

Abstract
While the pathways that permit IL-2 production and the full activation of T cells upon antigen encounter are fairly well defined, the negative regulatory circuits that limit these pathways are poorly understood. Here we show that the E3 ubiquitin ligase adaptor Ndfip1 directs one such negative regulatory circuit. T cells lacking Ndfip1 produce IL-2, upregulate IL-2 receptor alpha (IL-2Rα), and proliferate, in the absence of CD28 co-stimulation. Furthermore, T cells in mice lacking both Ndfip1 and CD28 become activated, produce IL-4, and drive inflammation at barrier surfaces. Ndfip1 constrains T cell activation by limiting the duration of IL-2 mRNA expression after TCR stimulation. Ndfip1 and IL-2 have a similar expression pattern and, following TCR stimulation, expression of both Ndfip1 and IL-2 require the activity of NFAT and Erk. Taken together, these data support a negative regulatory circuit in which factors that induce IL-2 expression downstream of TCR engagement also induce the expression of Ndfip1 to limit the extent of IL-2 production and, thus, dampen T cell activation.
INTRODUCTION
Upon T cell receptor (TCR) stimulation, various signaling cascades are initiated that instruct T cells towards the appropriate response. For example, when T cells see their cognate antigen in the presence of co-stimulation they produce and secrete IL-2 (1, 2). Autocrine IL-2 receptor signaling initiates a positive feedback loop that further increases IL-2 and IL-2Rα expression, and triggers proliferation (3). Co-stimulatory signals are key to this process by complementing the signals received from the T cell receptor, thus, boosting IL-2 production.
In contrast, T cells that receive signals only through their TCR produce poor amounts of IL-2 and do not proliferate (4,5). This is partly due to a lack of co-receptor signals that supplement the production of IL-2. This is also because, in the absence of co-stimulation, T cells activate mechanisms that actively suppress IL-2 expression (6–8). While the pathways downstream of T cell activation that promote IL-2 production have been characterized, less is known about pathways that actively repress IL-2 production.
One way to repress IL-2 production and secretion is by reducing the levels or functions of signaling proteins by E3 ubiquitin ligases. E3 ubiquitin ligases that restrain T cell activation include Casitas B cell lymphoma-b (Cbl-b), gene regulating anergy in lymphocytes (Grail) and Itch (6,9). These factors can dampen signaling downstream of the T cell receptor by blocking protein-protein interactions or by ubiquitylating and degrading signaling proteins (9–12). For example, Itch and Cbl-b have been shown to increase the rate of degradation of PKCθ and PLCγ1 in effector T cells stimulated in the absence of co-stimulation (9).
Itch is a homologous to the E6-AP carboxyl terminus (HECT)-type E3 ubiquitin ligase of the Neural-precursor cell expressed and developmentally downregulated 4 (Nedd4)-family. Nedd4-family E3 ubiquitin ligases have intrinsic catalytic activity and can directly mediate the transfer of ubiquitin to substrate proteins (13). While Itch, WWP2 and Nedd4 have known functions in T cells (9,14–16), a role for the other 6 Nedd4-family members in T cells has yet to be defined. In vitro, most members of this family have been shown to associate with the membrane-tethered adaptor Nedd4-family interacting protein 1 (Ndfip1) and its only relative Ndfip2 (17–19).
While Ndfip1 has been shown to bind most Nedd4-family members in vitro (18), to date it has only been shown to interact with Itch in primary T cells (17). Both Itchy-mutant (mice lacking Itch) and Ndfip1−/− mice develop TH2-mediated inflammation at barrier surfaces, including the skin, gastrointestinal (GI) tract and lung (14,17). This is in part because, in antigen experienced T cells, both Ndfip1 and Itch are required for ubiquitylation and degradation of JunB, a transcription factor that promotes IL-4 and IL-5 production (14,17). Accumulation of JunB in these cells leads to excessive IL-4 production and promotes the differentiation of T cells into TH2 cells (17). Moreover, IL-4 production by Itch or Ndfip1-deficient T cells leads to defective inducible T regulatory cell (iTreg) differentiation (20). These findings may help explain why both Ndfip1−/− and Itchy-mutant mice develop TH2-mediated inflammation.
In contrast to Itchy-mutant mice, which exhibit inflammation at 5 months of age, Ndfip1−/− mice develop inflammation by 6 weeks of age and do not survive beyond 13 weeks of age. Furthermore, T cells from 4–6 week old Ndfip1−/− mice display markers characteristic of activation (21), while T cells from Itchy-mutant mice do not (unpublished observation). This suggests that Ndfip1 might regulate other Nedd4-family members in T cells.
Due to the increased frequency of T cells with an activated phenotype in Ndfip1−/− mice we hypothesized that Ndfip1-deficient T cells lack a negative regulatory circuit that limits T cell activation. Here we show that naïve Ndfip1−/− T cells are hyperactive in response to TCR stimulation due to a T cell intrinsic defect. Loss of Ndfip1 leads to increased IL-2 production, elevated levels of CD25 expression, and proliferation in the absence of CD28 co-stimulation. Our data provide evidence that NFAT and Erk, which are essential for the expression of IL-2, also drive the expression of Ndfip1. Once expressed, Ndfip1 regulates the duration of IL-2 production and, thus, prevents T cells from becoming fully activated in the absence of co-stimulation.
MATERIALS AND METHODS
Mice
Ndfip1−/− and Itchy mutant mice have been described previously (14,17). CD45.1 (C57BL6. SJL-Ptprca Pepcb/BoyJ mice, #002014), IL-4−/− (B6.129P2-Il4tm1Cgn/J, #002253), CD28−/− (B6.129S2- Cd28tm1Mak/J, #002666) OT-II+ (B6. Cg-Tg (TcraTcrb) 425Cbn/J, #004194) and Rag1−/− (B6.129S7-Rag1tm1Mom/J, #002216) mice were purchased from the Jackson Laboratory. CD4-cre transgenic mice (B6. Cg-Tg (CD4-cre) 1Cwi N9, 4196) were purchased from Taconic. Ndfip1CD4-CKO mice were generated as described in Figure 2. All mice were housed in a barrier facility at the Children’s Hospital of Philadelphia in accordance with the Institutional Animal Care and Use Committee protocol. For genotyping, DNA from tail biopsies was amplified by PCR using the following primers: Ndfip1 WT Forward: 5′TAGGCCAAGGTGAAAACTGG3′; Ndfip1 WT Reverse: 5′AGAGGTGGGTTCAACAGTGG3′. Ndfip1 knockout Forward: 5′CGACTTCCAGTTCAACATCAGC3′; Ndfip1 knockout Reverse: 5′GTCTGTTGTGCCCAGTCATAGC3′. Primers for IL-4−/− CD28−/−, Rag1−/− and CD4-cre Tg mice are available on the Jackson Laboratories website (www.jaxmice.jax.org)
Figure 2. Activation of T cells in the absence of Ndfip1 is T cell intrinsic.

