

Abstract
Short stature, auditory canal atresia, mandibular hypoplasia, and skeletal abnormalities (SAMS) has been reported previously to be a rare, autosomal-recessive developmental disorder with other, unique rhizomelic skeletal anomalies. These include bilateral humeral hypoplasia, humeroscapular synostosis, pelvic abnormalities, and proximal defects of the femora. To identify the genetic basis of SAMS, we used molecular karyotyping and whole-exome sequencing (WES) to study small, unrelated families. Filtering of variants from the WES data included segregation analysis followed by comparison of in-house exomes. We identified a homozygous 306 kb microdeletion and homozygous predicted null mutations of GSC, encoding Goosecoid homeobox protein, a paired-like homeodomain transcription factor. This confirms that SAMS is a human malformation syndrome resulting from GSC mutations. Previously, Goosecoid has been shown to be a determinant at the Xenopus gastrula organizer region and a segment-polarity determinant in Drosophila. In the present report, we present data on Goosecoid protein localization in staged mouse embryos. These data and the SAMS clinical phenotype both suggest that Goosecoid is a downstream effector of the regulatory networks that define neural-crest cell-fate specification and subsequent mesoderm cell lineages in mammals, particularly during shoulder and hip formation. Our findings confirm that Goosecoid has an essential role in human craniofacial and joint development and suggest that Goosecoid is an essential regulator of mesodermal patterning in mammals and that it has specific functions in neural crest cell derivatives.
Main Text
During embryogenesis, the first and second branchial arches are derived from mesodermal and neural crest cells that develop into specific craniofacial and skeletal structures. During intramembranous ossification, mesenchymal cells of the cranial neural crest differentiate into osteoprogenitor cells, which further differentiate directly into osteoblasts.1 The mandibular process of the first branchial (or mandibular) arch develops into the skeletal, muscular, and neural elements of the mandible, whereas the dorsal edge of the first branchial (or hyomandibular) cleft forms the auditory meatus. Syndromes of the first and second branchial arches comprise a broad group of developmental disorders that are characterized by craniofacial and multiple congenital anomalies. These disorders are generally autosomal or X-linked dominant and have nearly complete penetrance but a wide spectrum of clinical variability.2 Branchio-oto-renal syndrome (BORS [MIM 113650]), for example, is characterized by a long and narrow facial shape, microtia and other malformations of the ear, and conductive hearing loss due to narrowing of the external ear canal. Short stature, auditory canal atresia, mandibular hypoplasia, and skeletal abnormalities (SAMS [MIM 602471]) is reported, in two previous published cases (individuals A and B, Table 1), to be a provisionally autosomal-recessive disorder with features of a first and second branchial arch syndrome.3,4 However, both individuals A and B had additional, unique rhizomelic skeletal anomalies (Figure S1 in the Supplemental Data available with this article online) that comprised bilateral humeral hypoplasia, humeroscapular synostosis, pelvic abnormalities (delayed ossification of the pubic rami and central dislocation of the hips), and proximal defects of the femora (Table 1). In addition to the previously published cases, we now report two further cases of SAMS, individual C (Table 1 and Figure 1) and individual D (Table 1), both presenting in the neonatal period.
Table 1.
