hydrogen bond (Fig. 4 B). This approach leads to the following three types of hydrogen bonds: i), standard hydrogen bonds, such as those that hold together α-helices, with a length close to 2.8 Å and a typical strength of ∼15 kJ/mol (43); ii), ionic hydrogen bonds, with a length close to 2.6 Å and a typical strength near 50 kJ/mol (7,45); and iii), LBHBs with a length close to 2.4 Å and a typical strength of ∼100 kJ/mol (5,7). Although the lengths of these three types of hydrogen bonds are quite well defined, their energies are likely to vary substantially depending on the chemical details and molecular environment of the hydrogen bond. Of these three types of hydrogen bonds only the LBHB acts as a single well system for proton transfer. This analysis implies that the presence of a short, ionic hydrogen bond is distinct from a LBHB. Thus, the strong short hydrogen bonds often referred to in various protein systems (e.g., (46)) can in fact involve two distinct types of hydrogen bonding.
Figure 4.
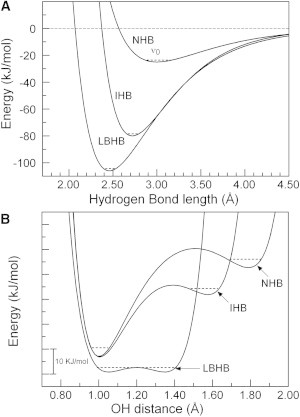
Distinction between three types of hydrogen bonds. The distance between heteroatoms in the hydrogen bond donor-acceptor pair (A) define the equilibrium length and strength of hydrogen bonds, whereas the position of the proton along this hydrogen bond (B) determines the potential energy for proton transfer between the hydrogen bond donor and acceptor. These two reaction coordinates lead to a distinction between normal hydrogen bonds (NHB) with a length of 3.00 (±0.20) Å, ionic hydrogen bond (IHB) with a length of 2.72 (±0.10) Å, and low-barrier hydrogen bonds (LBHB) with a length of 2.46 (±0.03) Å. The depicted potential energy curves are intended to reflect the qualitative differences between the three types of hydrogen bonds, while depicting physically reasonable values. Quantitative information on the shapes of these energy landscapes were obtained from (52–54). Although ionic hydrogen bonds and LBHBs are both considerably stronger than standard hydrogen bonds, the values for their strength are less well defined and likely to vary considerably depending on the details of the chemical nature of the donor-acceptor pair and their physical environment. Likewise, the exact values for the barriers and energy differences for proton transfer in panel B are intended to illustrate the differences between the three types of hydrogen bonds; the absolute energies of the right wells and the vibrational levels are for illustrative purposes. The depicted values were chosen to reflect hydrogen bonds involving O (and N) heteroatoms. Normal hydrogen bonds occur abundantly in proteins; ionic hydrogen bonds are found at some active sites and occur often on the surface of protein. LBHBs are rare and often are present at the active site of the protein.
Relevance of ionic hydrogen bonding for proteins
The value of the previous classification of hydrogen bonds is illustrated by the recent debate about a functionally important and short hydrogen bond at the active site of PYP: different research groups have reported that this hydrogen bond is (39) or is not (38,47) a LBHB. We propose that the truth is likely to be in between, namely that this active site hydrogen bond is a strong, ionic hydrogen bond without being a LBHB, i.e., the second class of hydrogen bonds depicted in Fig. 4. This proposal reconciles the short length of the hydrogen bond with vibrational spectroscopy showing that in the dark state of PYP the side chain of Glu-46 is protonated (14,15) whereas the pCA is ionized, and is in line with recent computational results (47).
Ionic hydrogen bonds in proteins are of considerable biological interest. They are substantially shorter and often much stronger than standard hydrogen bonds (7,45), and have been implicated to be important features at the active sites of a range of proteins, including green fluorescent protein (48), serine protease (49), HIV protease (50), acetylcholinesterase, ketosteroid isomerase (46), and PYP (39,47). However, until recently these structural and spectroscopic results have often been interpreted without taking into account the unusual properties of the ionic hydrogen bond. A relevant example is that the hydrogen bonds between the pCA and the side chains of Tyr-42 and Glu-46 have been described as unusually short (20,38). However, the length of these hydrogen bonds is what would be expected for an ionic hydrogen bond, and therefore is unusual only if the properties of these hydrogen bonds are considered within the framework of standard hydrogen bonds. The unique properties of ionic hydrogen bonds are also responsible for the inverted nature of the SIE reported here.