(A) Model illustrating T cell specific deletion of Ndfip1. The targeting vector was designed to introduce loxP sites on either side of exon 2 of the gene encoding Ndfip1. Correct insertion of the vector into the Ndfip1 locus is shown as ‘targeted Ndfip1 locus’. Selection cassettes were removed using flp recombinase. Mice harboring the floxed exon 2 were crossed to CD4-cre mice to generate mice with T cells lacking Ndfip1 as illustrated at bottom of panel. In this Ndfip1 locus, Cre-mediated deletion results in loss of Exon 2 and the remaining exons are out of frame (illustrated by frameshift). Ndfip2 is encoded on a separate chromosome and not directly affected by our knockout strategy. (B) Mice harboring the floxed allele were analyzed by PCR and representative results are shown in upper panel. The upper (slower migrating) band shows the WT locus and the faster migrating band shows the floxed locus. Presence of Cre was also determined by PCR and is shown in the lower panel. The slower migrating band is an internal positive control while the faster migrating band reflects the presence of Cre. Lane 3 shows the results indicating a mouse with a T cell specific deletion of Ndfip1 (designated by Ndfip1CD4-CKO). (C) Loss of Ndfip1 expression in Ndfip1CD4-CKO mice was analyzed using qPCR. WT and Ndfip1CD4- CKO CD4+ T cells were stimulated with anti-CD3 and anti-CD28 for 24 hours and analyzed using primers for Ndfip1. Ndfip1 mRNA levels were normalized to an internal control and data shown are relative to WT unstimulated levels. Bars represent the mean and lines show standard deviation of triplicate samples. Data are representative of 2 different experiments using a total of 4 mice per genotype. (D) Activation profiles of CD4+ gated T cells from spleens of mice of the indicated genotypes are shown. Gates indicate “activated” phenotype cells and numbers indicate the percentage of cells falling within that gate. Data are representative of mice from at least three independent experiments. At least 5 mice of each genotype were analyzed. (E) Sections of the esophagus of 6-week old mice were analyzed by histology using H and E stains. Images show representative sections from at least three different mice of each genotype. Bar illustrates 100 microns. (F) Cells were isolated from the esophagus and analyzed by flow cytometry for eosinophils (SiglecF+) or T cells (CD4+). Each dot represents percentages of cells from a single mouse, bars represent the mean. At least four mice were analyzed from each genotype. * represents a P value of <0.05.
Tissue processing and cell isolation
Spleen and lymph nodes were harvested and mashed through 70mm filters in cold Hank’s Balanced Salt Solution (HBSS). Cell suspensions from spleens were treated with ACK Lysis Buffer to lyse red blood cells.
Esophagus and a 3–4″ section of small bowel were flushed with cold PBS. Peyer’s patches were removed from small bowel. Organs were minced with scissors and treated with DNAse (20ug/ml, Sigma D5025), collagenase type I (.8mg/ml, Sigma C0130) and collagenase type Ia (.9mg/mL, Sigma C2674) in DMEM for 1 hour in end-over-end rotation at room temperature. Cell suspensions were filtered through a 100mm filter, then a 40mm filter and 10% FCS was added. Cells were incubated for 10 minutes at 4°C with Fc Block (2.4G2, BD Biosciences) prior to antibody staining.
Flow Cytometry, Cell Sorting and antibodies
Flow cytometry was performed using a FACSCalibur or a BD LSR II(BD Biosciences, San Diego, CA). For flow cytometry, cells were stained with fluorescently labeled antibodies in 3% fetal bovine serum in PBS for 30 minutes at 4°C and washed. Naïve CD4+ T cells were isolated by sorting spleen and lymph node cells for CD4+ CD25− CD44lo and CD62Lhi cells on the FACS Aria (BD Biosciences). CD25 (PC61.5) and CD62L (MEL-14) antibodies were obtained from eBiosciences (San Diego, CA). CD4 (GK1.5) and CD44 (IM7) antibodies were obtained from BioLegend (San Diego, CA). Siglec F (E502440) antibody was obtained from BD Biosciences (San Diego, CA).
Histology
Esophagus and sections of small bowel were dissected and fixed in 10% formalin for at least 24 hours. All organs were then embedded in paraffin, sectioned and stained with hematoxylin and eosin.
Enzyme-Linked Immunosorbent Assay (ELISA)
IL-2 and IL-4 ELISAs were performed on supernatants harvested at the indicated times from in vitro cell cultures. Assays were performed using Ready-Set-Go ELISA kits (eBiosciences) in Nunc-Immuno MicroWell 96 well solid plates (Thermo Scientific). Results were analyzed using a Synergy HT Microplate Reader (Bio Tek).
Co-Cultures, stimulation and CFSE
Naïve sorted CD4 T cells were stimulated with plate-bound anti-CD3 (145-2C11, BD Biosciences) with or without anti-CD28 (37.51, BD Biosciences) antibodies (as indicated) (5μg/mL) for time points as indicated. Percentage of live cells was determined using flow cytometry by live-cell gating of events on forward scatter by side scatter. For figure 7A, CD4 T cells (not sorted for naïve) were stimulated with plate-bound anti-CD3 and anti- CD28 (5μg/mL) for 3 days and left resting for 2 days in IL-2 (50 u/mL) (ro 23– 6019, Hoffman-LaRoche). Cells were then stimulated with ionomycin (0–3 uM) for 16 hours, rested for 4 hours and re-stimulated with anti-CD3 and CD28 antibodies (5μg/mL). Culture supernatants were collected 24 hours after re-stimulation. For figure 8, the following inhibitors were used: Cyclosporine A (NFAT inhibitor) (239835, EMD Millipore), PI3K inhibitor (LY294002) (PHZ1144, Invitrogen), JNK inhibitor (S5567, Sigma) and Erk inhibitor (#513001, Calbiochem). Inhibitors were added to cultures after the first 24 hours of stimulation. Carboxyfluorescein succinimidyl ester (CFSE) labeling: Cells were re-suspended at a 1×10^7/mL concentration in PBS at room temperature and mixed at a 1:1 ratio with CFSE (C-1157, Invitrogen) in PBS for 4 minutes with constant agitation. Labeling process was quenched with FCS. Co-culture assays: CD45.1 and CD45.2 cells were mixed in a 1:1 ratio and CFSE-labeled as described above. Cells were cultured in the presence of anti-IL-2 (S4B6, BD Biosciences) and IL-4 (11B11, Biolegend) antibodies where specified.
Figure 7. Ndfip1−/− T cells express more IL-2 mRNA upon TCR stimulation.
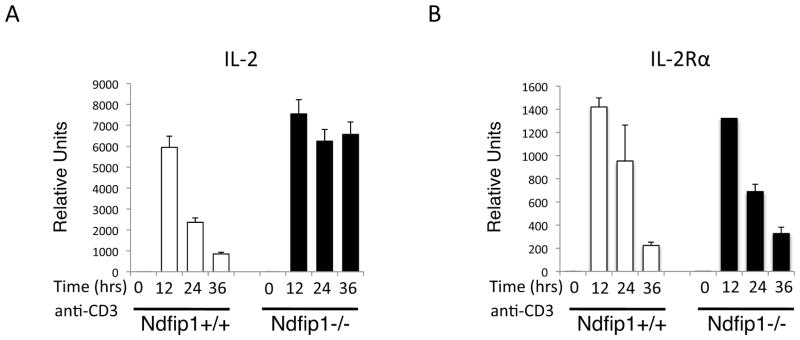
(A–B) Naïve CD4+ T cells were stimulated with anti-CD3 and mRNA expression for IL-2 (A) and IL-2Rα (B) was analyzed at the indicated time points. mRNA levels were normalized to an internal control and data shown are relative to WT unstimulated levels. Bars represent the mean and lines show standard deviation of triplicate samples. Data is representative of 2 different experiments using at least total of 4 mice per genotype.
Figure 8. Inhibitors for NFAT and Erk prevent the expression of Ndfip1 in TCR-stimulated cells.

(A–B) Naïve CD4+ T cells were stimulated with anti-CD3 and Ndfip1 mRNA levels were measured at the indicated time points (A) or after overnight stimulation (B). (B) Cells were cultured in the presence of the indicated inhibitors. Cyclosporine A (cspA) inhibits NFAT. (A–B) mRNA levels were normalized to an internal control and data shown are relative to unstimulated levels. Bars represent the mean and lines show standard deviation of triplicate samples. Data is representative of 3 different experiments using three or more mice per genotype. (C) Comparison of conserved non-coding sequences in the Ndfip1 promoter between mouse and man. The red and green box between CNS2 and CNS3 indicates the signal peptide and exon 1 (respectively) of the Ndfip1 gene.
qPCR
RNA from harvested cells was isolated with the RNeasy Mini Kit (Qiagen). RNA- to-cDNA reactions were done using the High Capacity RNA-to-cDNA Kit (Applied Biosystems). For qPCR reactions, TaqMan Gene Expression Master Mix was used (4370048, Applied Biosystems). The Ndfip1 primer/probe set has been previously described (20). FAM dye, MGB primer/probes sets for IL-2 (Mm00434256_m1), IL-2Rα (Mm01340213_m1) and ACTB (4352933E) were obtained from Applied Biosystems. Samples were amplified in triplicate using the 7500 Real-Time PCR system (Applied Biosytems). Data were analyzed using the 7500 software v2.0 (Applied Biosystems).
