

Background: It has been proposed that stepwise folding of the extracellular (passenger) domains of bacterial autotransporter proteins drives their secretion.
Results: Mutations in N-terminal or medial segments of a passenger domain that severely impair folding only moderately reduce secretion efficiency.
Conclusion: Stepwise folding only partially accounts for the energetics of autotransporter secretion.
Significance: Autotransporter secretion may involve the use of a novel energy source.
Keywords: Escherichia coli, Membrane Proteins, Protein Folding, Protein Translocation, Virulence Factors, Autotransporter
Abstract
Autotransporters are a superfamily of virulence proteins produced by Gram-negative bacteria. They consist of an N-terminal β-helical domain (“passenger domain”) that is secreted into the extracellular space and a C-terminal β-barrel domain (“β-domain”) that anchors the protein to the outer membrane. Because the periplasm lacks ATP, vectorial folding of the passenger domain in a C-to-N-terminal direction has been proposed to drive the secretion reaction. Consistent with this hypothesis, mutations that disrupt the folding of the C terminus of the passenger domain of the Escherichia coli O157:H7 autotransporter EspP have been shown to cause strong secretion defects. Here, we show that point mutations introduced at specific locations near the middle or N terminus of the EspP β-helix that perturb folding also impair secretion, but to a lesser degree. Surprisingly, we found that even multiple mutations that potentially abolish the stability of several consecutive rungs of the β-helix only moderately reduce secretion efficiency. Although these results provide evidence that the free energy derived from passenger domain folding contributes to secretion efficiency, they also suggest that a significant fraction of the energy required for secretion is derived from another source.
Introduction
Autotransporters are a large superfamily of proteins produced by Gram-negative bacteria that consist of two domains, an N-terminal extracellular domain (“passenger domain”) that mediates a virulence function and a C-terminal β-barrel domain (“β-domain”) that anchors the protein to the outer membrane (OM)2 (1). Passenger domains are variable in length but often exceed 100 kDa. Although passenger domains are also highly heterogeneous in sequence, they generally form a parallel right-handed β-helical structure (2–5). Some passenger domains also contain small globular domains that protrude from the middle of the β-helical core or that are tethered to the N terminus. Following their translocation across the OM, passenger domains either remain covalently attached to the β-domain or are released into the environment by a proteolytic cleavage. β-Domains are ∼30 kDa in length and fold into nearly superimposable 12-stranded β-barrels (6–8). After autotransporters are translocated across the inner membrane through the Sec machinery, they interact with periplasmic chaperones, which presumably maintain them in an assembly-competent conformation (9–11). Subsequently, they interact with components of the Bam complex, a hetero-oligomer that has been shown to catalyze the integration of β-barrel proteins into the OM (10–15).
Although it is now clear that passenger domains are translocated across the OM in a C-to-N-terminal direction, the mechanism of translocation has been hotly debated. The term “autotransporter” is based on an early proposal that passenger domains are secreted through the covalently linked β-domain, which functions as an autonomous translocation channel (16). Translocation through the β-domain would presumably involve the formation of a hairpin, followed by the progressive sliding of medial and N-terminal segments past a static C-terminal strand. At first glance, this proposal seems to be supported by crystal structures showing that the two domains are connected by an α-helical linker that traverses the β-domain pore (6–8). However, several lines of evidence have strongly suggested that the translocation mechanism is more complicated. The same structural studies have shown that the β-domain is ∼10 Å in diameter and is therefore only wide enough to accommodate an extended hairpin that lacks secondary and tertiary structure. Nevertheless, some passenger domains acquire limited tertiary structure in the periplasm, and at least some small folded polypeptides are secreted efficiently by the autotransporter pathway (17, 18). In addition, the linker appears to acquire an α-helical conformation inside the β-domain pore before translocation is complete (19). Finally, cross-linking studies have shown that the passenger domain interacts with one of the components of the Bam complex (BamA) when it traverses the OM (10). This and other recent observations suggest that the Bam complex facilitates both the integration of the β-domain into the OM and the secretion of the passenger domain, possibly in a concerted reaction.
