

Abstract
Background
Bacteriophage EC1-UPM is an N4-like bacteriophage which specifically infects Escherichia coli O78:K80, an avian pathogenic strain that causes colibacillosis in poultry. The complete genome sequence of bacteriophage EC1-UPM was analysed and compared with other closely related N4-like phage groups to assess their genetic similarities and differences.
Results
Bacteriophage EC1-UPM displays a very similar codon usage profile with its host and does not contain any tRNA gene. Comparative genomics analysis reveals close resemblance of bacteriophage EC1-UPM to three N4-like bacteriophages namely vB_EcoP_G7C, IME11 and KBNP21 with a total of 44 protein coding genes shared at 70% identity threshold. The genomic region coding for the tail fiber protein was found to be unique in bacteriophage EC1-UPM. Further annotation of the tail fiber protein using HHpred, a highly sensitive homology detection tool, reveals the presence of protein structure homologous to various polysaccharide processing proteins in its C-terminus. Leveraging on the availability of multiple N4-like bacteriophage genome sequences, the core genes of N4-like bacteriophages were identified and used to perform a multilocus phylogenetic analysis which enabled the construction of a phylogenetic tree with higher confidence than phylogenetic trees based on single genes.
Conclusion
We report for the first time the complete genome sequence of a N4-like bacteriophage which is lytic against avian pathogenic Escherichia coli O78:K80. A novel 928 amino acid residues tail fiber protein was identified in EC1-UPM which may be useful to further the understanding of phage-host specificity. Multilocus phylogenetic analysis using core genes of sequenced N4-like phages showed that the evolutionary relationship correlated well with the pattern of host specificity.
Keywords: Bacteriophage EC1-UPM, Tail fiber protein, Complete genome, Multilocus phylogenetic analysis
Background
Escherichia coli O78:K80 is one of the common serogroups of Avian Pathogenic Escherichia coli (APEC) which causes colibacillosis in all ages of chickens, turkeys and other avian species. The infection which is associated with respiratory infection (airsacculitis), followed by perihepatitis, pericarditis and septicaemia is a devastating infection as it may reduce growth and egg production of poultry. Under untreated condition, high mortality rates of birds are recorded and this incurs high economic losses to farmers and the poultry industry [1]. These bacteria enter the human food chain through contamination of the environment by fecal droppings from infected chickens and soiled poultry products [2]. Conventional treatment by using antibiotics has been reported less effective in recent years due to the emergence of antibiotic resistance in the causative agent [3].
In our previous studies, bacteriophage EC1-UPM which was isolated from chicken faecal sample is able to reduce the severity of infection caused by E. coli O78:K80 and has the potential to be used for the treatment of colibacillosis in chickens. Based on the results of our in vivo study, the total mortality rate of the chickens was reduced by 70% when infected chickens were treated with bacteriophage EC1-UPM. The body weights of treated chickens were 15.4% higher than those of the untreated chickens [4,5]. To further exploit the potential of bacteriophage EC1-UPM, it is essential to have an understanding on its genetic make-up, particularly the genes which are responsible for the infection and lysis of the host bacteria.
In this study, we report for the first time the genome of a bacteriophage EC1-UPM, an N4-like bacteriophage that infects E. coli O78:K80. The genetic components of bacteriophage EC1-UPM which may be responsible for its host specificity and virulence were identified and analyzed in-silico. We also identified the core genes from the available N4-like bacteriophage genomes and utilized this information to gain insights into the evolutionary relationship of N4-like bacteriophages using multilocus phylogenetic analysis.
