

Abstract
In antiviral RNA interference (RNAi), the DICER enzyme processes virus-derived double-stranded RNA (dsRNA) into small interfering RNAs (siRNAs) that guide ARGONAUTE proteins to silence complementary viral RNA. As a counterdefense, viruses deploy viral suppressors of RNAi (VSRs). Well-established in plants and invertebrates, the existence of antiviral RNAi remains unknown in mammals. Here, we show that undifferentiated mouse cells infected with encephalomyocarditis virus (EMCV) or Nodamura virus (NoV) accumulate ~22-nucleotide RNAs with all the signature features of siRNAs. These derive from viral dsRNA replication intermediates, incorporate into AGO2, are eliminated in Dicer knockout cells, and decrease in abundance upon cell differentiation. Furthermore, genetically ablating a NoV-encoded VSR that antagonizes DICER during authentic infections reduces NoV accumulation, which is rescued in RNAi-deficient mouse cells. We conclude that antiviral RNAi operates in mammalian cells.
Although mammalian viruses are susceptible to experimental RNA interference (RNAi) via synthetic small interfering RNAs (siRNAs) (1), the existence of a natural antiviral RNAi response in mammals is debated (2). First, in many infected somatic cells, viral double-stranded RNA (dsRNA) triggers the potent and non-sequence-specific interferon (IFN) response (3) that may have largely supplanted antiviral RNAi functions (4). Second, several mammalian viral proteins display viral suppressor of RNAi (VSR)–like activities still awaiting validation in authentic virus expression contexts (1). Third, diverse virus-infected mammalian cell types accumulate virus-derived small RNAs (vsRNAs), but these have unspecified functions (5) and lack the biochemical features, size, and distribution patterns of plant and invertebrate viral siRNAs (6–9). Ascertaining genetically the DICER-dependency of mammalian vsRNA is further complicated by the essential contribution of the mammalian RNAi machinery (one Dicer, four Ago) to the endogenous microRNA (miRNA) pathway (10). Pluri-potent mouse embryonic stem cells (mESCs) withstand the complete ablation of DICER (DCR) or ARGONAUTE (AGO) functions (11, 12) and support RNAi triggered by long dsRNA possibly because they lack an IFN response (13, 14); accordingly, DCR-dependent endogenous siRNAs are detected in these cells (15). We thus reasoned that mESCs constituted potentially valuable models to genetically validate both viral siRNA accumulation and VSR function in authentic mammalian infection contexts.
Several mESC lines were infected with purified virions of encephalomyocarditis virus (EMCV), a mammalian positive-sense single-stranded RNA (ssRNA) picornavirus producing high levels of dsRNA within its 8-hour infection cycle (16). All cells accumulated the EMCV-encoded VP1 capsid to varying degrees, with the highest levels displayed by line E14 (Fig. 1A and fig. S1, A and B). In two separate infections, 15- to 50-nt-long RNA was isolated from E14 mESCs and subjected to ILLUMINA deep-sequencing (table S1). Six hours postinfection (hpi), 0.4 and 0.7% of total reads mapped the EMCV genome, of which 33 and 27% were in the 21- to 23-nt size range of DCR products (Fig. 1B and table S1). For comparison, miR-134, miR-296, and miR-470, which functionally target the mESC pluripotency factors Nanog, Oct4, and Sox2 (17) represented respectively 0.11%, 0.02%, and 0.05% of total reads (table S2). The remaining EMCV reads, in a heterogeneous 24- to 44-nt size range, mapped nearly exclusively along the viral positive strand (Fig. 1C), which accumulates disproportionately more than the negative strand during positive-sense RNA virus replication, and were thus mostly viral breakdown products (5, 18). By contrast, 36 and 28% of 21- to 23-nt reads mapped to both viral strands within the first 200-nt of the EMCV 5′ untranslated region and so exhibited a ~2:1 (+):(−) strand ratio contrasting with the ~10:1 ratio of all other reads (Fig. 1C and table S1). A less-pronounced symmetrical reads distribution was also observed at the EMCV RNA 3′-end, whereas the remaining 21- to 23-nt reads originated from discrete positive-strand regions (Fig. 1C).