Here, we report and assign a SIE in the electronic absorbance spectrum of the bacterial blue light receptor PYP. Previously, small shifts in the absorbance peaks of both PYP (33) and green fluorescent protein (51) were reported as uninterpreted observations. We provide a conceptual framework for understanding such spectral isotope effects, report the experimental assignment of the inverted SIE to the ionic Glu-46-pCA hydrogen bond, and demonstrate how SIE effects can be used as a spectroscopic readout of the presence and strength of such ionic hydrogen bonds. Determining hydrogen bonding strength in proteins remains an important challenge in protein biophysics, and the values reported in Table 1 are tentative estimates. However, the results and analysis presented here allow a firm assignment and analysis of the SIE in PYP, and draw attention to the importance of distinguishing between ionic and LBHB interactions. The results are likely to be widely applicable to systems that exhibit predictable spectral shifts in an electronic transition upon changes in hydrogen bonding and indicate that the inverted SIE provides a novel, to our knowledge, tool to probe ionic hydrogen bonds. In general, a SIE is expected for all photoreceptors in which the λmax of the photoreceptor under study is affected by hydrogen bonds between the chromophore and groups in its binding pocket, or more precisely, that an ∼5% change in active site hydrogen bond strength leads to a measurable change in λmax. When the relationship between the strength of the hydrogen bond between the chromophore and this H/D exchanging group is known, the SIE provides a direct spectroscopic readout of the relative strength of the hydrogen bond versus deuterium bond involved. For ionic hydrogen bonds a weakening of the interaction strength is expected upon H/D exchange of the bridging hydrogen in the hydrogen bond.
Conclusions
Although a large body of published work exists on spectral tuning in proteins, the phenomenon of a spectral isotope effect in protein absorbance spectra has remained largely unstudied. The lack of data on the SIE stands in stark contrast to the wide use of isotope effects in proteins in both infrared spectroscopy to assign vibrational modes and via kinetic isotope effects in studies of enzyme reaction mechanisms. Here, we report the detection, assignment, and interpretation of a spectral isotope effect in PYP. Quantitative considerations, published information on the dependence of the λmax of PYP on the strength of the hydrogen bond between residue 46 and the pCA, and the observation that the E46Q mutation reduces the size of the SIE ∼twofold, provide strong evidence that H/D exchange of the side chain of Glu-46 plays a major role in causing this effect. The observation that the SIE in Sr PYP is larger than that for Hh PYP indicates that the Glu-46-pCA hydrogen bond in Sr PYP is even stronger than the corresponding hydrogen bond in Hh PYP. The direction of the observed SIE (toward the red) matches the ionic nature of the Glu-46-pCA hydrogen bond. These results illustrate the role that ionic hydrogen bonds can play in protein active sites, and underscore the importance of taking into account the qualitative and quantitative differences in the properties of standard, ionic, and low barrier hydrogen bonds.
Acknowledgments
This work was funded by National Science Foundation (NSF) grant MCB-1051590 to W.D.H. and Oklahoma Center for the Advancement of Science and Technology (OCAST) grant HR10-078 to A.X.
Footnotes
Sandip Kaledhonkar and Miwa Hara contributed equally to this work.