GSC Mutations and Associated Clinical Features in SAMS Individuals
| Individual ID | Aa | Ba | C | D |
|---|---|---|---|---|
| Karyotype | 46,XXb | arr CGH 14q32.13(95,204,793–95,511,597)x0 [hg19] | 46,XYb | 46,XYb |
| GSC nucleotide mutation | c.400C>T | chr14.hg19:g.95,204,793_95,511,597del | c.196_212del | c.355+1G>C |
| Predicted protein alteration | p.Gln134∗ | p.? | p.Gly66Argfs∗98 | p.? |
| Country of origin (ethnic group) | Canada (Mennonite) | Afghanistan | Pakistan (Mirpuri) | Bangladesh |
| Gender (present age) | F (26 years) | F (19 years) | M (6 years) | M (9 months) |
| Consanguinity (degree) | + (first cousin) | + (first cousin) | + (first cousin) | + (first cousin) |
| Height in cm (centile) | 142 (<3rd) | 157 (<3rd) | (<0.4th) | 52 (<0.4th) |
| Weight in kg (centile) | 37 (<3rd) | 42 (<2nd) | 1.95 (2nd)c | 5.52 (<0.4th) |
| Microcephaly (OFC centile) | + (5th) | − | − | + (<0.4th) |
| Neonatal respiratory insufficiency | − | − | + | − |
| Neonatal feeding difficulties | + | − | + | + |
| Bilateral auditory canal atresia | + | + | + | + |
| Abnormal/fused middle ear ossicles | + | − | + | ND |
| Conductive hearing loss (dB) | + | + (40) | + (30) | + |
| Simple or dysplastic pinnae | + | + | + | + |
| Preauricular pits (+) or tags (++) | − | + | ++ | − |
| Malar hypoplasia | + | − | + | − |
| Micrognathia | + | + | + | + |
| High-arched palate | + | − | + | ND |
| Scapulo-humeral synostosis | +L/+R | +L/+R | +L/−R | +L/+R |
| Lumbar hyperlordosis | + | + | − | − |
| Short humeri and distal metaphyseal flaring | + | + | + | − |
| Radial head dislocation | − | +L/+R | +L/+R | ND |
| Short ulnae | + | + | − | − |
| Delayed/absent ossification of pubic bones | + | + | + | + |
| Narrow sacrosciatic notches | + | + | − | + |
| Central dislocation of the hips | + | + | + | + |
| Proximal femoral defects | +L/+R | +R | − | + |
| Bilateral talipes | − | + | + | + |
| Cryptorchidism or other urogenital anomalies | + | + | + | + |
Abbreviations are as follows: +, present; −, absent; ND, not determined; M, male; F, female; L, left; R, right; OFC, occipitofrontal circumference; and ?, unknown.
Clinical information on individuals A and B has been published previously.3,4
Normal karyotype by high-resolution microarray analysis.
Birth weight.
Figure 1.
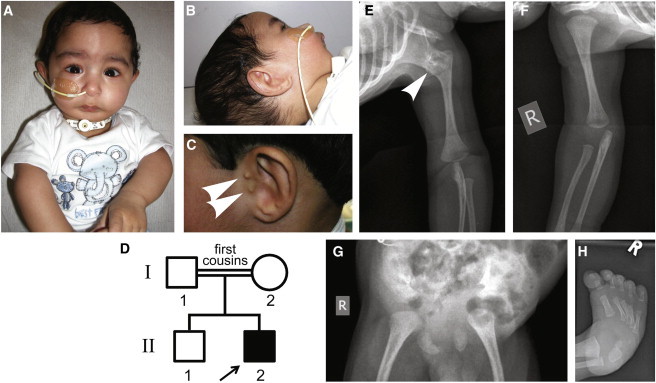
Clinical Features of SAMS in Individual C
(A–C) At the age of 1 month, individual C presented with malar hypoplasia, severe micrognathia, scaphocephaly with a prominent forehead, downward-slanting palpebral fissures, and rhizomelic shortening of the upper limbs (A). Rudimentary pinnae with bilateral atresia of the (B) right and (C) left external auditory canals with small preauricular tags (arrowheads) can be seen.
(D) Pedigree of individual C (arrow) and unaffected family members.
(E and F) Left humeroscapular synostosis (E, arrowhead) but normal right shoulder joint (F), with bilateral shortening of the humeri and flaring of the distal metaphyses.
(G) Immature ossification of the pelvis, with a flattened acetabulum and bilaterally dislocated hips, and absence of ossification of the pubic rami.
(H) Fixed talipes equinovarus of the right foot.
Individual C is a second, male infant (Figure 1A and individual II-2 in Figure 1D) born to unaffected first-cousin consanguineous UK-Pakistani parents (I-1 and I-2 in Figure 1D). The first child (II-1 in Figure 1D) is 5 years old and well. Individual C was born at 34 weeks of gestation by emergency caesarean section for fetal distress and failure to progress after an uneventful pregnancy with normal scans. He required no resuscitation at birth but developed mild respiratory distress and required nasal continuous positive airway pressure (CPAP) for respiratory support for 48 hr. The birth weight of individual C was 1.95 kg, which is in the second centile, and his occipitofrontal circumference (OFC) was in the 91st centile. Initial and subsequent postnatal examinations revealed a high-arched palate, malar hypoplasia, and severe micrognathia (Figure 1B) leading to respiratory compromise. He appeared to have rhizomelic shortening of upper limbs, and reduced pronation /supination of elbows with proximally placed thumbs. He also had bilateral fixed talipes equinovarus with reduced calf bulk, bilateral cryptorchidism, rudimentary pinnae, and bilateral external auditory canal atresia (Figures 1B and 1C). A computed-tomography scan showed that the auditory ossicles were fused to the respective atretric plates, but both internal auditory canals were well formed. An auditory brainstem response test was performed at a corrected gestational age (CGA) of 8 weeks and revealed that both sides were measured at less than 30 dBnHL. Other dysmorphic features included scaphocephaly with a prominent forehead; a slightly downward slant to the palpebral fissures; a short, upturned nose; and a small mouth.