Statistical Analysis
All statistical analyses were performed using Student’s t-tests unless stated otherwise. A P-value of equal or less than 0.05 was used to determine statistical significance.
RESULTS
T cells lacking Ndfip1 require antigen exposure to become CD44hi in vivo
We have shown previously that Ndfip1−/− mice develop inflammation at sites of environmental antigen exposure and die prematurely (17). In part, this is because Ndfip1 regulates JunB degradation and thus limits IL-4 production. While this accounts for the TH2 bias of these cells, it does not explain why T cells in Ndfip1−− mice become activated. Mice lacking Ndfip1 have increased percentages of T cells that are CD44hi (17,21), suggesting that these cells have been activated by antigen presenting cells (APCs). However, lymphopenic conditions, particular cytokine signals, and the absence of factors that maintain quiescence can also cause T cells to acquire this activated phenotype (22–25). Under these conditions, T cells will display increased CD44 levels even in the absence of cognate antigen. To determine whether T cells lacking Ndfip1 require antigen exposure to acquire elevated CD44 levels, we generated Ndfip1−/− mice that contain T cells specific for a peptide of ovalbumin (Ova) in the context of MHC class II. These mice, referred to as OTII, are Rag1-deficient and OTII T cell receptor (TCR) transgenic (Tg). When compared to control Ndfip1+/+ OTII animals, Ndfip1−/− OTII mice have similar life spans (data not shown). Additionally, Ndfip1−/− OTII mice do not develop the eosinophilic inflammation that is observed in Ndfip1−/− animals with a polyclonal T cell repertoire (data not shown). We analyzed CD44 levels from T cells isolated from the spleens of Ndfip1−/− OTII mice and Ndfip1+/+ OTII control animals that had not been exposed to Ovalbumin. For comparison, we have included data showing this same analysis on Ndfip1−/− and Ndfip1+/+ T cells. As shown previously (21), T cells from Ndfip1−/− mice were more likely to have an activated phenotype than T cells from Ndfip1+/+ control animals (Figure 1 upper right versus upper left panel and Figure 1B) as long as these mice are maintained on a Rag1−/− background. Importantly, T cells from both OTII Ndfip1+/+ and Ndfip1−/− mice remain CD44lo (Figure 1A lower panel and Figure 1B). Compared to Ndfip1−/− mice with a polyclonal T cell repertoire, T cells from Ndfip1−/− OTII mice show significantly reduced percentages of CD44hi T cells. Therefore, T cells lacking Ndfip1 do not acquire an activated phenotype (CD44hi) in the absence of antigen. By inference, these data suggest that T cells from Ndfip1−/− mice are CD44hi due to antigen-mediated activation.
Figure 1. Activated phenotype of T cells in mice lacking Ndfip1 is antigen-dependent.

(A, B) Spleen cells were isolated from mice and analyzed by flow cytometry. Representative plots (A) or combined data (B) of CD4+ T cells from 3–7 mice per genotype. (A) Gate shown in plot illustrates how we define the percent of “activated” T cells. Numbers represent percentage of cells in gate. (B) Graph representing ‘activated’ phenotype cells illustrated in A. Each dot represents the percentage of cells in the spleen of a single mouse. Lines represent mean for each sample population and P values are for samples connected by the line. *** represents a P value of <0.001. n. s. stand for non-significant, with a P value greater than 0.05 based on a paired T test. This analysis was performed in at least three independent experiments.
T cell specific deletion of Ndfip1 leads to increased percentages of activated T cells and eosinophilic inflammation
Having shown that activation of Ndfip1−/− T cells did not occur in OTII Tg T cells in the absence of antigen, we next sought to determine the basis of the T cell activation in Ndfip1−/− mice. Increased numbers of activated T cells in vivo could be due to cell intrinsic or cell extrinsic defects such as stimulation by innate cells (26). To determine whether these defects were T cell intrinsic, we generated mice lacking Ndfip1 only in cells of the T cell lineage (Ndfip1CD4-CKO). To delete Ndfip1 in these cells we inserted loxP sites on either side of exon 2 of the Ndfip1 gene. Cre-mediated recombination of these sites results in a deletion of exon 2 as well as a frame shift mutation for the remaining exons (Figure 2A). Ndfip1 floxed mice were crossed to CD4-Cre transgenic animals to generate mice lacking Ndfip1 in T cells. The resulting progeny were intercrossed and offspring were analyzed for both the presence of the floxed Ndfip1 alleles as well as the Cre transgene (Figure 2B). To analyze the effectiveness of Cre-mediated deletion of Ndfip1, T cells from mice homozygous for the floxed Ndfip1 and positive for Cre (i.e. Figure 2B lane 3) were tested for expression of Ndfip1 by qPCR (Figure 2C). Stimulation of WT CD4+ T cells induced expression of Ndfip1 by 24 hours. Ndfip1 mRNA expression in Ndfip1CD4-CKO mice was similar to levels in Ndfip1−/− T cells, indicating that Ndfip1CD4-CKO mice lack expression of Ndfip1 in T cells.
Constitutive Ndfip1 knockout mice contain increased percentages of activated T cells and develop inflammation in the esophagus, characterized by an influx of both CD4+ T cells and eosinophils, by 6 weeks of age (21). To determine whether Ndfip1-deficient T cells could drive these phenotypic changes, we first compared the activation status of the T cells from Ndfip1CD4-CKO and Ndfip1−/−mice. As described previously, spleens of Ndfip1−/− animals have increased percentages of activated (CD44hi) CD4+ T cells compared to Ndfip1+/+littermates (Figure 2D upper right). Importantly, we found that the frequency of activated T cells in spleens from Ndfip1CD4-CKO mice was comparable to the frequency observed in Ndfip1−/− mice (Figure 2D lower right). This shows that the activation of the Ndfip1-deficient T cells in vivo results from a T cell intrinsic defect.
We next analyzed inflammation in the GI tract of Ndfip1CD4-CKO mice. Histological analysis of the esophagus showed severe inflammation characterized by epithelial hypertrophy and inflammatory cell infiltrates (Figure 2E), similar to that previously observed in Ndfip1−/− mice (21). Analysis of cells isolated from the esophagus revealed increased percentages of CD4+ T cells and eosinophils (Figure 2F). Increased percentages of eosinophils were also observed in the small bowel and lung of Ndfip1CD4-CKO mice (data not shown). Interestingly, skin inflammation was less evident in the Ndfip1CD4-CKO mice. While these mice do develop inflammation of the skin, evidence of skin lesions occurs at approximately 9 weeks of age, several weeks later than lesions observed in Ndfip1−/− animals (data not shown). Furthermore, the extent of eosinophilia was reduced in Ndfip1CD4-CKO mice compared to Ndfip1−/− animals. Thus, the loss of Ndfip1 in cells other than T cells likely contributes to the severity of the inflammation in Ndfip1−/− mice. Nonetheless, these data show that loss of Ndfip1 specifically in T cells leads to increased T cell activation, infiltration of T cells into tissues, and eosinophilic inflammation.
Ndfip1−/− T cells are less dependent on CD28 co-stimulation than Ndfip1+/+counterparts
Data described thus far show that the loss of Ndfip1 leads to increased frequency of activated T cells in the mice due a T cell intrinsic defect. Based on this, we hypothesized that Ndfip1−/− T cells are hyperresponsive to T cell receptor stimulation. To test this, we isolated naïve CD4+ T cells from Ndfip1−/− mice and Ndfip1+/+ littermate controls and stimulated them ex vivo through their TCR (anti- CD3) in the presence or absence of CD28 co-stimulation (anti-CD28). We then measured their levels of CD44 (data not shown) and IL-2Rα (Figure 3A and B). We found that Ndfip1+/+ T cells increased their levels of IL-2Rα on day 1 when stimulated with anti-CD3 in the presence of CD28 co-stimulation (Figure 3A) and this still occurred, albeit to a lesser extent, in the absence of CD28 co-stimulation (Figure 3B). However, by day 3 in the absence of co-stimulation, the levels of IL-2Rα diminished. By day 5, Ndfip1+/+ cells that did not receive co-stimulation were mostly dead (data not shown). In contrast, Ndfip1+/+ cells that were stimulated in the presence of CD28 co-stimulation continued to display high levels of IL-2Rα and survived well over the course of the experiment. Supporting previously published results, these data show that in vitro CD28 co-stimulation is required to maintain levels of IL-2Rα and promote survival of cells in vitro (27).