Fig. 1. EMCV-derived siRNAs in infected mESCs.

(A) Western analysis of VP1 in E14 mESCs. NI, noninfected; ACTIN, protein-loading control. (B) Size distribution of vsRNA reads mapping to EMCV in samples from (A). (C) Distributions of 21- to 23-nt and 24- to 44-nt reads along EMCV (+) and (−) strands 6 hpi. (D) Same as (C), but along the first 5′-terminal 300 nt. Symmetrical reads are numbered. (Inset) Perfect duplexes formed by the abundant reads 1 to 2 and 3 to 4; 2-nt 3′ overhangs are indicated in red. Asterisk: Read sequence corresponding to the oligonucleotide probe used in (E). Radar plots: 21- and 23-nt reads in each of 22 possible registers mapping along the entire EMCV (+) and (−) strands; the 5′ first EMCV nucleotide defines register no. 1. Distance to the center indicates read percentage within each register. (E) Northern analysis of EMCV 5′-end siRNAs 6 hpi. Total RNA from SUC-SUL (SS) transgenic Arabidopsis run in parallel and hybridized secondarily provides a size marker for 21-nt and 24-nt siRNAs. U6, RNA-loading controls.
The symmetrical 5′ and 3′ EMCV reads mapped to the regions where dsRNA replication-intermediates (RIs) initiate during positive- and negative-strand synthesis. Similar to RI-derived siRNAs observed in virus-infected plants and invertebrates (6, 9), abundant (+) and (−) reads at the EMCV 5′ end formed contiguous and perfectly complementary duplexes with 2-nt 3′ overhangs (Fig. 1D). In addition, all EMCV-derived 21- to 23-nt reads defined a dominant, phased register initiated from the 5′ end at a ~22-nt periodicity, in which complementary (+) and (−) strands were offset by 2 nt (Fig. 1D and fig. S1, C to E). Northern analyses using oligonucleotide probes confirmed accumulation of the predicted 5′-end 22-nt siRNAs in EMCV-infected cells (Fig. 1E). Phased, perfect duplexes with 2-nt 3′ overhangs are signature products of sequential dicing of long dsRNA (19, 20). The DCR-dependency of EMCV-derived vsRNAs was thus explored in Dcr knockout (Dcr−/−) mESCs (Fig. 2A and fig. S2A), which, due to pleiotropy and reduced division rates (11), accumulated less EMCV than control DcrFlx/Flx cells. Viral inocula were thus precalibrated to produce comparable infection levels in both cell types (Fig. 2A and fig. S2A). Detected, as expected, in infected DcrFlx/Flx mESCs, EMCV-derived 5′-end vsRNAs were below Northern detection limit in Dcr−/− mESCs (Fig. 2A), which confirmed that they were bona fide siRNAs. Moreover, in wild-type (WT) mESCs, these were detected by Northern analysis of RNA from FLAG- and hemagglutinin (HA)–tagged human AGO2 (FLAG-HA-hAGO2) immunoprecipitates (IPs) (Fig. 2B and fig. S2B) and by deep-sequencing RNA from endogenous mAGO2 IPs also containing cellular miRNAs (Fig. 2, C and D, and fig. S2, C and D). Abundant positive-strand reads also coincided with several peaks already observed in total RNA sequencing (Fig. 2C, *), of which one mapped to the internal ribosomal entry site (IRES: nt 577 to 680) and another to a predicted dsRNA fold back (nt 1497 to 1670) (fig. S2E). Therefore, the AGO2-loading landscape of EMCV-infected mESCs comprised RI-derived and DCR-dependent siRNA duplexes, as well as other 21- to 23-nt vsRNAs generated from positive-strand secondary structures via mechanisms that, as in plants and invertebrates, await clarification (6, 9).
Fig. 2. AGO2-loaded EMCV siRNAs are reduced in Dcr−/− mESCs and following differentiation.