In contrast to individuals A and B, individual C had severe neonatal respiratory insufficiency necessitating a tracheostomy and nasogastric feeding because of poor coordination with no suck, swallow, or gag (Table 1). A skeletal survey of individual C revealed left humeroscapular synostosis, a feature that appears to be pathognomonic for SAMS, but the right shoulder joint appeared normal (Figures 1E and 1F). There was flaring of the distal metaphysis (Figure 1E), immature ossification of the pelvis (Figure 1G), and resulting flattened acetabulum and bilaterally dislocated hips. There was bilateral shortening of clavicles and absence of ossification of the pubic rami. The spine appeared normal. His talipes (Figure 1H) were initially managed by serial Ponsetti casting, and at the age of 2 months (CGA) he underwent bilateral percutaneous Achilles tenotomies followed by serial casting. This procedure went well, and at the time of this report individual C has progressed to ankle-foot orthoses.
Individual D, the male propositus for a consanguineous UK-Bangladeshi family, was born at 34 weeks of gestation after prolonged rupture of membranes at 32 weeks. Birth weight was 2.25 kg, and head circumference was in the 9th centile. He had contractures involving all four limbs at birth, rhizomelic skeletal abnormalities, and talipes equinovarus. He had microphthalmia, micrognathia, bilateral external auditory canal atresia, and no testes or scrotal sac. He experienced feeding difficulties from birth and required a nasogastric tube. A skeletal survey of individual D demonstrated scapulohumeral synostosis bilaterally, mild scapula hypoplasia, and markedly abnormal pelvic ossification, including small iliac bones, narrow sacrosciatic notches, an absence of the ischial bodies, deficient medial acetabular walls, and an absence of pubic bones (Table 1).
Ethical approval for molecular genetics research studies was obtained from the South Yorkshire Research Ethics Committee (REC ref. no. 11/H1310/1), and we obtained informed consent from all participating families or individuals. Genomic DNA was isolated from peripheral venous blood by standard salt extraction, and subsequent high-resolution array CGH studies excluded putative pathogenic DNA copy-number variants (CNVs) in individuals A, C, and D. To identify the causative mutation for SAMS, we used whole-exome sequencing (WES) of individual C (II-2 in Figure 1D), his unaffected brother (II-1), and his parents (I-1 and I-2) so that we could perform segregation analysis of genetic variants, excluding variants that did not follow the appropriate Mendelian patterns of recessive inheritance in the family members. This analysis also excluded the possibility of de novo mutations as a cause of SAMS. We used the Agilent SureSelectXT Human All Exon v4 Kit to enrich exonic DNA from genomic DNA of individual C, his unaffected brother, and both parents. Three micrograms of genomic DNA was sheared, and Illumina paired-end adapters ligated according to Agilent’s SureSelect Library Prep protocol. Samples were prepared for sequencing according to Illumina’s standard amplification, linearization, blocking, and primer hybridization protocols. The flow cell was then transferred to the Illumina HiSeq2500 for paired-end sequencing for 100 cycles. Whole-genome alignment of FASTQ files to the February 2009 human reference sequence (GRCh37) was performed with Novoalign short-read alignment software (Novocraft Technologies). We processed alignments in the SAM and BAM formats5 with SAMtools,5 Picard, and the Genome Analysis Toolkit (GATK)6,7 java programs in order to correct alignments around indel sites and mark potential PCR duplicates. Indel and single-nucleotide variants were called in the VCF format via the Unified Genotyper function of GATK. Variants were filtered with GATK on the basis of mapping quality, strand bias, and genotype quality.