Results and discussion
Genomic features of bacteriophage EC1-UPM
The genome of bacteriophage EC1-UPM consists of 70,912 bp (GC content of 42.9%). A total of 80 protein coding genes were predicted with an average length of 816 bp (92.5% coding density). Annotation of the majority of predicted CDS showed confident hits against proteins of the N4-like bacteriophages, indicating that bacteriophage EC1-UPM may be a member of the N4-like bacteriophages (Additional file 1: Table S1). N4-like bacteriophages are bacteriophages of the Podoviridae family. Members of this group are lytic against their hosts. The host specificity of N4-like bacteriophages is rather diverse, ranging from enterobacteria such as E. coli and Pseudomonas aeruginosa to marine bacteria such as Sulfitobacter sp. EE-36 and Silicibacter pomeroyi DSS-3 [6-9]. Bacteriophage N4 which infects E. coli K-12 is currently the most studied strain for this group of bacteriophages [10]. In addition to having its complete genome sequenced, the identity and locations of several of its structural proteins have been established through comparisons of three-dimensional, cryo-electron microscopic structures of wild-type N4 and its mutants [11]. Based on our analysis, the annotated proteins of bacteriophage EC1-UPM can be categorized into the following functional groups: bacteriophage structure and packaging (portal protein, major coat protein, tail protein, tailspike protein, structural protein, capsid decorating protein), DNA replication/modification (DNA helicase, DNA polymerase, endonuclease, terminase), signal transduction and regulatory function (ssDNA-binding protein, RNA polymerase), nucleotide metabolism (thymidilate synthase, dCTP deaminase) and host lysis (holin, N-acetylmuramidase). The presence of lysis gene but not lysogeny-related gene indicates that bacteriophage EC1-UPM is a lytic bacteriophage. The largest gene encodes for the virion polymerase and is 10,839 bp, which is approximately 15% of the whole genome length (Figure 1). The presence of bacteriophage-encoded RNA polymerase of such size is a signature of N4 bacteriophage. It was demonstrated in N4 bacteriophage that this particularly large RNA polymerase was packaged into its capsid and ejected into the host cell thus eliminating the need to rely on host RNA polymerase for the transcription of its early genes [11]. No tRNA was identified in the genome of bacteriophage EC1-UPM, thus indicating that upon entry into the host, it is completely reliant on the host tRNA makeup for its protein synthesis.
Figure 1.
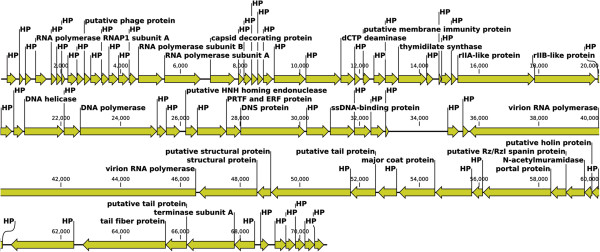
Linear genome visualization of bacteriophage EC1-UPM. Direction of the arrow represents transcription orientation. HP, hypothetical protein; PTF, pathogenesis-related transcription factor; DNS, extracellular deoxyribonuclease.
Codon usage of bacteriophage EC1-UPM
Comparison of the codon usage in E. coli O78:K80 and bacteriophage EC1-UPM revealed a rather similar codon frequency (Figure 2). Compatibility of phage codon-usage with that of its host will confer an evolutionary advantage as this will aid in the energy-demanding and laborious mechanism of protein synthesis. Three codons however were overrepresented in bacteriophage EC1-UPM, namely, codons ACT, GTA and GCT that correspond to the amino acids threonine, valine and alanine, respectively. Interestingly, bacteriophage EC1-UPM does not adapt to the codon usage dissimilarity as evidence by the absence of tRNA gene in its genome. In the evolution of dsDNA phages, the presence of tRNAs particularly those that are abundant in the phage and less common in the host allows the phage to outcompete its potential competitors in the host by translating its proteins more effectively, speeding up the reproduction rate and reducing its latency time [12]. In T4 phages, deletion of the tRNA genes was shown to decrease burst sizes and protein synthesis rates, demonstrating the growth advantage of carrying tRNA genes in phages [13]. The complete absence of tRNA in bacteriophage EC1-UPM may imply that its ability to infect E. coli O78:K80 could have been a recent acquisition.
Figure 2.
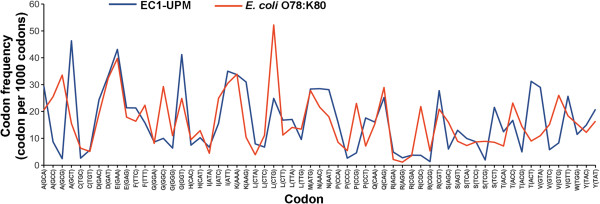
Codon frequency of bacteriophage EC1-UPM and its host E. coli O78:K80. Non-bracketed letters on the X-axis represent the amino acid associated with the codon.