(A) Western and Northern analysis of VP1 (top), EMCV 5′-end siRNAs (middle) and miR-16/pre-miR-16 (bottom) in DcrFlx/Flx and Dcr−/− mESCs infected (+) or not (−) with EMCV. SS, as in Fig. 1E. ACTIN and U6: protein- and RNA-loading controls. (B) Northern analysis of EMCV 5′-end siRNAs 6 hpi in FLAG-specific immunoprecipitates isolated from WT mESCs or mESCs stably expressing human FLAG-HA hAGO2, infected (+) or not (−) with EMCV. SS as in (A). Western analyses show comparable infection levels (VP1) and confirm FHA-hAGO2 immunoprecipitation with miR-16. Total: Coomassie-stained protein loading control. (C) Distributions of 21- and 23-nt reads along EMCV (+) and (−) strands after deep-sequencing of RNA from endogenous mAGO2 IP 6 hpi. Asterisks: reads further analyzed in fig. S2E. (D) Same as (C), but along the first 5′-terminal 150 nt. (E) Western and Northern analyses of the pluripotency markers OCT4, VP1, EMCV 5′-end siRNAs and miR-16 in undifferentiated mESCs on day 0 (d0) or after 10 days of differentiation (d10), infected (+) or not (−) with EMCV. Total as in (B); U6 and SS are as above. (F) Size distribution of vsRNA deep-sequencing reads mapping to EMCV in samples from (E). Abundance was normalized to the total number of reads mapping to EMCV. (G) Reads mapping to EMCV in infected day 0 and day 10 mESCs, as in Fig. 1D. Note the scale change in counts, highlighted in red. (Inset) siRNA duplex 1 to 2 remains detectable in day 10 cells.
The use of mESCs granted an investigation of viral siRNA accumulation in genetically identical cells but under distinct differentiation states. Differentiation of E14-derived embryoid body was confirmed at day 10 by the loss of expression of pluripotency markers Nanog and Oct4 and gain in expression of the ectoderm-specific marker Fgf5 (Fig. 2E and fig. S2F). At 6 hpi, 5′-end siRNAs were below Northern detection in EMCV-infected day 10 compared with day 0 E14 cells, despite their similar infection levels (Fig. 2E and fig. S2F). Accordingly, EMCV-derived reads represented 0.15% of total deep-sequencing reads in infected day 10 cells, a nearly fivefold decrease compared to infected day 0 cells (Fig. 2F). The 21- to 23-nt reads were also 10 times less abundant in day 10 cells as in day 0 cells but were still detectable, including in the first 5′-terminal 200 nt, representing 16% of all EMCV-derived reads (Fig. 2G and table S1). Therefore, EMCV siRNA accumulation was significantly reduced, albeit not abolished, by differentiation, unlike that of miRNAs (Fig. 2E).
Demonstrating the antiviral activity of siRNAs entails the genetic rescue of VSR-deficient viruses in RNAi-compromised host cells (6, 7, 21), an approach not possible with EMCV for which a potential VSR awaits identification. The dsRNA-binding B2 protein encoded by the bipartite, positive-sense ssRNA Nodamura virus (NoV) inhibits DCR activity during experimental mammalian RNAi (22), a property shared by its ortholog in the NoV-related Flock house nodavirus (FHV) in Drosophila (6). NoV or its B2-deficient counterpart, NoVΔB2, were titrated to similar levels in stable B2-expressing BHK-21 cells (23) and subsequently used to infect E14 mESCs. NoVΔB2 accumulated considerably less than NoV at 3 dpi, and only the former infection was able to generate virus-derived 21- to 23-nt deep-sequencing reads (Fig. 3, A and B, and fig. S3, A and B), whereas global miRNA levels remained unchanged in both infections (tables S1 and S2). NoV-derived reads, heterogeneous in size, mapped mostly along the RNA1 positive strand (Fig. 3C, fig. S3C, and table S1). By contrast, those from NoVΔB2, nearly exclusively 21 to 23 nt in length, derived mainly from the 5′ and 3′ ends of RNA1 (+) and (−) strands (Fig. 3, C and D), which resembled the FHVΔB2 siRNA pattern in Drosophila (6). Furthermore, NoVΔB2 5′ end reads had a ~22-nt periodicity and formed contiguous, perfect duplexes with 2- to 3-nt 3′ overhangs reminiscent of the DCR-dependent EMCV siRNAs (Fig. 3, E and F). An identical set of phased, perfect duplexes was detected in both NoVΔB2-infected BHK-21 somatic cells and limbs of newborn mice but not upon infection with NoV (Fig. 3F) (23). Likewise, reads from the B2-proficient NoV displayed none of these features in mESCs despite their much higher abundance (Fig. 3, E and F, and fig. S3, D and E). Thus, mirroring the action of the FHV-encoded B2 VSR in Drosophila (6), DCR-dependent processing of RI-derived dsRNA was suppressed by the NoV-encoded B2 protein in mESCs. Furthermore, this B2-restricted mechanism operated almost identically in mESCs and suckling mice.