Figure 3. T cells lacking Ndfip1 can produce IL-2 and proliferate in the absence of CD28 co-stimulation.

(A–D) Naïve (CD44loCD62LhiCD25−) CD4+ T cells were isolated from Ndfip1−/− mice or Ndfip1+/+ littermate controls and stimulated in vitro with anti-CD3 and anti-CD28 (A) or anti-CD3 only (B–E). (A and B) Cells were analyzed for levels of IL-2Rα by flow cytometry at the indicated time points. Plots are representative of three independent experiments from 3 mice of each genotype. (C) Supernatants were analyzed for IL-2 by ELISA at time points indicated. Graphs are of representative data from three independent experiments, showing a single comparison of genotypes performed in triplicate. White bars represent cultures of Ndfip1+/+ T cells and black bars show Ndfip1−/− T cells. Bars represent the mean and lines show the standard deviation of the triplicate samples. (D) Cells described in panel B were labeled with CFSE immediately prior to stimulation and analyzed at day three for proliferation. Histograms showing CFSE dilution are representative of at least three independent experiments. (E) IL-2 levels in cultures of Ndfip1+/+CD28−/− and Ndfip1−/−CD28−/− T cells stimulated with various concentrations of anti-CD3 as noted and analyzed at day three. White bars are Ndfip1+/+CD28−/− T cells and black bars are Ndfip1−/−CD28−/− T cells. Bars represent the mean and lines show the standard deviation of the triplicate samples.
Levels of IL-2Rα on T cells lacking Ndfip1 looked strikingly similar to Ndfip1+/+ counterparts when stimulated with both anti-CD3 and anti-CD28 (Figure 3A). Additionally, after one day of stimulation by anti-CD3 only, IL-2Rα levels on Ndfip1−/− T cells were equivalent to those on Ndfip1+/+ cells. However, after 3 days of stimulation with anti-CD3 only, Ndfip1−/− T cells showed increased levels of IL- 2Rα, and by day 5 these cells looked similar to cells that received CD28 co- stimulation (Figure 3B). These data suggest that T cells lacking Ndfip1 are hyper- responsive to TCR stimulation and thus less dependent on CD28 co-stimulation.
In Ndfip1−/− T cells, IL-2Rα levels increased following TCR signaling even in the absence of CD28 co-stimulation. IL-2Rα expression levels are known to increase further after IL-2 receptor signaling, due to a positive feedback loop (3). The further upregulation of IL-2Rα on Ndfip1−/− T cells between days 1 and 3 after anti-CD3 stimulation suggested that these cells were producing IL-2 despite the lack of co-stimulation. Therefore, we measured the levels of IL-2 in the culture supernatants (Figure 3C). While Ndfip1+/+ T cells stimulated through their TCR alone produced little IL-2 over the course of the assay, T cells lacking Ndfip1 showed significant levels of IL-2 by day 3 and by day 5 (Figure 3C). Additionally, by day 3 after anti-CD3 only treatment, T cells lacking Ndfip1 were proliferating, as indicated by their loss of CFSE (Figure 3D). In contrast, no proliferation was observed in the Ndfip1+/+ cultures during this period.
The increased levels of IL-2 could be due to enhanced IL-2 production or increased cell number due to improved survival. To determine whether the IL-2 production at day 3 could be accounted for by increased survival of Ndfip1−/− T cells, we analyzed the percentage of live cells in the cultures described in Figure 3A and B. At day 3, the frequency of live cells did not differ significantly between Ndfip1+/+ and Ndfip1−/− cells regardless of whether the cells were stimulated in the presence or absence of anti-CD28 (data not shown). However by day 5, the Ndfip1+/+ cells stimulated with anti-CD3 only were mostly dead, while a significantly higher percentage of the Ndfip1−/− cells survived (data not shown). This is likely due to the well-characterized effects of IL- 2 on T cell survival (27).
The hyperresponsiveness of T cells lacking Ndfip1 might only occur following a certain threshold of stimulation or it could occur over a range of strengths of TCR stimulation. Thus, we analyzed naïve Ndfip1+/+ and Ndfip1−/− T cells stimulated with different amounts of anti-CD3 in the absence of CD28 co-stimulation. To ensure that these cells were devoid of all CD28 co-stimulatory signals, we crossed Ndfip1−/− mice to CD28−/− animals and used T cells from the double knockout animals (Ndfip1−/− CD28−/− or mice lacking only CD28 (Ndfip1+/+CD28−/− T cells were stimulated with increasing amounts of anti-CD3 and analyzed for IL- 2 production and IL-2Rα levels on day 3. We found that, at all concentrations of anti-CD3 tested, T cells lacking Ndfip1 produced more IL-2 (Figure 3E) than controls. Furthermore, the frequency of cells expressing high levels of IL-2Rα increased in cultures of T cells lacking Ndfip1 (data not shown). Interestingly, while both Ndfip1+/+ and Ndfip1−/− T cells showed peak IL-2 production at 5μg/ml of anti-CD3, the amount of IL-2 produced by Ndfip1−/− T cells far exceeded that of Ndfip1+/+ cells at all concentrations of anti-CD3 tested (Figure 3E). Together, these data suggest that Ndfip1 negatively regulates IL-2 production following T cell activation.
Ndfip1−/− T cells become activated and differentiate into IL-4 producing cells in the absence of CD28 co-stimulation in vivo
Having demonstrated that Ndfip1−/− T cells were less dependent on CD28 co- stimulation in vitro, we sought to test whether this was also true in vivo. Hence, we analyzed Ndfip1−/− CD28−/− mice for signs of T cell activation and pathology. By 8 weeks of age, mice lacking both Ndfip1 and CD28 contained significantly increased percentages of CD4+ T cells that were CD44hi (Figure 4A). When splenocytes isolated from the Ndfip1−/− CD28−/− animals were stimulated with anti-CD3, they produced significant amounts of IL-4 (Figure 4B). In contrast, little or no IL-4 was detectable in supernatants from Ndfip1+/+CD28−/− cells stimulated in the same manner. Furthermore, elevated frequencies of T cells were evident in both the esophagi and lungs of mice lacking both Ndfip1 and CD28 compared to CD28-deficient controls (Figure 4C and data not shown).
Figure 4. T cells in mice lacking both Ndfip1 and CD28 become activated, make IL-4 and migrate into the gastrointestinal tract.

(A) Percentages of activated phenotype cells among the CD4+ cells in the spleens of 5–10 wk old Ndfip1−/− CD28−/− mice or Ndfip1+/+CD28−/− littermate controls. Each dot represents a single mouse and data are representative of 4 independent experiments. Dots connected by lines show age matched littermate pairs in each experiment. * represents a P value of <0.05 based on a paired T test. (B) Cells were isolated from spleens of mice and stimulated overnight with anti-CD3. IL-4 in the supernatants was determined using ELISA. Data are representative of three independent experiments from three mice of each genotype. Bars represent the mean and error bars indicate standard deviation of three triplicate samples. (C and D) Cells isolated from esophagus (C) and small bowel (D) following collagenase treatment were analyzed by flow cytometry for eosinophils (SiglecF+) or T cells (CD4+). Representative plots from at least three independent experiments are shown. (E-H) Sections of esophagus from Ndfip1+/+CD28−/− mice (E and G) or Ndfip1−/−CD28−/− mice (F and H). E and F were taken at 20X magnification, while G and H are insets depicted by boxed regions in E and F. Epithelial thickening can be seen in G and H. L indicates the lumen for visual reference. Representative of three mice of each genotype from three independent experiments. (I-L) Sections of small bowel from Ndfip1+/+CD28−/− mice (I and K) or Ndfip1−/−CD28−/− mice (J and L). I and J were taken at 20X magnification, while K and L are insets depicted by boxed regions in I and J. Eosinophils can be seen in insets and are indicated by arrows. Representative of three mice of each genotype from three independent experiments.