The functional consequences of variants were determined with Ensembl’s SNP Effect Predictor.8 Filtering variants via Bespoke Perl scripts removed those variants that were present in dbSNP129 or that had a minor allele frequency of 1% or higher in dbSNP, the 1000 Genomes Project database, or the NHLBI GO Exome Sequencing Project (ESP) database. Further filtering used a collection of locally sequenced exomes and alignment files from 70 Gujarati Indians from the 1000 Genomes Project. After confirming the absence of potentially pathogenic de novo variants in individual C, we processed the remaining variants with a Bespoke Perl script to identify either compound heterozygous variants or homozygous variants that were predicted to be damaging and segregated appropriately in the remaining family members (Table S1). Perl scripts are available on request. After this analysis, only two variants remained (Table S1): a nonsense mutation in LAG3 (MIM 153337; RefSeq accession number NM_002286.5; c.1570C>T [p.Gln524∗]) and a homozygous frameshift mutation in GSC (MIM 138890; RefSeq accession number NM_173849.2; c.196_212del [p.Gly66Argfs∗98]) (Figure 2A and Table 1). The nonsense variant in LAG3 only resulted in the loss of two amino acids and was therefore considered unlikely to be responsible for the SAMS phenotype. The GSC 17 bp deletion was confirmed to be absent in the 1000 Genomes Project and NHLBI GO ESP databases and, by Sanger sequencing, in 300 unrelated ethnically matched control chromosomes.
Figure 2.
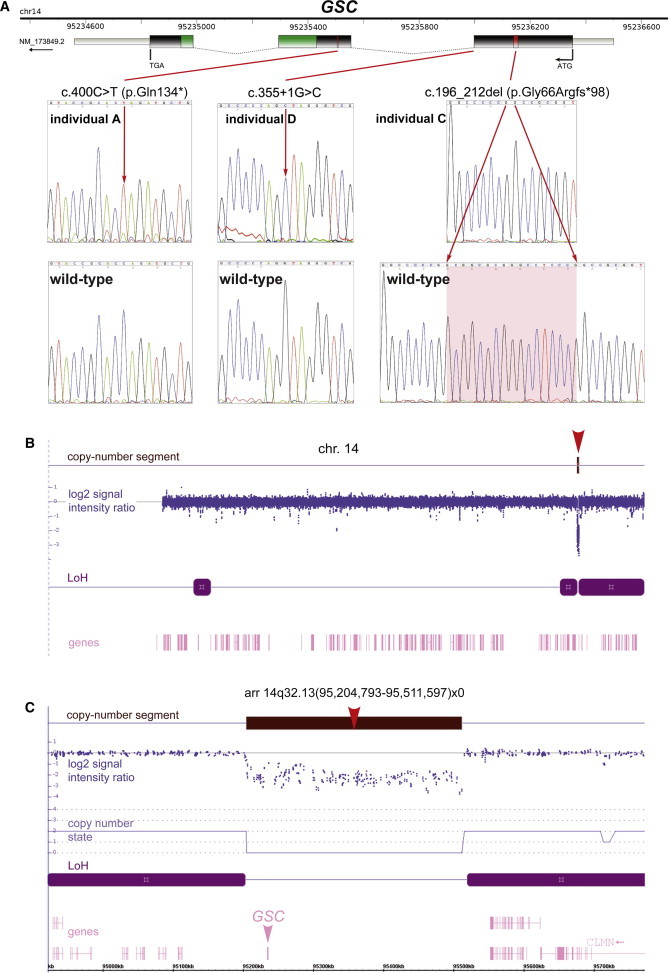
Homozygous-Null Mutations and Microdeletion of GSC Cause SAMS
(A) fancyGENE representation9 of human GSC and homozygous point mutations identified in SAMS individuals A, C, and D. The homeobox domain is shown in green. Mutations are marked on the gene in red, and corresponding electropherograms are shown in the top panels, as indicated (red arrows). Wild-type normal sequence is shown below for comparison.
(B) Affymetrix CytoScan HD Microarray SNP genotyping of individual B for chromosome 14. The top track identifies a copy-number segment (brown, indicated by a red arrowhead). Other tracks show the log2 ratio signal intensity (blue), genes (pink), and stretches of contiguous loss of heterozygosity (LoH; purple) consistent with identity-by-descent from the consanguineous parents.
(C) Array result for individual B of chromosomal region 14q32.13 shows a 306 kb copy-number segment loss arr 14q32.13(95,204,793-95,511,597)x0 (brown, indicated by a red arrowhead) with a copy-number state (light purple) of CN = 0, indicating a homozygous deletion. The Goosecoid gene (GSC) is indicated on the gene track (pink arrowhead), and the physical location in kb is shown at the bottom.