Fig. 3. B2 antagonizes NoV-derived siRNA production.

(A) Northern analysis of genomic RNA1 and subgenomic (sg)RNA3 in mESCs 72 hpi with NoV or NoVΔB2. Ribosomal RNA (rRNA) indicates rRNA ethidium bromide staining; NI, noninfected.(B)Normalized size distribution of deep-sequencing reads mapping the NoV or NoVΔB2 genome in samples from (A). (C) Distributions of 21- to 23-nt and 24- to 44-nt reads along the (+) and (−) strands of NoV (left) or NoVΔB2 (right) RNA1. (D) Same as (C), but along the first 5′-terminal 300 nt. (E) Radar plots as in Fig. 1D, but for NoV and NoVΔB2; RNA1 5′ first nucleotide defines register no. 1. (F) Read sequences along the first 180 nt of the 5′-terminus of NoVΔB2 RNA1 (+) and (−) strands. Read counts (in bold italic), genomic position, and sequence variants are indicated. Nonsequenced reads within the main ~22-nt vsRNA periodicity register are indicated with XXX. Reads detected identically in NoVΔB2-infected BHK-21 cells and suckling mice (23) are depicted in blue. 2-nt3′ overhangs are in red.
FHV B2 inhibits both siRNA processing and incorporation into AGO (24). Therefore, to explore antiviral RNAi in NoV-infected mESCs and to avoid functional redundancy with AGO1, AGO3, or AGO4, we used the quadruple Ago1,2,3,4 KO mESC line E7, in which an ectopically expressed hAgo2 transgene is removable by tamoxifen application (12). hAgo2 depletion was confirmed 5 days after tamoxifen treatment (Fig. 4A), upon which mESCs were infected with NoV or NoVΔB2 for 3 days. In two separate experiments, the NoV and NoVΔB2 RNA1 levels were respectively ~2 and ~8 times as high in tamoxifen-induced as in untreated mESCs (Fig. 4B). Northern analyses further showed that NoVΔB2 accumulation was rescued in AGO2-deficient mESCs similarly to FHVΔB2 accumulation in Ago2-deficient Drosophila cells (6, 25); the lower impact of AGO2 depletion on virulent NoV confirmed B2 VSR activity in authentic infections (Fig. 4C). Combined with those obtained with EMCV, the results demonstrate that antiviral RNAi operates in mammalian cells.
Fig. 4. Rescue of NoVΔB2 accumulation in AGO2-deficient mESCs.