Comparative genomics of bacteriophage EC1-UPM and closely related N4-like bacteriophages
Initial whole genome BLAST of bacteriophage EC1-UPM against the NCBI database showed that bacteriophage EC1-UPM is closely related to three N4-like viruses namely KBNP21, vB_EcoP_G7C and IME11 (data not shown). Bacteriophages KBNP21, vB_EcoP_G7C and IME11 were isolated from chicken farm, horse feces and hospital sewage, respectively [6,14,15]. Bacteriophage IME11 could infect 13 of 31 enteropathogenic clinical E. coli strains [6]. Bacteriophage vB_EcoP_G7C could infect E. coli strain 4 while bacteriophage KBNP21 could infect E. coli strains KBP21 and KBP135 [14,15]. However, the lack of O and K antigen data of the tested E. coli strains severely impeded host specificity determination based on surface antigen. Using Blast Ring Image generator (BRIG), it was shown that the majority of genomes are conserved across the four bacteriophages (Figure 3), except in the genomic region encompassing the genes coding for tail fiber and tail spike proteins. Tail fiber and tail spike proteins are often associated with the binding and degradation of host cell surface antigen [14]. Given the differences in the isolation source of each E. coli-infecting N4-like bacteriophage, it is likely that their E. coli hosts are of different strains that lack similarity in surface antigen profile. The gene coding for tail spike protein in bacteriophage EC1-UPM is conserved only in bacteriophage KBNP21 which was similarly isolated from avian source. Although, InterProscan annotation of the tail spike protein did not show any significant protein domain (Additional file 1: Table S1), augmentation of the protein annotation with HHpred indicates three major domains associated with carbohydrate binding and degradation between residues 120 to 190, 230 to 500 and 550 to 700 (Figure 4). Tail spike has been shown to be a vital component particularly in the infection of bacteria possessing both O and K antigens [14]. Some K antigens could prevent the direct recognition of receptor on the O antigen by the phage tail due to the large K-antigen size (up to 4000 A). The tail spike protein functions to overcome such limitation by creating a tunnel in the cell capsule and exposing the receptor to the tail protein [15,16].
Figure 3.

Blast ring image using the complete genome of bacteriophage EC1-UPM as reference. Outer rings represent the genomes of three N4-like bacteriophages closely related to EC1-UPM.
Figure 4.
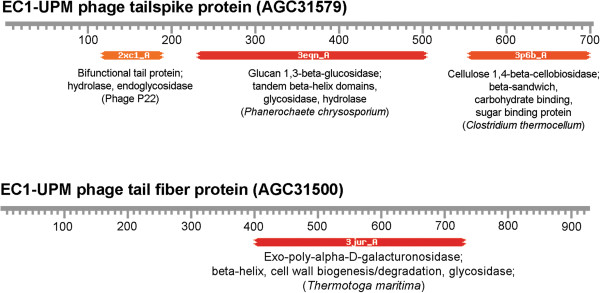
Identification of protein regions homologous to polysaccharide degrading enzymes in the tailspike and tail fiber proteins of bacteriophage EC1-UPM via HHpred. Protein IDs are indicated by the bracketed text.
Novel region in the C-terminus of tail fiber protein
Based on BRIG visualization, the genomic region corresponding to the C-terminus of bacteriophage EC1-UPM tail fiber protein was not shared by any member of the closely related N4-like bacteriophages (Figure 3). Similar to tail spike protein, the tail fiber of bacteriophage has also been implicated in host specificity. The C-terminus region of tail fiber protein has been reported to alter the host range of a bacteriophage [17]. Divergences in the C-terminus may therefore represent the phage adaptation towards the huge variation of O and K antigens in E. coli[18]. Expanding the BLAST search towards the whole NR database did not reveal any significant hit in the C-terminus region. Based on HHpred annotation, the C-terminus of tail fiber protein showed significant degree of structural homology with various polysaccharide degrading enzymes possessing the right-handed β-helix structure (Figure 4 and Additional file 2: Table S2). Notably, residues 402 to 731 of the tail fiber displayed significant similarity to a β-helix structure-containing exo-poly-α-D-galacturonosidase of Thermotoga maritima, a thermophilic bacteria (E-value = 4.44 × 10-44) (Figure 4 and Additional file 2: Table S2). Exo-poly-α-D-galacturonosidase is involved in the hydrolysis of pectic from the non-reducing to release digalacturonate. Initially identified in pectate lyase C, the right-handed β-helix is commonly found in the catalytic domains of various proteins that process oligo- and polysaccharides [19]. Given the structural homology of the tail fiber C-terminus to various polysaccharide degrading enzymes (Additional file 2: Table S2), it is plausible that the tail fiber may be associated with binding and/or degradation of the polysaccharide present on the bacterial antigens. The presence of hydrolytic phage tail fiber proteins has been shown to allow bacteriophage ΦK1-5 to attach and degrade both K1 and K5 capsular polysaccharides [20]. Further enzymatic study is necessary to validate the function of bacteriophage EC1-UPM tail fiber protein and its substrate range. This novel tail fiber protein consisted of 928 amino acid residues with molecular mass and pI of 99.3 kDa and 4.85, respectively.