Increased T cell activation and effector differentiation correlated with increased tissue inflammation in the esophagus and lung (Figure 4C, 4E, and data not shown). Ndfip1−/− CD28−/− mice showed histologic evidence of epithelial hypertrophy in the esophagus and showed increased infiltration of inflammatory cells. While eosinophils were not significantly increased in the esophagus (Figure 4C), these cells could be readily detected in the small bowel by histology and flow cytometry (Figure 4D and F). Although we observed a delay in the onset of inflammation by approximately 3 weeks, the pathology that developed in Ndfip1−/− CD28−/− mice was comparable to that in Ndfip1−/− mice. Thus, T cells lacking Ndfip1 can become activated in vivo in the absence of CD28 co-stimulation, differentiate into IL-4 producing effector cells, migrate into tissues and promote the recruitment of eosinophils and inflammation.
Negative regulation of IL-2 production by Ndfip1 is not dependent on the E3 ubiquitin ligase Itch
We have shown previously that Ndfip1 interacts with the HECT-type E3 ubiquitin ligase known as Itch, and that this helps to promote the ubiquitylation and degradation of JunB to dampen IL-4 production by T cells (17,20). Thus, Ndfip1 might promote other functions of Itch to dampen IL-2 production. This seemed particularly likely since Itchy-mutant T cells have been shown to produce more IL-2 under certain conditions (14,28). To test this, we analyzed IL-2 production by naïve T cells lacking either Ndfip1 or Itch following anti-CD3 stimulation. As shown previously, T cells lacking Ndfip1 produce more IL-2 at day 3 and day 5 following stimulation with anti-CD3 alone (Figure 5A). T cells lacking Itch produced no more IL-2 than their WT counterparts. These data were surprising considering previously published data showing that upon TCR-only stimulation, Itch-deficient T cells become activated (28), However, these prior experiments were performed using cells that had been previously activated in vitro or using differentiated effector cells. Thus we performed experiments as the ones described by Venuprassad and colleagues using Ndfip1−/− or Itchy-mutant cells differentiated in vitro. As shown in Figure 5B, WT T cells differentiated in vitro produced less IL-2 upon TCR-only stimulation with ionomycin. In contrast, effector cells lacking either Ndfip1 or Itch were still able to make IL-2 after ionomycin treatment (Figure 5B). Taken together, these data suggest that there are different mechanisms that negatively regulate T cell activation in naïve and effector T cells. Our data support a role for Itch and Ndfip1 in controlling IL-2 production in effector T cells, whereas Ndfip1 negatively regulates activation and IL-2 production in naïve T cells via an Itch independent mechanism.
Figure 5. Ndfip1 limits IL-2 production after activation of naïve T cells via an Itch independent mechanism.
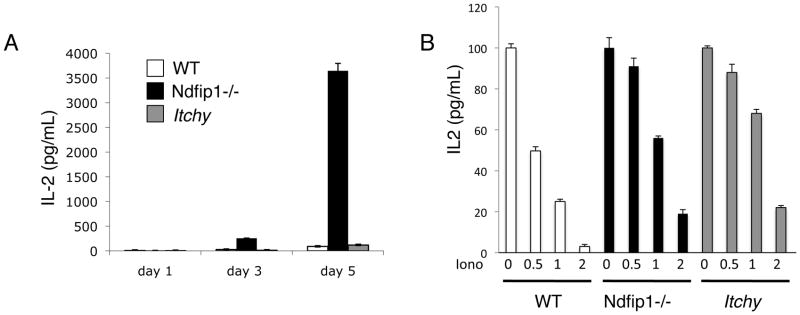
(A) Naïve T cells from WT, Ndfip1−/− and Itch-deficient mice (Itchy) were sorted and stimulated with anti-CD3. Supernatants were analyzed for IL-2 at the various time points by ELISA. (A–B) Data are representative of at least three independent experiments. White bars represent cultures of Ndfip1+/+ T cells, black bars show Ndfip1−/− T cells and gray bars represent Itch-deficient T cells. Bars represent the mean and error bars indicate standard deviation of three triplicate samples. (B) Sorted naïve T cells from the various genotypes were differentiated into effectors as described in methods and then stimulated with the various concentrations of ionomycin. Cells were rested for 24 hours and then restimulated with anti-CD3 and anti-CD28. The amount of IL-2 in the supernatants was analyzed by ELISA.
Ndfip1 restricts T cell activation by limiting the expression of IL-2
Following TCR stimulation, T cells increase the levels of expression of IL-2Rα, a subunit of the high affinity IL-2 receptor. When cells also receive CD28 co-stimulation, IL-2 production increases and acts in an autocrine manner via the IL-2R to further increase IL-2 production and IL-2Rα expression in a positive regulatory loop that results in proliferation. T cells lacking Ndfip1 show both increased IL-2 production as well as elevated levels of IL-2Rα. Thus, the hyperresponsive phenotype of Ndfip1−/− T cells could be directly due to increased IL-2 production or a consequence of increased IL-2R signaling. To test this, we co-cultured naïve CD4+ T cells from either Ndfip1−/− or Ndfip1+/+ littermates (both express CD45.2) together with equal numbers of WT T cells (expressing CD45.1) and analyzed IL-2Rα levels and proliferation. Supporting data shown in Figure 2D, T cells lacking Ndfip1 proliferate more than Ndfip1+/+ counterparts (Figure 6A upper left versus lower left panel). Interestingly, WT T cells cultured with Ndfip1−/− T cells proliferated more than cells cultured with Ndfip1+/+ cells (Figure 6A upper right versus lower right panel), suggesting that Ndfip1-deficient T cells secrete a soluble factor that drives proliferation of WT cells. We therefore repeated the experiment but added anti-IL-2 into the cultures to block the binding of IL-2 to the IL-2R. In the presence of anti-IL-2, proliferation of Ndfip1+/+ T cells was abrogated, as was proliferation of the WT T cells in the same cultures (Figure 6B upper left and upper right panels). Furthermore, the proliferation of WT T cells cultured in the presence of Ndfip1−/− T cells was comparably diminished in the presence of anti-IL-2 (Figure 6B lower right panel). Regardless of whether IL-2 blocking antibodies were present, Ndfip1−/− T cells proliferate more than the WT cells in the same cultures (Figure 6A and B lower left versus lower right, respectively).
Figure 6. IL-2 production by Ndfip1−/− T cells can increase the IL-2Rα levels and allow proliferation of WT T cells.

(A–D) Naïve Ndfip1+/+ or Ndfip1−/− T cells (both are CD45.2) were mixed with equal numbers of WT (CD45.1) T cells, labeled with CFSE, and cultured in the presence of anti-CD3. Data are representative of two independent experiments, which together represent 4 mice from each genotype. (A) After 3 days, cells were analyzed for proliferation as determined by CFSE dilution and results are shown in histograms. (B) Anti-IL-2 was added to the cultures at day 0 and cells were again analyzed at day three for proliferation as described for panel A. (C) IL-2Rα levels were analyzed on the cells in panel A. Mean fluorescence intensities (MFI) for the population are shown within each histogram. (D) IL-2Rα levels were analyzed on the cells in panel B. The MFI for the population are shown within each histogram.