We next used microarray-based molecular karyotyping for CNV and loss-of-heterozygosity (LoH) analysis of genomic DNA from individual B. We used the Affymetrix high-resolution CytoScan high-density (HD) platform (Affymetrix) and a total of 2.67 million probes (1.9 million nonpolymorphic CNV and 0.75 million SNP probes) with median inter-marker distance of ca. 1 kb for genome-wide coverage. Genomic DNA was fragmented, processed, and hybridized to arrays with standard protocols recommended by the manufacturer. Arrays were washed and stained via a GeneChip Fluidics Station 450 and scanned with an Affymetrix GeneChip Scanner 3000 7G. Scanned data files were generated with Affymetrix GeneChip Command Console Software v.1.2 and analyzed for detection of CNV segments with Affymetrix Chromosome Analysis Suite v.1.2 (ChAS) (Affymetrix). To calculate copy numbers, we normalized the data from individual B to baseline reference intensities by using an in-house reference set from 50 mixed-sex unaffected individuals. In separate analyses, we used thresholds of log2 ratio ≥ +0.58 and ≤ −1 to categorize altered regions as CNV gains (amplification) and losses (deletions), respectively. To prevent false-positive calls, only alterations detected by ≥10 consecutive probes and having a length of >50 kbp were considered. Common normal CNVs were excluded by interrogation of the Database of Genomic Variants (DGV), and CNV breakpoints were mapped to UCSC hg19/NCBI.37 builds and Ensembl Release 71 positions. High-resolution CNV and LoH analysis of DNA from individual B revealed a homozygous 306 kb deletion, arr CGH 14q32.13(95,204,793–95,511,597)x0 [hg19], that entirely encompassed GSC on chromosomal region 14q32.13 (Figures 2B and 2C), whereas the parents and unaffected brother were heterozygous 14q32.13(95,204,793–95,511,597)x1 (data not shown). The deletion was surrounded by an LoH block, 14q32.12(92,220,868-95,203,002) (Figure 2C), consistent with a haplotype that is identical-by-descent in a consanguineous individual.
For mutation analysis of individuals A and D, we used Primer3 to design PCR primers that flanked all three coding exons of GSC (Table S2). We performed direct sequencing by using the dideoxy chain termination method (ABI BigDye 3.0 system) on an ABI 3730 DNA Sequencer and analyzed sequences with 4Peaks software. Consistent with autosomal-recessive inheritance, individual A had a homozygous nonsense mutation, c.400C>T (p.Gln134∗) (Figure 1A and Table 1) in GSC, but the parents were unavailable for investigation, and segregation could not be determined. Individual D had a homozygous c.355+1G>C (p.?) mutation at the splice donor site of GSC intron 1 (Figure 1A and Table 1). All mutations were verified bidirectionally. We did not detect these mutations in the 1000 Genomes Project database, ESP database, or in-house exomes. Mutations in GSC have previously been excluded as a cause of cerebro-costo-mandibular syndrome10 and as a cause of familial and sporadic hemifacial microsomia,11 a common birth defect involving derivatives of the first and second branchial arches. This report provides evidence that malformations in humans result from GSC mutations.
GSC encodes Goosecoid homeobox protein (RefSeq accession number NP_776248.1), a highly conserved paired-like homeodomain transcription factor that was first identified as a determinant at the Xenopus gastrula organizer region (Spemann’s organizer) required for mesodermal patterning during gastrulation.12 Mammalian Goosecoid has identical DNA-binding specificity and a homeodomain similar to that of the segment polarity determinant encoded by gooseberry and the anterior morphogen encoded by bicoid, both essential for anteroposterior axis formation in Drosophila.13 A later study has suggested that Goosecoid regulates patterning of the primitive steak by TGFβ signaling during early mammalian embryogenesis.14 However, two separate mouse knockouts of Gsc had normal gastrulation and no early axial midline defects.15,16 In mouse embryos, a later phase of transcript expression, starting from embryonic age E10.5 and continuing during organogenesis, occurred in a number of neural-crest derivatives, such as the first branchial arch and cleft, and subsequent tissue derivatives, such as the mandible and auditory meatus.17 Expression in the proximal limb buds also persisted during the formation of proximal limb structures. This later pattern of Gsc expression correlated with craniofacial and limb defects in Gsc knockout mice,15,16 which had defects in the shoulder and hip joints along with malformations of the head, humeri, and femora.18 A subsequent study confirmed the expression of Gsc in the appendicular skeleton during mid- to late organogenesis, including in the shoulder and hip joint region, and in several ligaments and adductor muscles.19 At E14.5, Gsc expression was also detected in the labioscrotal swellings, which in males give rise to the scrotal sac and in females develop into the labia major, and correlated with later genital abnormalities in Gsc mutant mice.19 Therefore, there is good correlation between the sites of Gsc expression in mouse mid-embryogenesis and organogenesis and the phenotypic alterations observed in human individuals with GSC mutations (Table 1). However, Gsc mutant mice have additional minor skeletal abnormalities, such as rib fusion and sternum anomalies,16,18 that are not observed in humans.