Host lysis
It is widely accepted that lysis of the host is indispensable for the release and dispersion of progenies in all bacteriophages, except in filamentous bacteriophages [21,22]. In bacteriophage EC1-UPM, genes involved in the holin-endolysin system are located adjacent to each other. The N-acetylmuramidase (NCBI accession number: AGC31576) which may be responsible for the endolysis activity contains a domain implicated in glycosyl hydrolase (PF05838) from residues 22 to 113. The glycosyl hydrolase family functions via the hydrolysis of β-1,4 bond between N-acetylmuramic acid and N-acetylglucosamine in the peptidoglycan layer of bacteria. The presence of a peptidoglycan binding domain (PF09374) from residue 116 to 195 after the glycosyl hydrolase domain may further enhance the activity of N-acetylmuramidase by localizing the protein to its target region. Membrane topology visualization of bacteriophage EC1-UPM holin protein indicates that it contains two transmembrane domains with both N-and C-terminus (data not shown) located within the cytoplasmic region, thus classifying it as a class II holin [21,22]. The genes coding for the holin-endolysin system are well-conserved in bacteriophages KBNP21, vB_EcoP_G7C, IME11 and EC1-UPM (Figure 3), indicating a common mechanism for host lysis.
Orthologs identification and multilocus phylogenetic analysis of N4-like bacteriophages
The recent steady increase and availability of the complete genomes of various N4-like bacteriophages in the GenBank provides an opportunity to identify the orthologs that are well-conserved in all N4-like bacteriophages. Initial orthologs identification in bacteriophage EC1-UPM and its closely related strains vB_EcoP_G7C, IME11 and KBNP21 revealed that at 70% identity and 70% aligned cutoffs, a total of 44 proteins were conserved across the bacteriophages (Table 1). The average identities of the orthologs shared in bacteriophages vB_EcoP_G7C, IME11 and KBNP21 as compared to that of EC1-UPM were 91%, 93% and 95%, respectively. No orthologs can be identified when the analysis was expanded to include the more distant N4-like bacteriophages (Table 2) using the similar setting. Nevertheless, by lowering the identity cutoff to 40%, seven orthologous clusters were identified namely DNA polymerase I, N4 gp42-like protein, DNA primase, N4 gp44-like protein, portal protein, terminase and N4 gp69-like protein. The functional assignment of proteins in the orthologous clusters is in agreement with the notion that viruses share homologous “hallmark genes” encoding for proteins involved in genome replication and virion structure formation [23]. The phylogenetic tree analysis utilizing the trimmed and concatenated alignment of proteins in these orthologous clusters showed that the evolutionary relationship of the N4-like bacteriophage is very much correlated with their host specificity. Comparison of several phylogenetic trees constructed using individual orthologous clusters with known function (DNA polymerase, terminase, portal protein and DNA primase) showed that phylogenetic tree analysis based solely on sequence alignment of DNA polymerase resulted in a slightly different tree topology with a lower bootstrap support as compared to the multi-locus phylogenetic tree (Figure 5). This demonstrates that although DNA polymerase is a good indicator of evolutionary relationship in general, caution should be taken when resolving closely related N4-like bacteriophages. In contrast, phylogenetic tree construction based on DNA primase is highly comparable with the multi-locus phylogeny approach in terms of tree topology and bootstrap support value.
Table 1.