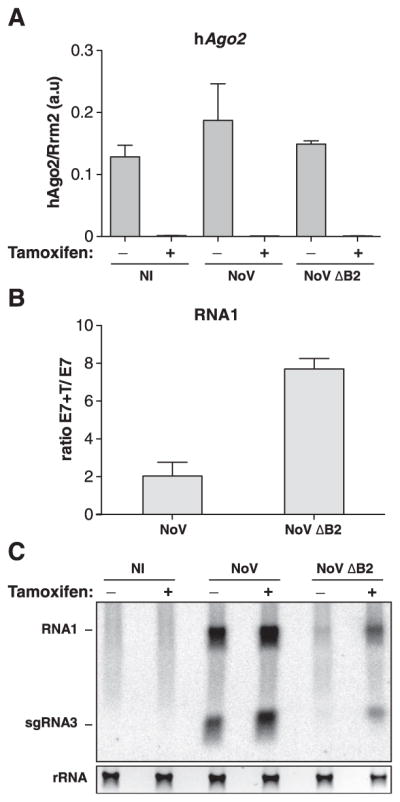
(A) Quantitative reverse transcription polymerase chain reaction (RT-PCR) analysis of the hAgo2 transgene mRNA levels in noninfected (NI) and NoV- and NoVΔB2-infected E7 mESCs previously treated (+) or not (−) with tamoxifen for 5 days. Results show the mean and standard deviation of two independent experiments; a.u., arbitrary units. (B) Relative accumulation of NoV or NoVΔB2 RNA1 72 hpi in E7 mESC treated (+T) or not with tamoxifen, assessed by quantitative RT-PCR on samples used in (A). Results show the mean of the ratio and the standard deviation calculated from two independent experiments. (C) Northern analysis of NoV and NoVΔB2 genomic RNA1 and sgRNA3 72 hpi of the cells used in (A). rRNA, as in Fig. 3A.
The biogenesis and distribution patterns of small RNA derived from ssRNA viruses are thus conserved among infections of plant, invertebrate, and mammalian cells; orthologous VSRs of insect- and mammalian-infecting viruses also suppress DCR action in genetically indistinguishable ways. Therefore, defensive, in addition to possible regulatory, functions likely underpin the evolutionary persistence of catalytic RNAi in mammals. Our results and those of Li et al. (23) provide clues as to why mammalian antiviral RNAi has remained elusive thus far. First, previous studies invariably involved virulent viruses, of which some probably encode VSRs that, like the NoV-encoded B2, prevent production of siRNAs, the diagnostic molecules of antiviral RNAi. Second, virus-derived siRNA levels were at least one order of magnitude higher in undifferentiated than in differentiated mESCs or BHK-21 cells (23). This probably relates to the distinctive efficacy of long dsRNA-triggered RNAi in undifferentiated cells derived from embryonic or adult tissues, which is possibly underpinned by their generally reduced ability to mount non–sequence specific immune responses, including the IFN response, against long dsRNA (4). Alternatively, or coincidently, DCR siRNA-processing activity might decrease during cell differentiation, perhaps via modification of its internal autoinhibitory helicase domain (20). In this context, the identical distribution, relative abundance, and biochemical features of NoVΔB2 siRNAs observed in mESCs and suckling mice suggest that multipotent progenitor cells, which abound in various mammalian tissues, might form the primary and most potent sites of antiviral RNAi in vivo. Nonetheless, long dsRNA-triggered RNAi was reported in somatic myoblasts, or even in fully differentiated myotubes and neural cells despite the possible activation of an IFN response (13, 26, 27); it would thus be premature to confine the antiviral functions of RNAi to undifferentiated or IFN-deficient cellular states. Tools developed in this and the accompanying study (23) now allow a thorough investigation of these fundamental questions in developing and adult mammals.
Supplementary Material
Acknowledgments
We thank the Voinnet laboratory for advice and discussions and L. Bakkali-Kassimi, G. Meister, E. Brocchi, and J.-C. Paillart for reagents. Supported by a Prospective Researchers Fellowship from the Swiss National Foundation to P.V.M., a fellowship from the Federation of European Biochemical Societies to C.C., and an award from the Bettencourt Foundation to O.V.