This difference between Ndfip1−/− and WT T cells was not limited to proliferation, but was also evidenced by IL-2Rα levels. WT T cells cultured with Ndfip1−/− T cells displayed higher levels of IL-2Rα than cells cultured with Ndfip1+/+ cells (Figure 6C middle panel versus left panel). However, when stimulated in the presence of anti-IL-2, levels of IL-2Rα on CD45.1 WT T cells were significantly lower (middle panel, Figure 6C versus 6D) and were similar regardless of whether stimulated in the presence of Ndfip1+/+ or Ndfip1−/− cells (Figure 6D middle panel versus left panel). While the addition of anti-IL-2 to cultures decreased IL-2Rα expression on T cells lacking Ndfip1 (right panel, Figure 6C versus 6D), these IL-2Rα levels were higher than those on WT T cells in the same conditions (Figure 6D middle panel versus right panel). Increased IL-2Rα levels on Ndfip1−/− T cells (6D right panel) may account for the proliferation observed in the presence of anti-IL-2 (6B lower left). This could indicate that Ndfip1−/− T cells have increased IL-2Rα signaling even in the absence of IL-2. However, considering that the IL- 2R has a much higher affinity for IL-2 than the blocking antibody, it is possible that anti-IL-2 failed to completely block autocrine IL-2 signaling. Nevertheless, it is clear that overproduction of IL-2 by Ndfip1−/− cells can promote increased IL-2Rα expression and proliferation of WT T cells in trans.
In an effort to determine whether Ndfip1 directly controls IL-2 production and/or IL-2Rα surface levels, we assessed the mRNA expression of IL-2 and IL-2Rα in Ndfip1+/+ and Ndfip1−/− naïve T cells at early time points following TCR-stimulation. As shown in Figure 7A, by 12 hours after TCR stimulation IL-2 transcription was evident in Ndfip1+/+ T cells. However, by 24 hours levels of IL-2 transcripts were significantly decreased, suggesting that transcription was being terminated. While IL-2 levels declined by 24 hours of stimulation in Ndfip1+/+ T cells, under the same conditions IL-2 was still highly expressed in Ndfip1−/− cells. Hence, Ndfip1 is not regulating initial IL-2 production in T cells but rather is limiting its duration. Conversely, the levels of IL-2Rα expression in WT and Ndfip1−/− T cells were similar at all measured time points (Figure 7B). Therefore, Ndfip1 regulates IL-2 production by restricting its transcription. However, under the same conditions, Ndfip1 is not directly affecting the expression of IL-2Rα. This would appear to contradict our data in Figure 2B, in which we show that on day 3 of stimulation the levels of IL-2Rα are increased; however, the RNA expression data shows that this likely due to the downstream consequences of increased IL-2 production. Thus, these data support a model in which Ndfip1 prevents the full activation of T cells by limiting IL-2 production at the transcriptional level.
NFAT and Erk induce the expression of Ndfip1 to limit IL-2 production in the absence of co-stimulation
Having shown that Ndfip1 limits the duration of IL-2 production following initial IL-2 expression (Figure 7A), we next wanted to determine how Ndfip1 expression is regulated in T cells. Thus, we stimulated Ndfip1+/+ T cells through the TCR and analyzed expression of Ndfip1 at different time points. Prior to stimulation of naïve T cells little, if any, Ndfip1 was expressed. However, expression of Ndfip1 was upregulated after 12 hours of TCR-stimulation (Figure 8A) dropped after 24 hours of TCR signaling and continued declining by 36 hours. Interestingly, the expression pattern of Ndfip1 was strikingly similar to that of IL-2 in TCR-stimulated T cells (Figure 7A). The similarity between the transcriptional patterns of Ndfip1 and IL-2 suggested that factors that induce IL-2 expression upon TCR-stimulation might also play a role in regulating the expression of Ndfip1, to limit IL-2 transcription.
TCR signaling promotes IL-2 expression through the cooperation of various factors, including Jnk, NFAT, Erk and PI3K (reviewed in 29). While co-stimulatory signals, such as those delivered from CD28, can significantly enhance signaling, TCR-stimulation alone can support IL-2 expression to some extent (30). It is not known, however, how Ndfip1 expression is affected by TCR signaling and whether the factors that promote IL-2 expression also play a role in its expression.
To determine whether Jnk, NFAT, Erk or PI3K also regulate Ndfip1 expression, we stimulated naïve Ndfip1+/+ T cells through the TCR in the presence of inhibitors for these different factors. We then analyzed Ndfip1 mRNA levels after overnight stimulation. Ndfip1 expression increased following TCR stimulation (Figure 8B) but this was somewhat reduced when either Jnk or PI3K were inhibited. Importantly, the expression of Ndfip1 was almost completely abrogated in the presence of inhibitors of either NFAT or Erk. Thus, NFAT and Erk are required for Ndfip1 expression. Taken together, these data suggest that two key factors that induce IL-2 production, NFAT and Erk, are also inducers of Ndfip1, a factor that attenuates IL-2 expression. This suggests that NFAT and Erk induce Ndfip1 upon T cell stimulation to create a negative feedback loop that restricts IL-2 transcription. Supporting this, comparing the region within 5kb of the mouse and human Ndfip1 promoter, we found several conserved non-coding sequences with NFAT and AP-1 binding sites (Figure 8C).
Increased IL-2 production by Ndfip1−/− T cells is independent of IL-4
We have shown previously that Ndfip1−/− T cells aberrantly produce IL-4 after T cell activation (20, 31) and that these cells are biased towards TH2 differentiation (17). While IL-4 signaling has not been shown to directly impact IL-2 production, IL-4 could increase cell survival and thus alter IL-2 production indirectly. To test whether the increased IL-2 was due to IL-4 production by Ndfip1−/− cells, we analyzed T cells from mice lacking both Ndfip1 and IL-4. Naïve T cells from Ndfip1−/− IL-4−/− mice or IL-4−/− littermate controls were stimulated with anti-CD3 and we analyzed the amount of IL-2 in the supernatants by ELISA. We found that IL-2 production by Ndfip1−/− IL-4−/− T cells was significantly greater than in IL-4−/−controls (Figure 9A), suggesting that exposure to elevated IL-4 signals cannot account for the hyperresponsiveness of these cells in vitro.
Figure 9. Ndfip1−/− IL-4−/− T cells produce high levels of IL-2 and promote inflammation.

(A) IL-2 production from Ndfip1+/+IL-4−/− or Ndfip1−/−IL-4−/− T cells stimulated with anti-CD3 as measured by ELISA. Data are representative of three independent experiments from at least three mice of each genotype. (B–D) Cells isolated from the esophagus of Ndfip1+/+IL-4−/− and Ndfip1−/−IL-4−/− mice analyzed by flow cytometry. (B and C) Representative sections of esophagus (B) and lung (C) from Ndfip1+/+IL-4−/− and Ndfip1−/−IL-4−/− mice. Bars illustrate 100 microns and lower panels represent inset shown in upper panels. Similar results were observed in the sections from three mice analyzed from each genotype. (D) Cells were isolated from esophagi from Ndfip1+/+IL-4−/− and Ndfip1−/−IL-4−/− mice. Cells were analyzed using flow cytometry for percentages of T cells among live-gated cells. Data were combined from three independent experiments using a total of three mice of each genotype. Bars are mean values of T cells (CD4+) (E). Cells isolated from lungs from Ndfip1+/+IL-4−/− and Ndfip1−/−IL-4−/− mice were analyzed by flow cytometry for percentages of T cells among live-gated cells. Data were combined from three independent experiments using a total of four mice of each genotype. Bars are mean values of T cells (CD4+) Each dot represents a single mouse. * represents a P value of <0.05 based on a paired T test while ** represents P<0.01.