Orthologous group of proteins conserved across bacteriophages EC1-UPM, vB_EcoP_G7C (E), IME11 (I) and KBNP21 (K) as identified by PanOCT (>70% Identity and > 70% aligned length)
| Protein ID | Product Description |
% Identity |
InterProscan ID | ||
|---|---|---|---|---|---|
| E | I | K | |||
|
AGC31510 |
hypothetical protein |
87 |
95 |
97 |
|
|
AGC31513 |
RNA polymerase RNAP1 subunit A |
96 |
94 |
98 |
|
|
AGC31517 |
hypothetical protein |
94 |
93 |
96 |
|
|
AGC31519 |
putative phage protein |
74 |
90 |
84 |
|
|
AGC31522 |
hypothetical protein |
82 |
99 |
99 |
|
|
AGC31523 |
hypothetical protein |
91 |
88 |
91 |
|
|
AGC31525 |
RNA polymerase subunit B |
95 |
96 |
99 |
|
|
AGC31526 |
RNA polymerase subunit A |
88 |
96 |
97 |
IPR002092 |
|
AGC31527 |
capsid decorating protein |
79 |
91 |
93 |
IPR003599; IPR007110; IPR013783 |
|
AGC31528 |
hypothetical protein |
98 |
97 |
97 |
|
|
AGC31531 |
hypothetical protein |
95 |
97 |
95 |
|
|
AGC31533 |
hypothetical protein |
95 |
95 |
95 |
IPR027417 |
|
AGC31534 |
hypothetical protein |
95 |
97 |
99 |
IPR018698; IPR025154 |
|
AGC31537 |
putative membrane immunity protein |
86 |
98 |
94 |
IPR016410 |
|
AGC31539 |
hypothetical protein |
88 |
94 |
95 |
|
|
AGC31540 |
thymidilate synthase |
84 |
90 |
95 |
IPR003669 |
|
AGC31543 |
hypothetical protein |
76 |
80 |
82 |
|
|
AGC31545 |
rIIA-like protein |
86 |
91 |
94 |
IPR003594 |
|
AGC31546 |
rIIB-like protein |
88 |
89 |
87 |
|
|
AGC31547 |
hypothetical protein |
90 |
96 |
95 |
|
|
AGC31549 |
DNA helicase |
98 |
99 |
99 |
IPR027417; IPR027785 |
|
AGC31550 |
hypothetical protein |
97 |
99 |
98 |
|
|
AGC31551 |
DNA polymerase |
98 |
98 |
97 |
IPR001098; IPR002298; IPR012337 |
|
AGC31552 |
hypothetical protein |
92 |
91 |
99 |
|
|
AGC31555 |
hypothetical protein |
97 |
95 |
98 |
|
|
AGC31557 |
DNS protein |
97 |
96 |
100 |
IPR014820 |
|
AGC31558 |
hypothetical protein |
99 |
99 |
99 |
|
|
AGC31559 |
ssDNA-binding protein |
95 |
97 |
97 |
|
|
AGC31560 |
hypothetical protein |
99 |
99 |
98 |
|
|
AGC31561 |
hypothetical protein |
76 |
92 |
94 |
|
|
AGC31567 |
putative structural protein |
88 |
85 |
85 |
|
|
AGC31568 |
hypothetical protein |
92 |
93 |
94 |
|
|
AGC31569 |
putative tail protein |
94 |
82 |
96 |
|
|
AGC31570 |
hypothetical protein |
96 |
97 |
99 |
|
|
AGC31571 |
major coat protein |
97 |
97 |
100 |
|
|
AGC31572 |
hypothetical protein |
95 |
93 |
96 |
|
|
AGC31573 |
hypothetical protein |
99 |
96 |
100 |
|
|
AGC31574 |
portal protein |
97 |
97 |
99 |
|
|
AGC31576 |
N-acetylmuramidase |
95 |
97 |
98 |
IPR008565; IPR018537 |
|
AGC31502 |
terminase subunit A |
98 |
98 |
100 |
IPR004921 |
|
AGC31503 |
hypothetical protein |
98 |
98 |
100 |
|
|
AGC31504 |
hypothetical protein |
80 |
91 |
85 |
|
|
AGC31505 |
hypothetical protein |
81 |
76 |
77 |
|
| AGC31506 | hypothetical protein | 87 | 74 | 80 | |
Table 2.
General features and genome accession numbers of the N4-like bacteriophages involved in the multilocus phylogenetic study
| Phage | Isolation source | Host | Accession number | Reference |
|---|---|---|---|---|
| EC-UPM |
Chicken fecal material |
E. coli |
KC206276 |
This study |
| KBNP21 |
Chicken farm |
E. coli |
JX415535 |
[24] |
| vB_EcoP_G7C |
Horse fecal material |
E. coli |
HQ259105 |
[25] |
| IME11 |
Hospital sewage |
E. coli |
JX880034 |
[6] |
| N4 |
Sewer |
E. coli |
EF056008 |
[11] |
| EamP-S6 |
Fruit production environment |
Erwinia amylovora |
NC_019514 |
[17] |
| DSS3P2 |
Seawater sample |
Silicibacter pomenyi |
Nc_012697 |
[7] |
| EE36P1 |
Seawater sample |
Sulfitobacter sp. |
NC_012696 |
[7] |
| LIT1 |
Pond |
Pseudomonas aeruginosa |
NC_013692 |
[9] |
| LUZ7 | Hospital waste sample | Pseudomonas aeruginosa | NC_013691 | [9] |
Figure 5.