Footnotes
References and Notes
- 1.Haasnoot J, Westerhout EM, Berkhout B. Nat Biotechnol. 2007;25:1435–1443. doi: 10.1038/nbt1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Umbach JL, Cullen BR. Genes Dev. 2009;23:1151–1164. doi: 10.1101/gad.1793309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goubau D, Deddouche S, Reis C, Sousa E. Immunity. 2013;38:855–869. doi: 10.1016/j.immuni.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chalupnikova K, Nejepinska J, Svoboda P. Methods Mol Biol. 2013;942:291–314. doi: 10.1007/978-1-62703-119-6_16. [DOI] [PubMed] [Google Scholar]
- 5.Parameswaran P, et al. PLoS Pathog. 2010;6:e1000764. doi: 10.1371/journal.ppat.1000764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aliyari R, et al. Cell Host Microbe. 2008;4:387–397. doi: 10.1016/j.chom.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deleris A, et al. Science. 2006;313:68–71. doi: 10.1126/science.1128214. [DOI] [PubMed] [Google Scholar]
- 8.Ding SW, Voinnet O. Cell. 2007;130:413–426. doi: 10.1016/j.cell.2007.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang XB, et al. Plant Cell. 2011;23:1625–1638. doi: 10.1105/tpc.110.082305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim VN, Han J, Siomi MC. Nat Rev Mol Cell Biol. 2009;10:126–139. doi: 10.1038/nrm2632. [DOI] [PubMed] [Google Scholar]
- 11.Murchison EP, Partridge JF, Tam OH, Cheloufi S, Hannon GJ. Proc Natl Acad Sci USA. 2005;102:12135–12140. doi: 10.1073/pnas.0505479102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Su H, Trombly MI, Chen J, Wang X. Genes Dev. 2009;23:304–317. doi: 10.1101/gad.1749809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paddison PJ, Caudy AA, Hannon GJ. Proc Natl Acad Sci USA. 2002;99:1443–1448. doi: 10.1073/pnas.032652399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Billy E, Brondani V, Zhang HD, Müller U, Filipowicz W. Proc Natl Acad Sci USA. 2001;98:14428–14433. doi: 10.1073/pnas.261562698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Babiarz JE, Ruby JG, Wang Y, Bartel DP, Blelloch R. Genes Dev. 2008;22:2773–2785. doi: 10.1101/gad.1705308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weber F, Wagner V, Rasmussen SB, Hartmann R, Paludan SR. J Virol. 2006;80:5059–5064. doi: 10.1128/JVI.80.10.5059-5064.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tay Y, Zhang J, Thomson AM, Lim B, Rigoutsos I. Nature. 2008;455:1124–1128. doi: 10.1038/nature07299. [DOI] [PubMed] [Google Scholar]
- 18.Pfeffer S, et al. Nat Methods. 2005;2:269–276. doi: 10.1038/nmeth746. [DOI] [PubMed] [Google Scholar]
- 19.Allen E, Xie Z, Gustafson AM, Carrington JC. Cell. 2005;121:207–221. doi: 10.1016/j.cell.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 20.Ma E, MacRae IJ, Kirsch JF, Doudna JA. J Mol Biol. 2008;380:237–243. doi: 10.1016/j.jmb.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Diaz-Pendon JA, Li F, Li WX, Ding SW. Plant Cell. 2007;19:2053–2063. doi: 10.1105/tpc.106.047449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sullivan CS, Ganem D. J Virol. 2005;79:7371–7379. doi: 10.1128/JVI.79.12.7371-7379.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Y, Lu J, Han Y, Fan X, Ding SW. Science. 2013;342:231–234. doi: 10.1126/science.1241911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chao JA, et al. Nat Struct Mol Biol. 2005;12:952–957. doi: 10.1038/nsmb1005. [DOI] [PubMed] [Google Scholar]
- 25.Wang XH, et al. Science. 2006;312:452–454. doi: 10.1126/science.1125694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yi CE, Bekker JM, Miller G, Hill KL, Crosbie RH. J Biol Chem. 2003;278:934–939. doi: 10.1074/jbc.M205946200. [DOI] [PubMed] [Google Scholar]
- 27.Gan L, et al. J Neurosci Methods. 2002;121:151–157. doi: 10.1016/s0165-0270(02)00230-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.