We recently showed that T cells lacking Ndfip1 were defective in iTreg cell differentiation and that this was due to increased IL-4 production (20). While mice lacking Ndfip1 showed fewer Foxp3+ T cells in their small bowel, mice lacking both Ndfip1 and IL-4 contained normal numbers of these cells. Based on the data in Figure 9A, we hypothesized that Ndfip1−/− T cells would still be activated in vivo under conditions where iTreg cell differentiation was restored (i.e. in Ndfip1−/− IL-4−/− animals). Thus, we analyzed Ndfip1−/− IL-4−/− mice for signs of T cell activation, T cell migration into tissues, and inflammation. Ndfip1−/− IL-4−/−animals do not show signs of inflammation at 6 weeks of age, a time when Ndfip1−/− animals show pathology developing in the skin, lung and GI tract (20). However, by 12 weeks of age Ndfip1−/− IL-4−/− mice begin to develop disease and these mice ultimately die prematurely of inflammatory consequences (data not shown). Histological examination of the esophagi and lungs from Ndfip1−/− IL-4−/−mice revealed epithelial hyperplasia and infiltration of inflammatory cells (Figure 9B and C). Supporting this, mice lacking both Ndfip1 and IL-4 showed increased percentages of T cells in mucosal tissues, such as esophagus and lung (Figure 9D and E). These mucosal barrier sites also showed increased percentages of eosinophils and neutrophils (data not shown). Additionally, while we saw a trend towards increased percentages of activated T cells in the spleens of these mice, it did not reach statistical significance (data not shown). This may be because these cells emigrated to tissues following activation. Thus, while IL-4 overproduction clearly increases the number of activated T cells in Ndfip1−/− mice and exacerbates disease, even in the absence of IL-4 and with restored iTreg cell differentiation, T cells become activated move into tissues and drive inflammation leading to premature death. Taken together, our data support that T cell hyperresponsiveness is likely underlying the inflammation in Ndfip1−/− IL-4−/−mice.
DISCUSSION
In this study we show that Ndfip1, an adaptor for E3 ligases of the Nedd4-family, negatively regulates IL-2 production, thereby preventing the activation of T cells in the absence of CD28 co-stimulation. T cells lacking Ndfip1 produce IL-2, increase surface expression of the high affinity IL-2Rα subunit, and proliferate in the absence of CD28 co-stimulation in vitro. Additionally, activation in the absence of this negative regulator has severe pathologic consequences in vivo, since mice lacking both Ndfip1 and CD28 develop a TH2-mediated inflammation at barrier surfaces much like mice lacking only Ndfip1. These pathologic consequences are due to intrinsic defects in T cells lacking Ndfip1 since mice lacking Ndfip1 only in T cells (Ndfip1CD4-CKO) show a similar expression profile of activation markers.
We have shown previously that Ndfip1 promotes Itch mediated ubiquitylation and degradation of JunB, thus dampening IL-4 production (17). Overproduction of IL-4 explains the TH2 bias of cells lacking Ndfip1, however, this mechanism does not account for the increased IL-2 production. Supporting this, Ndfip1−/− T cells lacking IL-4 to produce IL-2 following TCR stimulation in the absence of CD28 co-stimulation. Additionally, in the absence of IL-4, Ndfip1−/− mice develop a delayed, yet ultimately fatal, inflammatory disease. We propose that hyperactive IL-2 producing T cells promote this inflammation.
Increased IL-2 production is not seen in naïve T cells lacking the E3 ubiquitin ligase Itch. Thus, Ndfip1 regulates IL-2 production by naïve T cells independent of its ability to promote Itch function. On the other hand, when T cells are differentiated into effectors in vitro, both Ndfip1−/− and Itch-deficient cells show more IL-2 production than their WT counterparts. Thus, Itch and Ndfip1 both regulate IL-2 production in effector differentiated T cells. Based on our prior studies, we propose that Itch and Ndfip1 collaborate in this task. Additionally, these results suggest that there are different mechanisms used by Ndfip1 to control IL-2 expression in naïve and antigen-experienced T cells. This is not surprising as naïve cells have different requirements for co-stimulatory signals than their antigen-experienced counterparts (32). As Ndfip1 can modulate the activity of many E3 ligases of the Nedd4 family in vitro, future studies will be needed to identify other Nedd4 family members whose function is regulated by Ndfip1 after TCR stimulation of naïve T cells.
Our data show that Ndfip1 limits IL-2 expression. Interestingly, Ndfip1 does not regulate initial expression of IL-2, rather, Ndfip1 restricts the duration of expression. Calcium-induced NFAT and signaling via Erk induce both IL-2 and Ndfip1 expression following TCR stimulation. Given that IL-2 and Ndfip1 have similar expression patterns after TCR signaling and that they rely on common transcription factors suggests a negative feedback loop through which TCR signaling activates IL-2 production but also limits IL-2 expression via Ndfip1.
It is known that in T cells stimulated in the absence of co-receptor signaling, NFAT opposes T cell activation. It does so by inducing the expression of genes that promote unresponsiveness, such as Grail (8) and Ikaros (33), among others. Our results suggest that in addition to these factors, NFAT interferes with T cell activation by increasing expression of Ndfip1. The Ndfip1 promoter contains multiple putative sites for NFAT binding (Figure 8C), supporting that NFAT may directly regulate Ndfip1 expression.
CD28 co-stimulation can supplement TCR signaling and thus can enhance the signaling cascade downstream of the T cell receptor that allows for IL-2 production. However, the activation of both NFAT and Erk pathways in T cells is less dependent on CD28 co-stimulatory signals than that of other pathways such as NF-κB (32, 33). Based on these data, we propose that the negative regulatory factor Ndfip1 limits IL-2 production to enforce a requirement for co-stimulation, thereby preventing T cells from responding to low affinity signals such as those coming from self-peptides or environmental antigens. Such factors are prime candidates for therapeutic strategies designed to either augment or dampen T cell signaling to promote tumor rejection or prevent autoimmune or allergic disorders, respectively.
Acknowledgments
We would like to thank Claire O’Leary and G. Scott Worthen for critical reading of this manuscript. Technical support was provided by the University of Pennsylvania flow cytometry core and the Children’s Hospital of Philadelphia histology core. N.R-H, H.E.R., A.M.B., A.L., E.A.N. and P.M.O. performed experiments, analyzed and interpreted the data as well as assisted with writing the manuscript. H.E.R. initiated the project and P.M.O. supervised the work.
This work was supported by NIH grants R01AI093566 and 1F32AI085837.
References
- 1.Linsley PS, Brady W, Grosmaire L, Aruffo A, Damle NK, Ledbetter JA. Binding of the B cell activation antigen B7 to CD28 costimulates T cell proliferation and interleukin 2 mRNA accumulation. J Exp Med. 1991;173:721–730. doi: 10.1084/jem.173.3.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thompson CB, Lindsten T, Ledbetter JA, Kunkel SL, Young HA, Emerson SG, Leiden JM, June CH. CD28 activation pathway regulates the production of multiple T-cell-derived lymphokines/cytokines. Proc Natl Acad Sci U S A. 1989;86:1333–1337. doi: 10.1073/pnas.86.4.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim HP, Kelly J, Leonard WJ. The basis for IL-2-induced IL-2 receptor alpha chain gene regulation: importance of two widely separated IL-2 response elements. Immunity. 2001;15:159–172. doi: 10.1016/s1074-7613(01)00167-4. [DOI] [PubMed] [Google Scholar]
- 4.Jenkins MK, Chen CA, Jung G, Mueller DL, Schwartz RH. Inhibition of antigen-specific proliferation of type 1 murine T cell clones after stimulation with immobilized anti-CD3 monoclonal antibody. J Immunol. 1990;144:16–22. [PubMed] [Google Scholar]