References
- 1.Pauling L., Corey R.B., Branson H.R. The structure of proteins; two hydrogen-bonded helical configurations of the polypeptide chain. Proc. Natl. Acad. Sci. USA. 1951;37:205–211. doi: 10.1073/pnas.37.4.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kabsch W., Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 3.Gutteridge A., Thornton J.M. Understanding nature’s catalytic toolkit. Trends Biochem. Sci. 2005;30:622–629. doi: 10.1016/j.tibs.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 4.Shan S.O., Loh S., Herschlag D. The energetics of hydrogen bonds in model systems: implications for enzymatic catalysis. Science. 1996;272:97–101. doi: 10.1126/science.272.5258.97. [DOI] [PubMed] [Google Scholar]
- 5.Cleland W.W., Kreevoy M.M. Low-barrier hydrogen bonds and enzymic catalysis. Science. 1994;264:1887–1890. doi: 10.1126/science.8009219. [DOI] [PubMed] [Google Scholar]
- 6.Manikandan K., Ramakumar S. The occurrence of C-H center dot center dot center dot O hydrogen bonds in alpha-helices and helix termini in globular proteins. Proteins Struct. Funct. Bioinf. 2004;56:768–781. doi: 10.1002/prot.20152. [DOI] [PubMed] [Google Scholar]
- 7.Perrin C.L., Nielson J.B. “Strong” hydrogen bonds in chemistry and biology. Annu. Rev. Phys. Chem. 1997;48:511–544. doi: 10.1146/annurev.physchem.48.1.511. [DOI] [PubMed] [Google Scholar]
- 8.Cusanovich M.A., Meyer T.E. Photoactive yellow protein: a prototypic PAS domain sensory protein and development of a common signaling mechanism. Biochemistry. 2003;42:4759–4770. doi: 10.1021/bi020690e. [DOI] [PubMed] [Google Scholar]
- 9.Hellingwerf K.J., Hendriks J., Gensch T. Photoactive Yellow Protein, a new type of photoreceptor protein: Will this “yellow lab” bring us where we want to go? J. Phys. Chem. A. 2003;107:1082–1094. [Google Scholar]
- 10.Meyer T.E. Isolation and characterization of soluble cytochromes, ferredoxins and other chromophoric proteins from the halophilic phototrophic bacterium Ectothiorhodospira halophila. Biochim. Biophys. Acta. 1985;806:175–183. doi: 10.1016/0005-2728(85)90094-5. [DOI] [PubMed] [Google Scholar]
- 11.Hoff W.D., Düx P., Hellingwerf K.J. Thiol ester-linked p-coumaric acid as a new photoactive prosthetic group in a protein with rhodopsin-like photochemistry. Biochemistry. 1994;33:13959–13962. doi: 10.1021/bi00251a001. [DOI] [PubMed] [Google Scholar]
- 12.Kumauchi M., Hara M.T., Hoff W.D. Identification of six new photoactive yellow proteins—diversity and structure-function relationships in a bacterial blue light photoreceptor. Photochem. Photobiol. 2008;84:956–969. doi: 10.1111/j.1751-1097.2008.00335.x. [DOI] [PubMed] [Google Scholar]
- 13.Getzoff E.D., Gutwin K.N., Genick U.K. Anticipatory active-site motions and chromophore distortion prime photoreceptor PYP for light activation. Nat. Struct. Biol. 2003;10:663–668. doi: 10.1038/nsb958. [DOI] [PubMed] [Google Scholar]
- 14.Xie A.H., Hoff W.D., Hellingwerf K.J. Glu46 donates a proton to the 4-hydroxycinnamate anion chromophore during the photocycle of photoactive yellow protein. Biochemistry. 1996;35:14671–14678. doi: 10.1021/bi9623035. [DOI] [PubMed] [Google Scholar]
- 15.Xie A.H., Kelemen L., Hoff W.D. Formation of a new buried charge drives a large-amplitude protein quake in photoreceptor activation. Biochemistry. 2001;40:1510–1517. doi: 10.1021/bi002449a. [DOI] [PubMed] [Google Scholar]
- 16.Kroon A.R., Hoff W.D., Hellingwerf K.J. Spectral tuning, fluorescence, and photoactivity in hybrids of photoactive yellow protein, reconstituted with native or modified chromophores. J. Biol. Chem. 1996;271:31949–31956. doi: 10.1074/jbc.271.50.31949. [DOI] [PubMed] [Google Scholar]
- 17.Memmi S., Kyndt J., Van Beeumen J. Photoactive yellow protein from the halophilic bacterium Salinibacter ruber. Biochemistry. 2008;47:2014–2024. doi: 10.1021/bi701486n. [DOI] [PubMed] [Google Scholar]
- 18.Nielsen I.B., Boyé-Péronne S., Andersen L.H. Absorption spectra of photoactive yellow protein chromophores in vacuum. Biophys. J. 2005;89:2597–2604. doi: 10.1529/biophysj.105.061192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rocha-Rinza T., Christiansen O., Nielsen M.B. Gas phase absorption studies of photoactive yellow protein chromophore derivatives. J. Phys. Chem. A. 2009;113:9442–9449. doi: 10.1021/jp904660w. [DOI] [PubMed] [Google Scholar]