To further understand why loss of the Goosecoid protein causes the unique and specific SAMS phenotype, we used immunofluorescence confocal microscopy in staged wild-type mouse embryo sections (Figure 3; see also Figure S2) to investigate the colocalization of Goosecoid with Pax6, a marker of neural-crest derivatives. Wild-type mouse embryos (embryonic ages E11.5–E15.5) were fixed in 2% [w/v] para-formaldehyde and embedded in paraffin. Thin sections (4 μm) were mounted onto slides, deparaffinized, and rehydrated by standard methods.20 For fresh-frozen cryosections, embryos were embedded in OCT Compound (Tissue-Tek) and snap frozen in 2-methylbutane chilled on dry ice. Thin fresh-frozen sections (4 μm) were cut onto slides, allowed to air dry, and fixed in ice-cold methanol (5 min at 4°C) or 2% paraformaldehyde (20 min at room temperature). Permeabilization, blocking methods, and immunofluorescence staining were essentially as described previously.20 The following primary antibodies were used: goat polyclonal anti-GSC (N-12; Santa Cruz Biotechnology) and mouse monoclonal anti-PAX3 (Developmental Studies Hybridoma Bank, University of Iowa) at final dilutions between 1:1,000 and 1:200. Confocal images were obtained with a Nikon Eclipse TE2000-E system, controlled, and processed by EZ-C1 3.50 (Nikon) software. Images were assembled with Adobe Illustrator CS4.
Figure 3.
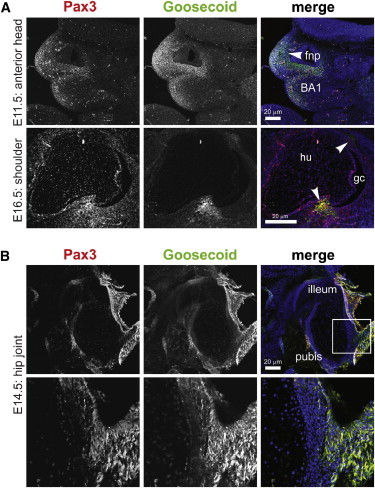
Goosecoid Colocalizes with Pax3 in Neural-Crest Derivatives and the Developing Pelvis
(A) Mouse embryonic midline sagittal and horizontal sections showing Goosecoid (green) and Pax3 (red) colocalization in neural-crest-tissue derivatives in the frontonasal prominence (fnp) and the first branchial arch (BA1) and bony attachment points in the embryonic shoulder joint (arrowheads). Scale bars represent 20 μm. Abbreviations are as follows: gc, glenoid cavity; and hu, humerus.
(B) Goosecoid and Pax3 colocalization in the developing pubic bone for a midline sagittal section of an E14.5 mouse embryo. The scale bar represents 20 μm. A magnified inset (bottom) is indicated by a white frame.