Elucidation of the evolutionary relationship of various N4-like bacteriophages based on multilocus and single-gene phylogenetic analyses. A total of seven orthologous groups were used for the multilocus phylogenetic analysis. Four out of the seven orthologous groups contained functional assignment and were subsequently chosen for the construction of an individual single-gene phylogenetic tree.
Conclusions
We reported for the first time the genome sequence of a N4-like bacteriophage which is capable of infecting the avian pathogenic Escherichia coli O78:K80. The analysis of its genome identified a novel genomic region which may be responsible for its host specificity. This study also identified the orthologous groups of protein shared by all sequenced N4-like phages and showed that the evolutionary relationship of N4-like bacteriophages correlated with the pattern of host specificity.
Material and methods
Genome sequencing and annotation of bacteriophage EC1-UPM
Methods for propagation, purification and genomic DNA extraction of bacteriophage EC1-UPM followed those described by Lau et al. [4]. The purified DNA was sequenced on the Illumina Hiseq 2000 (100-bp paired-end reads). The reads were assembled de novo using Velvet v1.1.07. Subsequent scaffolding and gap filling were performed to obtain a complete genome of bacteriophage EC1-UPM. Protein coding sequences were predicted using combined gene model from GeneMarkS and Glimmer3 [26]. The predicted proteins were annotated by BLAST against the NCBI non-redundant database. Additional annotation was subsequently performed using Blast2Go and InterProscan [27,28]. Possible presence of tRNA in the genome was determined using tRNAscan-SE v1.23.
Genome visualization of bacteriophage EC1-UPM and comparison with other closely related bacteriophage genomes
The annotated complete genome of bacteriophage EC1-UPM was visualized using CLC Genomics Workbench 6.0 (CLC Bio, Denmark). To identify the closely related bacteriophage, BLASTN was performed on the complete genome of bacteriophage EC1-UPM against the NCBI non-redundant database. Blast Ring Image Generator [29] was used to provide the genomic comparison of bacteriophage EC1-UPM against its closely related bacteriophage (40% identity cut-off).
In-silico analysis of bacteriophage EC1-UPM tailspike and tail fiber proteins
Additional annotation of the tail fiber protein was performed using the HHpred interactive server [30]. Additionally, the computation of various physical and chemical parameters of the tail fiber protein was performed using ProtParam [31].
Classification of bacteriophage EC1-UPM holin
Protein sequence of the putative holin was subjected to a transmembrane helices prediction using the TMHMM Server v. 2.0. The text output from the prediction was subsequently piped into to TMRPres2D [32] to create uniform, two-dimensional high analysis graphical images of the putative transmembrane protein.
Evolutionary relationship of the N4-like bacteriophages
The currently available genomes of N4-like bacteriophage were retrieved from NCBI in Genbank format. PanOCT [33] was used to identify the genes that are conserved across the N4-like bacteriophages (40% or 70% identity cut-off, at least 70% protein length overlap). Each orthologs cluster was aligned using MUSCLE [34]. Then, the individual alignment was trimmed with TrimAl using the “strictplus” parameter which is optimized for NeighborJoining tree construction [35] and concatenated for phylogenetic analysis using MEGA. A phylogenetic tree was constructed using the NeighborJoining method with 1000 bootstrap repetitions.
Nucleotide sequence accession number
The Genbank accession number for the complete genome sequence of bacteriophage EC1-UPM is KC206276.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
HMG carried out genetic analysis, drafted the manuscript. CCS designed the whole process and helped editing the manuscript. SGHT participated in the experiments. ARO and YWH provided the facilities and reviewed the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Blast2Go and InterProscan annotation of all predicted proteins in bacteriophage EC1-UPM.
HHpred annotation of the tail fiber protein of bacteriophage EC1-UPM.
Contributor Information
Han Ming Gan, Email: gan.han.ming@monash.edu.
Chin Chin Sieo, Email: sieo@upm.edu.my.
Shirley Gee Hoon Tang, Email: psjt85@hotmail.com.
Abdul Rahman Omar, Email: aro@upm.edu.my.
Yin Wan Ho, Email: ywho@upm.edu.my.
Acknowledgements
This work was supported by an Institute of Bioscience, Higher Institution Centre of Excellence (IBS HICoE) grant from the Ministry of Higher Education, Government of Malaysia. We wish to thank G. L. Lau for her guidance in preparation of the genomic material and Y. L. Tay (Bioeasy SB) for bioinformatics assistance in the construction of the multilocus phylogenetic tree.