- 20.Anderson S., Crosson S., Moffat K. Short hydrogen bonds in photoactive yellow protein. Acta Crystallogr. D Biol. Crystallogr. 2004;60:1008–1016. doi: 10.1107/S090744490400616X. [DOI] [PubMed] [Google Scholar]
- 21.Sigala P.A., Tsuchida M.A., Herschlag D. Hydrogen bond dynamics in the active site of photoactive yellow protein. Proc. Natl. Acad. Sci. USA. 2009;106:9232–9237. doi: 10.1073/pnas.0900168106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sugishima M., Tanimoto N., Fukuyama K. Structure of photoactive yellow protein (PYP) E46Q mutant at 1.2 A resolution suggests how Glu46 controls the spectroscopic and kinetic characteristics of PYP. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2305–2309. doi: 10.1107/S0907444904024084. [DOI] [PubMed] [Google Scholar]
- 23.Genick U.K., Devanathan S., Getzoff E.D. Active site mutants implicate key residues for control of color and light cycle kinetics of photoactive yellow protein. Biochemistry. 1997;36:8–14. doi: 10.1021/bi9622884. [DOI] [PubMed] [Google Scholar]
- 24.Mihara K., Hisatomi O., Tokunaga F. Functional expression and site-directed mutagenesis of photoactive yellow protein. J. Biochem. 1997;121:876–880. doi: 10.1093/oxfordjournals.jbchem.a021668. [DOI] [PubMed] [Google Scholar]
- 25.Philip A.F., Eisenman K.T., Hoff W.D. Functional tuning of photoactive yellow protein by active site residue 46. Biochemistry. 2008;47:13800–13810. doi: 10.1021/bi801730y. [DOI] [PubMed] [Google Scholar]
- 26.Philip A.F., Nome R.A., Hoff W.D. Spectral tuning in photoactive yellow protein by modulation of the shape of the excited state energy surface. Proc. Natl. Acad. Sci. USA. 2010;107:5821–5826. doi: 10.1073/pnas.0903092107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Imamoto Y., Ito T., Tokunaga F. Reconstitution photoactive yellow protein from apoprotein and p-coumaric acid derivatives. FEBS Lett. 1995;374:157–160. doi: 10.1016/0014-5793(95)01096-w. [DOI] [PubMed] [Google Scholar]
- 28.Benjamin L., Benson G.C. A deuterium isotope effect on the excess enthalpy of methanol—water solutions. J. Phys. Chem. 1963;67:858–861. [Google Scholar]
- 29.Scheiner S., Čuma M. Relative stability of hydrogen and deuterium bonds. J. Am. Chem. Soc. 1996;118:1511–1521. [Google Scholar]
- 30.Scheiner S. Calculation of isotope effects from first principles. Biochim. Biophys. Acta. 2000;1458:28–42. doi: 10.1016/s0005-2728(00)00058-x. [DOI] [PubMed] [Google Scholar]
- 31.Benedict H., Limbach H.H., Janoschek R. Solid state N-15 NMR and theoretical studies of primary and secondary geometric H/D isotope effects on low-barrier NHN-hydrogen bonds. J. Am. Chem. Soc. 1998;120:2939–2950. [Google Scholar]
- 32.Kita Y., Kamikubo H., Tachikawa M. Theoretical analysis of the geometrical isotope effect on the hydrogen bonds in photoactive yellow protein with multi-component density functional theory. Chem. Phys. 2013;419:50–53. [Google Scholar]
- 33.Hendriks J., van Stokkum I.H.M., Hellingwerf K.J. Deuterium isotope effects in the photocycle transitions of the photoactive yellow protein. Biophys. J. 2003;84:1180–1191. doi: 10.1016/S0006-3495(03)74932-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Philip A.F., Kumauchi M., Hoff W.D. Robustness and evolvability in the functional anatomy of a PER-ARNT-SIM (PAS) domain. Proc. Natl. Acad. Sci. USA. 2010;107:17986–17991. doi: 10.1073/pnas.1004823107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krantz B.A., Moran L.B., Sosnick T.R. D/H amide kinetic isotope effects reveal when hydrogen bonds form during protein folding. Nat. Struct. Biol. 2000;7:62–71. doi: 10.1038/71265. [DOI] [PubMed] [Google Scholar]
- 36.Borgstahl G.E.O., Williams D.R., Getzoff E.D. 1.4 A structure of photoactive yellow protein, a cytosolic photoreceptor: unusual fold, active site, and chromophore. Biochemistry. 1995;34:6278–6287. doi: 10.1021/bi00019a004. [DOI] [PubMed] [Google Scholar]