- 5.Quill H, Schwartz RH. Stimulation of normal inducer T cell clones with antigen presented by purified Ia molecules in planar lipid membranes:specific induction of a long-lived state of proliferative nonresponsiveness. J Immunol. 1987;138:3704–3712. [PubMed] [Google Scholar]
- 6.Macian F, Im SH, Garcia-Cozar FJ, Rao A. T-cell anergy. Curr Opin Immunol. 2004;16:209–216. doi: 10.1016/j.coi.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 7.Jeon MS, Atfield A, Venuprasad K, Krawczyk C, Sarao R, Elly C, Yang C, Arya S, Bachmaier K, Su L, Bouchard D, Jones R, Gronski M, Ohashi P, Wada T, Bloom D, Fathman CG, Liu YC, Penninger JM. Essential role of the E3 ubiquitin ligase Cbl-b in T cell anergy induction. Immunity. 2004;21:167–177. doi: 10.1016/j.immuni.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 8.Soto-Nieves N, Puga I, Abe BT, Bandyopadhyay S, Baine I, Rao A, Macian F. Transcriptional complexes formed by NFAT dimers regulate the induction of T cell tolerance. J Exp Med. 2009;206:867–876. doi: 10.1084/jem.20082731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heissmeyer V, Macian F, Im SH, Varma R, Feske S, Venuprasad K, Gu H, Liu YC, Dustin ML, Rao A. Calcineurin imposes T cell unresponsiveness through targeted proteolysis of signaling proteins. Nat Immunol. 2004;5:255–265. doi: 10.1038/ni1047. [DOI] [PubMed] [Google Scholar]
- 10.Huang H, Jeon MS, Liao L, Yang C, Elly C, Yates JR, Liu YC. K33-linked polyubiquitination of T cell receptor-zeta regulates proteolysis- independent T cell signaling. Immunity. 2010;33:60–70. doi: 10.1016/j.immuni.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chiang YJ, Kole HK, Brown K, Naramura M, Fukuhara S, Hu RJ, Jang IK, Gutkind JS, Shevach E, Gu H. Cbl-b regulates the CD28 dependence of T-cell activation. Nature. 2000;403:216–220. doi: 10.1038/35003235. [DOI] [PubMed] [Google Scholar]
- 12.Krawczyk CM, Jones RG, Atfield A, Bachmaier K, Arya S, Odermatt B, Ohashi PS, Penninger JM. Differential control of CD28-regulated in vivo immunity by the E3 ligase Cbl-b. J Immunol. 2005;174:1472–1478. doi: 10.4049/jimmunol.174.3.1472. [DOI] [PubMed] [Google Scholar]
- 13.Gay DL, Ramon H, Oliver PM. Cbl- and Nedd4-family ubiquitin ligases: balancing tolerance and immunity. Immunol Res. 2008;42:51–64. doi: 10.1007/s12026-008-8034-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fang D, Elly C, Gao B, Fang N, Altman Y, Joazeiro C, Hunter T, Copeland N, Jenkins N, Liu YC. Dysregulation of T lymphocyte function in itchy mice: a role for Itch in TH2 differentiation. Nat Immunol. 2002;3:281–287. doi: 10.1038/ni763. [DOI] [PubMed] [Google Scholar]
- 15.Chen A, Gao B, Zhang J, McEwen T, Ye SQ, Zhang D, Fang D. The HECT-type E3 ubiquitin ligase AIP2 inhibits activation-induced T-cell death by catalyzing EGR2 ubiquitination. Mol Cell Biol. 2009;19:5348–5356. doi: 10.1128/MCB.00407-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang B, Gay DL, MacLeod MK, Cao X, Hala T, Sweezer EM, Kappler J, Marrack P, Oliver PM. Nedd4 augments the adaptive immune response by promoting ubiquitin-mediated degradation of Cbl-b in activated T cells. Nat Immunol. 2008;9:1356–1363. doi: 10.1038/ni.1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oliver PM, Cao X, Worthen GS, Shi P, Briones N, MacLeod M, White J, Kirby P, Kappler J, Marrack P, Yang B. Ndfip1 protein promotes the function of itch ubiquitin ligase to prevent T cell activation and T helper 2 cell-mediated inflammation. Immunity. 2006;25:929–940. doi: 10.1016/j.immuni.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harvey KF, Shearwin-Whyatt LM, Fotia A, Parton RG, Kumar S. N4WBP5, a potential target for ubiquitination by the Nedd4 family of proteins, is a novel Golgi-associated protein. J Biol Chem. 2002;277:9307–9317. doi: 10.1074/jbc.M110443200. [DOI] [PubMed] [Google Scholar]
- 19.Mund T, Pelham HR. Regulation of PTEN/Akt and MAP kinase signaling pathways by the ubiquitin ligase activators Ndfip1 and Ndfip2. Proc Natl Acad Sci U S A. 2010;107:11429–11434. doi: 10.1073/pnas.0911714107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beal AM, Ramos-Hernandez N, Riling CR, Nowelsky EA, Oliver PM. TGF-beta induces the expression of the adaptor Ndfip1 to silence IL-4 production during iTreg cell differentiation. Nat Immunol. 2011;13:77–85. doi: 10.1038/ni.2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramon HE, Riling CR, Bradfield J, Yang B, Hakonarson H, Oliver PM. The ubiquitin ligase adaptor Ndfip1 regulates T cell-mediated gastrointestinal inflammation and inflammatory bowel disease susceptibility. Mucosal Immunol. 2011;4:314–324. doi: 10.1038/mi.2010.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weinreich MA, Takada K, Skon C, Reiner SL, Jameson SC, Hogquist KA. KLF2 transcription-factor deficiency in T cells results in unrestrained cytokine production and upregulation of bystander chemokine receptors. Immunity. 2009;31:122–130. doi: 10.1016/j.immuni.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gagnon J, Ramanathan S, Leblanc C, Cloutier A, McDonald PP, Ilangumaran S. IL-6, in synergy with IL-7 or IL-15, stimulates TCR- independent proliferation and functional differentiation of CD8+ T lymphocytes. J Immunol. 2008;180:7958–7968. doi: 10.4049/jimmunol.180.12.7958. [DOI] [PubMed] [Google Scholar]
- 24.Murali-Krishna K, Ahmed R. Cutting edge: naive T cells masquerading as memory cells. J Immunol. 2000;165:1733–1737. doi: 10.4049/jimmunol.165.4.1733. [DOI] [PubMed] [Google Scholar]
- 25.Yamada T, Park CS, Mamonkin M, Lacorazza HD. Transcription factor ELF4 controls the proliferation and homing of CD8+ T cells via the Kruppel-like factors KLF4 and KLF2. Nat Immunol. 2009;10:618–626. doi: 10.1038/ni.1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hammer GE, Turer EE, Taylor KE, Fang CJ, Advincula R, Oshima S, Barrera J, Huang EJ, Hou B, Malynn BA, Reizis B, DeFranco A, Criswell LA, Nakamura MC, Ma A. Expression of A20 by dendritic cells preserves immune homeostasis and prevents colitis and spondyloarthritis. Nat Immunol. 2011;12:1184–1193. doi: 10.1038/ni.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Noel PJ, Boise LH, Green JM, Thompson CB. CD28 costimulation prevents cell death during primary T cell activation. J Immunol. 1996;157:636–642. [PubMed] [Google Scholar]
- 28.Venuprasad K, Elly C, Gao M, Salek-Ardakani S, Harada Y, Luo JL, Yang C, Croft M, Inoue K, Karin M, Liu YC. Convergence of Itch-induced ubiquitination with MEKK1-JNK signaling in Th2 tolerance and airway inflammation. J Clin Invest. 2006;116:1117–1126. doi: 10.1172/JCI26858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. 2009;27:591–619. doi: 10.1146/annurev.immunol.021908.132706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Diehn M, Alizadeh AA, Rando OJ, Liu CL, Stankunas K, Botstein D, Crabtree GR, Brown PO. Genomic expression programs and the integration of the CD28 costimulatory signal in T cell activation. Proc Natl Acad Sci U S A. 2002;99:11796–11801. doi: 10.1073/pnas.092284399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramon HE, Beal AM, Liu Y, Worthen GS, Oliver PM. The E3 ubiquitin ligase adaptor Ndfip1 regulates Th17 differentiation by limiting the production of proinflammatory cytokines. J Immunol. 2012;188:4023–4031. doi: 10.4049/jimmunol.1102779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adachi K, Davis MM. T-cell receptor ligation induces distinct signaling pathways in naive vs. antigen-experienced T cells. Proc Natl Acad Sci U S A. 2011;108:1549–1554. doi: 10.1073/pnas.1017340108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Macian F, Garcia-Cozar F, Im SH, Horton HF, Byrne MC, Rao A. Transcriptional mechanisms underlying lymphocyte tolerance. Cell. 2002;109:719–731. doi: 10.1016/s0092-8674(02)00767-5. [DOI] [PubMed] [Google Scholar]
- 34.Matsuda S, Koyasu S. Mechanisms of action of cyclosporine. Immunopharmacology. 2000;47:119–125. doi: 10.1016/s0162-3109(00)00192-2. [DOI] [PubMed] [Google Scholar]