References
- Gibbs PS, Petermann SR, Wooley RE. Comparison of several challenge models for studies in avian colibacillosis. Avian Dis. 2004;10:751–758. doi: 10.1637/7176-030404R. [DOI] [PubMed] [Google Scholar]
- Lutful Kabir SM. Avian colibacillosis and salmonellosis: a closer look at epidemiology, pathogenesis, diagnosis, control and public health concerns. Int J Environ Res Public Health. 2010;10:89–114. doi: 10.3390/ijerph7010089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casewell M, Friis C, Marco E, McMullin P, Phillips I. The European ban on growth-promoting antibiotics and emerging consequences for human and animal health. J Antimicrob Chemother. 2003;10:159–161. doi: 10.1093/jac/dkg313. [DOI] [PubMed] [Google Scholar]
- Lau GL, Sieo CC, Tan WS, Hair-Bejo M, Jalila A, Ho YW. Efficacy of a bacteriophage isolated from chickens as a therapeutic agent for colibacillosis in broiler chickens. Poult Sci. 2010;10:2589–2596. doi: 10.3382/ps.2010-00904. [DOI] [PubMed] [Google Scholar]
- Lau GL, Sieo CC, Tan WS, Ho YW. Characteristics of a phage effective for colibacillosis control in poultry. J Sci Food Agric. 2012;10:2657–2663. doi: 10.1002/jsfa.5683. [DOI] [PubMed] [Google Scholar]
- Fan H, Fan H, An X, Huang Y, Zhang Z, Mi Z, Tong Y. Complete genome sequence of IME11, a new N4-like bacteriophage. J Virol. 2012;10:13861. doi: 10.1128/JVI.02684-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Wang K, Jiao N, Chen F. Genome sequences of two novel phages infecting marine roseobacters. Environ Microbiol. 2009;10:2055–2064. doi: 10.1111/j.1462-2920.2009.01927.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Born Y, Fieseler L, Marazzi J, Lurz R, Duffy B, Loessner MJ. Novel virulent and broad-host-range Erwinia amylovora bacteriophages reveal a high degree of mosaicism and a relationship to Enterobacteriaceae phages. Appl Environ Microbiol. 2011;10:5945–5954. doi: 10.1128/AEM.03022-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceyssens PJ, Brabban A, Rogge L, Lewis MS, Pickard D, Goulding D, Dougan G, Noben JP, Kropinski A, Kutter E, Lavigne R. Molecular and physiological analysis of three Pseudomonas aeruginosa phages belonging to the “N4-like viruses”. Virology. 2010;10:26–30. doi: 10.1016/j.virol.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazmierczak KM, Rothman-Denes LB. In: The bacteriophages. Calendar R, editor. New York, NY: Oxford University Press; 2006. Bacteriophage N4; pp. 302–314. [Google Scholar]
- Choi KH, McPartland J, Kaganman I, Bowman VD, Rothman-Denes LB, Rossmann MG. Insight into DNA and protein transport in double-stranded DNA viruses: the structure of bacteriophage N4. J Mol Biol. 2008;10:726–736. doi: 10.1016/j.jmb.2008.02.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailly-Bechet M, Vergassola M, Rocha E. Causes for the intriguing presence of tRNAs in phages. Genome Res. 2007;10:1486–1495. doi: 10.1101/gr.6649807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss SB, Hsu WT, Foft JW, Schernerg NH. Transfer RNA coded by the T4 bacteriophage genome. Proc Natl Acad Sci USA. 1968;10:114–121. doi: 10.1073/pnas.61.1.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiman PG, Molineux IJ. Evolution of a new enzyme activity from the same motif fold. Mol Microbiol. 2008;10:287–290. doi: 10.1111/j.1365-2958.2008.06241.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander GC, Tang L, Casjens SR, Gilcrease EB, Prevelige P, Poliakov A, Potter CS, Carragher B, Johnson JE. The structure of an infectious P22 virion shows the signal for headful DNA packaging. Science. 2006;10:1791–1795. doi: 10.1126/science.1127981. [DOI] [PubMed] [Google Scholar]
- Leiman PG, Battisti AJ, Bowman VD, Stummeyer K, Muhlenhoff M, Gerardy-Schahn R, Scholl D, Molineux IJ. The structures of bacteriophages K1E and K1-5 explain processive degradation of polysaccharide capsules and evolution of new host specificities. J Mol Biol. 2007;10:836–849. doi: 10.1016/j.jmb.2007.05.083. [DOI] [PubMed] [Google Scholar]