- 37.Craven C.J., Derix N.M., Kaptein R. Probing the nature of the blue-shifted intermediate of photoactive yellow protein in solution by NMR: hydrogen-deuterium exchange data and pH studies. Biochemistry. 2000;39:14392–14399. doi: 10.1021/bi001628p. [DOI] [PubMed] [Google Scholar]
- 38.Fisher S.Z., Anderson S., Schultz A.J. Neutron and X-ray structural studies of short hydrogen bonds in photoactive yellow protein (PYP) Acta Crystallogr. D Biol. Crystallogr. 2007;63:1178–1184. doi: 10.1107/S0907444907047646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamaguchi S., Kamikubo H., Kataoka M. Low-barrier hydrogen bond in photoactive yellow protein. Proc. Natl. Acad. Sci. USA. 2009;106:440–444. doi: 10.1073/pnas.0811882106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim M., Mathies R.A., Hellingwerf K.J. Resonance Raman evidence that the thioester-linked 4-hydroxycinnamyl chromophore of photoactive yellow protein is deprotonated. Biochemistry. 1995;34:12669–12672. doi: 10.1021/bi00039a024. [DOI] [PubMed] [Google Scholar]
- 41.Okamoto K., Hamada N., Yamamoto H. Color regulation and stabilization of chromophore by Cys69 in photoactive yellow protein active center. Org. Biomol. Chem. 2009;7:3782–3791. doi: 10.1039/b905835d. [DOI] [PubMed] [Google Scholar]
- 42.Okamoto K., Hamada N., Yamamoto H. Investigation of the effect of the NH···OC hydrogen bond from Cys69 to PYP chromophore using novel active-center model compound. Chem. Lett. 2009;38:456–457. [Google Scholar]
- 43.Steiner T. The hydrogen bond in the solid state. Ang. Chem. Intl. Ed. 2002;41:49–76. doi: 10.1002/1521-3773(20020104)41:1<48::aid-anie48>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 44.Guthrie J.P. Short strong hydrogen bonds: can they explain enzymic catalysis? Chem. Biol. 1996;3:163–170. doi: 10.1016/s1074-5521(96)90258-6. [DOI] [PubMed] [Google Scholar]
- 45.Meot-Ner M. The ionic hydrogen bond. Chem. Rev. 2005;105:213–284. doi: 10.1021/cr9411785. [DOI] [PubMed] [Google Scholar]
- 46.Mildvan A.S., Massiah M.A., Kovach I.M. Short, strong hydrogen bonds on enzymes: NMR and mechanistic studies. J. Mol. Struct. 2002;615:163–175. [Google Scholar]
- 47.Saito K., Ishikita H. Energetics of short hydrogen bonds in photoactive yellow protein. Proc. Natl. Acad. Sci. USA. 2012;109:167–172. doi: 10.1073/pnas.1113599108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brejc K., Sixma T.K., Remington S.J. Structural basis for dual excitation and photoisomerization of the Aequorea victoria green fluorescent protein. Proc. Natl. Acad. Sci. USA. 1997;94:2306–2311. doi: 10.1073/pnas.94.6.2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fuhrmann C.N., Daugherty M.D., Agard D.A. Subangstrom crystallography reveals that short ionic hydrogen bonds, and not a His-Asp low-barrier hydrogen bond, stabilize the transition state in serine protease catalysis. J. Am. Chem. Soc. 2006;128:9086–9102. doi: 10.1021/ja057721o. [DOI] [PubMed] [Google Scholar]
- 50.Das A., Mahale S., Hosur M.V. X-ray snapshot of HIV-1 protease in action: observation of tetrahedral intermediate and short ionic hydrogen bond SIHB with catalytic aspartate. J. Am. Chem. Soc. 2010;132:6366–6373. doi: 10.1021/ja100002b. [DOI] [PubMed] [Google Scholar]
- 51.Shi X., Abbyad P., Boxer S.G. Ultrafast excited-state dynamics in the green fluorescent protein variant S65T/H148D. 2. Unusual photophysical properties. Biochemistry. 2007;46:12014–12025. doi: 10.1021/bi700904a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Freindorf M., Kraka E., Cremer D. A comprehensive analysis of hydrogen bond interactions based on local vibrational modes. Int. J. Quantum Chem. 2012;112:3174–3187. [Google Scholar]
- 53.Herzog E., Frigato T., Lancaster C.R. Energy barriers of proton transfer reactions between amino acid side chain analogs and water from ab initio calculations. J. Comput. Chem. 2006;27:1534–1547. doi: 10.1002/jcc.20442. [DOI] [PubMed] [Google Scholar]
- 54.Sadhukhan S., Muñoz D., Scuseria G.E. Predicting proton transfer barriers with density functional methods. Chem. Phys. Lett. 1999;306:83–87. [Google Scholar]