Goosecoid was localized specifically to nuclei (Figure S2) and colocalized with Pax3. Colocalization was limited to the postotic cranial neural crest (PONC) cells, the frontonasal prominence, the first branchial arch and cleft, and specific regions of large joints (Figure 3; see also Figure S3). Muscles of the shoulder girdle and their bony attachment points are derivatives of PONC cells21 and, consistent with this model, we observed specific Goosecoid localization in these tissue derivatives (Figure 3; see also Figures S2B–S2D). Because both the scapula and humerus develop from a single mesodermal condensation (the scleroblastema),22 we suggest that Goosecoid localization in the PONC cells and their derivatives is essential for this differentiation process, as well as for the subsequent formation of the muscles and connective tissues of the shoulder joint. Goosecoid and Pax3 also colocalized in the embryonic hip joint (Figure 3), suggesting that trunk neural-crest cell derivatives might contribute to human hip development. This suggestion is supported by the observation that trunk neural-crest cells had the potential to generate skeletal derivatives such as bone and cartilage.23 However, humans affected with SAMS not only have affected derivatives of the neural crest but also have affected intermediate-mesoderm derivatives, such as the urogenital system,24 and affected derivatives of the somatopleuric layer of the lateral-plate mesoderm, which gives rise to the pelvic bone primordia.25
Goosecoid therefore appears to be a downstream effector gene of the regulatory networks that define neural-crest cell-fate specification and determine mesoderm cell lineages in mammals. We suggest that dysregulation of Goosecoid-mediated gene expression in SAMS causes connective tissues to undergo pathological differentiation and adopt new cartilaginous or osseous fates. The unique skeletal anomalies therefore delineate SAMS as an unusual developmental condition with some neurocristopathy features that are distinct from those of other branchial-arch syndromes. Our findings confirm the essential role of Goosecoid in human craniofacial and joint development. Our segregation analysis of WES data demonstrates that even small nuclear families with a singleton individual affected with a rare recessive condition can contribute to and benefit from research studies. Our demonstration that predicted null mutations in GSC are causal for SAMS emphasizes the importance of data repositories of normal variants from specific ethnic groups to aid filtering strategies in gene-discovery projects.
Acknowledgments
The authors thank the individuals and families for participating in this study. We thank N. Lahiri, Great Ormond Street Hospital, for help with recruiting research participants. Z.A.A. received an Egyptian Government Scholarship. This work was supported by a Sir Jules Thorn Award for Biomedical Research (JTA/09 to C.A.J., E.S., and D.T.B.) The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Contributor Information
Angus Dobbie, Email: angus.dobbie@leedsth.nhs.uk.
Colin A. Johnson, Email: c.johnson@leeds.ac.uk.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org/
DGV (Database of Genomic Variants), http://projects.tcag.ca/variation/
fancyGENE, http://bio.ieo.eu/fancygene/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Picard, http://picard.sourceforge.net/
References
- 1.Erlebacher A., Filvaroff E.H., Gitelman S.E., Derynck R. Toward a molecular understanding of skeletal development. Cell. 1995;80:371–378. doi: 10.1016/0092-8674(95)90487-5. [DOI] [PubMed] [Google Scholar]
- 2.Passos-Bueno M.R., Ornelas C.C., Fanganiello R.D. Syndromes of the first and second pharyngeal arches: A review. Am. J. Med. Genet. A. 2009;149A:1853–1859. doi: 10.1002/ajmg.a.32950. [DOI] [PubMed] [Google Scholar]
- 3.Lemire E.G., Hildes-Ripstein G.E., Reed M.H., Chudley A.E. SAMS: provisionally unique multiple congenital anomalies syndrome consisting of short stature, auditory canal atresia, mandibular hypoplasia, and skeletal abnormalities. Am. J. Med. Genet. 1998;75:256–260. doi: 10.1002/(sici)1096-8628(19980123)75:3<256::aid-ajmg5>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 4.ter Heide H., Bulstra S.K., Reekers A., Schrander J.J., Schrander-Stumpel C.T. Auditory canal atresia, humeroscapular synostosis, and other skeletal abnormalities: confirmation of the autosomal recessive “SAMS” syndrome. Am. J. Med. Genet. 2002;110:359–364. doi: 10.1002/ajmg.10441. [DOI] [PubMed] [Google Scholar]