- Nguyen HA, Tomita T, Hirota M, Kaneko J, Hayashi T, Kamio Y. DNA Inversion in the tail fiber gene alters the host range specificity of carotovoricin Er, a phage-tail-like bacteriocin of phytopathogenic Erwinia carotovora subsp.carotovora Er. J Bacteriol. 2001;10:6274–6281. doi: 10.1128/JB.183.21.6274-6281.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnaitman CA. In: Molecular medical microbiology. Volume 1. Sussman M, editor. New York: Academic press; 2001. The genetics and biosynthesis of lipopolysaccharides; pp. 93–136. [Google Scholar]
- Yoder MD, Keen NT, Jurnak F. New domain motif: the structure of pectate lyase C, a secreted plant virulence factor. Science. 1993;10:1503–1507. doi: 10.1126/science.8502994. [DOI] [PubMed] [Google Scholar]
- Scholl D, Rogers S, Adhya S, Merril CR. Bacteriophage K1-5 encodes two different tail fiber proteins, allowing it to infect and replicate on both K1 and K5 strains of Escherichia coli. J Virol. 2001;10:2509–2515. doi: 10.1128/JVI.75.6.2509-2515.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundling A, Manson MD, Young R. Holins kill without warning. Proc Natl Acad Sci USA. 2001;10:9348–9352. doi: 10.1073/pnas.151247598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang IN, Smith DL, Young R. Holins: the protein clocks of bacteriophage infections. Annu Rev Microbiol. 2000;10:799–825. doi: 10.1146/annurev.micro.54.1.799. [DOI] [PubMed] [Google Scholar]
- Koonin EV, Senkevich TG, Dolja VV. The ancient virus world and evolution of cells. Biol Direct. 2006;10:29. doi: 10.1186/1745-6150-1-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nho S-W, Ha M-A, Kim K-S, Kim T-H, Jang H-B, Cha I-S, Park S-B, Kim Y-K, Jung T-S. Complete genome sequence of the bacteriophages ECBP1 and ECBP2 isolated from two different Escherichia coli strains. J Virol. 2012;10:12439–12440. doi: 10.1128/JVI.02141-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulikov E, Kropinski AM, Golomidova A, Lingohr E, Govorun V, Serebryakova M, Prokhorov N, Letarova M, Manykin A, Strotskaya A, Letarov A. Isolation and characterization of a novel indigenous intestinal N4-related coliphage vB_EcoP_G7C. Virology. 2012;10:93–99. doi: 10.1016/j.virol.2012.01.027. [DOI] [PubMed] [Google Scholar]
- Besemer J, Borodovsky M. GeneMark: web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res. 2005;10:W451–W454. doi: 10.1093/nar/gki487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A, Gotz S. Blast2GO: a comprehensive suite for functional analysis in plant genomics. Int J Plant Genomics. 2008;10:619832. doi: 10.1155/2008/619832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zdobnov EM, Apweiler R. InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001;10:847–848. doi: 10.1093/bioinformatics/17.9.847. [DOI] [PubMed] [Google Scholar]
- Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA. BLAST ring image generator (BRIG): simple prokaryote genome comparisons. BMC Genomics. 2011;10:1471–2164. doi: 10.1186/1471-2164-12-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;10:W244–W248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, Appel RD, Hochstrasser DF. Protein identification and analysis tools in the ExPASy server. Methods Mol Biol. 1999;10:531–552. doi: 10.1385/1-59259-584-7:531. [DOI] [PubMed] [Google Scholar]
- Spyropoulos IC, Liakopoulos TD, Bagos PG, Hamodrakas SJ. TMRPres2D: high quality visual representation of transmembrane protein models. Bioinformatics. 2004;10:3258–3260. doi: 10.1093/bioinformatics/bth358. [DOI] [PubMed] [Google Scholar]
- Fouts DE, Brinkac L, Beck E, Inman J, Sutton G. PanOCT: automated clustering of orthologs using conserved gene neighborhood for pan-genomic analysis of bacterial strains and closely related species. Nucleic Acids Res. 2012;10:16. doi: 10.1093/nar/gkr1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;10:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T. TrimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;10:1972–1973. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Blast2Go and InterProscan annotation of all predicted proteins in bacteriophage EC1-UPM.
HHpred annotation of the tail fiber protein of bacteriophage EC1-UPM.