- 5.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McLaren W., Pritchard B., Rios D., Chen Y., Flicek P., Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26:2069–2070. doi: 10.1093/bioinformatics/btq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rambaldi D., Ciccarelli F.D. FancyGene: Dynamic visualization of gene structures and protein domain architectures on genomic loci. Bioinformatics. 2009;25:2281–2282. doi: 10.1093/bioinformatics/btp381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Su P.H., Chen J.Y., Chiang C.L., Ng Y.Y., Chen S.J. Exclusion of MYF5, GSC, RUNX2, and TCOF1 mutation in a case of cerebro-costo-mandibular syndrome. Clin. Dysmorphol. 2010;19:51–55. doi: 10.1097/MCD.0b013e328335c133. [DOI] [PubMed] [Google Scholar]
- 11.Kelberman D., Tyson J., Chandler D.C., McInerney A.M., Slee J., Albert D., Aymat A., Botma M., Calvert M., Goldblatt J. Hemifacial microsomia: progress in understanding the genetic basis of a complex malformation syndrome. Hum. Genet. 2001;109:638–645. doi: 10.1007/s00439-001-0626-x. [DOI] [PubMed] [Google Scholar]
- 12.Cho K.W., Blumberg B., Steinbeisser H., De Robertis E.M. Molecular nature of Spemann’s organizer: The role of the Xenopus homeobox gene goosecoid. Cell. 1991;67:1111–1120. doi: 10.1016/0092-8674(91)90288-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blum M., Gaunt S.J., Cho K.W., Steinbeisser H., Blumberg B., Bittner D., De Robertis E.M. Gastrulation in the mouse: the role of the homeobox gene goosecoid. Cell. 1992;69:1097–1106. doi: 10.1016/0092-8674(92)90632-m. [DOI] [PubMed] [Google Scholar]
- 14.Kalisz M., Winzi M., Bisgaard H.C., Serup P. EVEN-SKIPPED HOMEOBOX 1 controls human ES cell differentiation by directly repressing GOOSECOID expression. Dev. Biol. 2012;362:94–103. doi: 10.1016/j.ydbio.2011.11.017. [DOI] [PubMed] [Google Scholar]
- 15.Yamada G., Mansouri A., Torres M., Stuart E.T., Blum M., Schultz M., De Robertis E.M., Gruss P. Targeted mutation of the murine goosecoid gene results in craniofacial defects and neonatal death. Development. 1995;121:2917–2922. doi: 10.1242/dev.121.9.2917. [DOI] [PubMed] [Google Scholar]
- 16.Rivera-Pérez J.A., Mallo M., Gendron-Maguire M., Gridley T., Behringer R.R. Goosecoid is not an essential component of the mouse gastrula organizer but is required for craniofacial and rib development. Development. 1995;121:3005–3012. doi: 10.1242/dev.121.9.3005. [DOI] [PubMed] [Google Scholar]
- 17.Gaunt S.J., Blum M., De Robertis E.M. Expression of the mouse goosecoid gene during mid-embryogenesis may mark mesenchymal cell lineages in the developing head, limbs and body wall. Development. 1993;117:769–778. doi: 10.1242/dev.117.2.769. [DOI] [PubMed] [Google Scholar]
- 18.Belo J.A., Leyns L., Yamada G., De Robertis E.M. The prechordal midline of the chondrocranium is defective in Goosecoid-1 mouse mutants. Mech. Dev. 1998;72:15–25. doi: 10.1016/s0925-4773(97)00204-9. [DOI] [PubMed] [Google Scholar]
- 19.Zhu C.C., Yamada G., Nakamura S., Terashi T., Schweickert A., Blum M. Malformation of trachea and pelvic region in goosecoid mutant mice. Dev. Dyn. 1998;211:374–381. doi: 10.1002/(SICI)1097-0177(199804)211:4<374::AID-AJA8>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 20.Abdelhamed Z.A., Wheway G., Szymanska K., Natarajan S., Toomes C., Inglehearn C., Johnson C.A. Variable expressivity of ciliopathy neurological phenotypes that encompass Meckel-Gruber syndrome and Joubert syndrome is caused by complex de-regulated ciliogenesis, Shh and Wnt signalling defects. Hum. Mol. Genet. 2013;22:1358–1372. doi: 10.1093/hmg/dds546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsuoka T., Ahlberg P.E., Kessaris N., Iannarelli P., Dennehy U., Richardson W.D., McMahon A.P., Koentges G. Neural crest origins of the neck and shoulder. Nature. 2005;436:347–355. doi: 10.1038/nature03837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gardner E., Gray D.J. Prenatal development of the human shoulder and acromioclavicular joints. Am. J. Anat. 1953;92:219–276. doi: 10.1002/aja.1000920203. [DOI] [PubMed] [Google Scholar]
- 23.McGonnell I.M., Graham A. Trunk neural crest has skeletogenic potential. Curr. Biol. 2002;12:767–771. doi: 10.1016/s0960-9822(02)00818-7. [DOI] [PubMed] [Google Scholar]
- 24.Staack A., Donjacour A.A., Brody J., Cunha G.R., Carroll P. Mouse urogenital development: a practical approach. Differentiation. 2003;71:402–413. doi: 10.1046/j.1432-0436.2003.7107004.x. [DOI] [PubMed] [Google Scholar]
- 25.Sadler T.W. Langman’s Medical Embryology. Twelfth Edition. Lippincott Williams& Wilkins; Baltimore, MD, USA: 2012. Limbs; pp. 151–161. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.