The source of energy used to drive passenger domain translocation is also unknown. Protein translocation reactions generally require ATP hydrolysis and/or energy derived from a membrane potential. The periplasm lacks ATP, however, and to date, no protein has been identified that drives passenger domain translocation by harnessing the membrane potential across the inner membrane. To resolve this paradox, it has been proposed that the stepwise folding of the passenger domain in the extracellular space drives translocation by a Brownian ratchet-type mechanism (20). Consistent with this hypothesis, a conserved ∼20-kDa C-terminal segment of several different passenger domains has been shown to form a protease-resistant structure that, in principle, might nucleate subsequent folding steps (5, 19, 21). Furthermore, mutations that perturb the folding of the C-terminal segment of the passenger domain of the Escherichia coli O157:H7 autotransporter EspP strongly impair secretion (19). Some of the available evidence, however, appears to be inconsistent with the vectorial folding model. One study that examined the purified passenger domain of the Bordetella pertussis autotransporter pertactin suggested that the β-helix folds in a concerted process involving the entire protein rather than in a stepwise fashion (22). Another study showed that although mutations that destabilize the C terminus of the passenger domain of the E. coli Pet protein hinder secretion, mutations that destabilize its N-terminal globular domain increase secretion efficiency (23). As noted above, there is also evidence that polypeptides that fold prior to (rather than after) their translocation across the OM can be secreted efficiently (17). Finally, experimental analysis has revealed that ∼5,000 ATP molecules are required to move a protein through the bacterial Sec machinery in the absence of a protonmotive force (24) and that even the less costly transport of proteins across the chloroplast envelope requires ∼650 ATP molecules (25). Given that even the lower value represents ΔG = 30,000 kJ/mol, whereas the ΔG associated with protein folding is typically ∼20–60 kJ/mol (26), it seems likely that the folding of the passenger domain does not fully account for the energetics of secretion.
In this study, we used EspP as a model protein to further explore the role of passenger domain folding in driving the translocation reaction. EspP is a member of the SPATE (serine protease autotransporters of Enterobacteriaceae) family of autotransporters. Following their translocation across the OM, the SPATE passenger domains are released in an intrabarrel autoproteolytic cleavage reaction (27). Like those of other members of the SPATE family, the EspP passenger domain consists of a small globular domain (residues 56–313) and a long β-helix (residues 313–1023) that is capped at both ends by β-hairpins (see Fig. 1A) (28). The core of the β-helix consists mostly of bulky aliphatic and aromatic side chains. We found that mutations introduced into the N-terminal and medial segments of the EspP β-helix that perturb folding also impair secretion, but more moderately than mutations that perturb the folding of the C terminus. Our results show that the progressive folding of the β-helix maximizes secretion efficiency but is not absolutely essential for secretion. The results therefore raise the intriguing possibility that energy derived from a source other than passenger domain folding plays a significant role in driving the translocation reaction.
FIGURE 1.

Structural properties of EspP. A, EspP is composed of a signal peptide (SP; residues 1–55), a passenger domain (residues 56–1023), and a β-domain (residues 1024–1300). The passenger domain is divided into a major globular subdomain (residues 56–313) and a long β-helix (residues 314–1023). The passenger domain and β-domain are covalently linked in a precursor form of the protein designated proEspP prior to the completion of passenger domain translocation. The location of a peptide insertion encoding a TEV protease recognition site that was used in this study is shown. B, the locations of the 15 hydrophobic residues that were converted to alanine in our initial screen are depicted on the crystal structure of the EspP passenger domain (28).
EXPERIMENTAL PROCEDURES
Bacterial Strains, Reagents, and Growth Conditions
E. coli strain AD202 (MC4100 ompT::kan) (29) was used in all experiments. Cultures were grown at 37 °C in LB or M9 medium containing 0.2% glycerol and all the l-amino acids except methionine and cysteine. Plasmids were maintained by the addition of ampicillin (100 μg/ml). Antisera against EspP C-terminal and N-terminal peptides and the purified EspP passenger domain have been described previously (30, 31). Purified recombinant tobacco etch virus (TEV) protease (hyperactive S219V mutant) was obtained from Susan Buchanan.
Plasmid Construction
A plasmid that harbors wild-type espP under the control of the trc promoter (pRLS5) has been described previously (30). Structurally important hydrophobic residues in the EspP passenger domain were predicted based on sequence conservation and an analysis of the EspP passenger domain crystal structure (28) using PyMOL Molecular Graphics System software (Schrödinger LLC). Mutations were introduced into pRLS5 using a QuikChange mutagenesis kit (Stratagene). A TEV protease recognition site was inserted after EspP residue 214 by PCR using oligonucleotides 5′-CGACAATCTCGAGAATCTGTATTTTCAGGGACGCATAACTCCATTTAATGAGGTTTCTTA and 5′-ATACAGATTCTCGAGATTTTTTTTAACGGATACCACACCAGAGCCGGCTCTGAATGCG and pRLS5 as a template. The PCR product was digested with XhoI and religated to create pWK15. An identical TEV site was also introduced at the same position of the EspP double mutant F349A/I355A to create pWK16.
Pulse-Chase Labeling and Quantitative Analysis
AD202 cells transformed with a plasmid encoding wild-type or mutant EspP were grown overnight in M9 medium. Overnight cultures were washed and diluted into fresh medium at OD550 = 0.02. When the cultures reached OD550 = 0.2, espP expression was induced by the addition of 10 μm isopropyl β-d-thiogalactopyranoside (IPTG). After 30 min, cells were pulse-labeled for 30 s with 30 μCi/ml Tran35S-label (MP Biomedical), and a chase was conducted by adding 1 mm methionine and cysteine. At various time points, aliquots of radiolabeled cultures were withdrawn and pipetted into tubes containing an equal volume of preformed ice. In most experiments, samples were divided in half, and one half was incubated on ice for 20 min with 200 μg/ml proteinase K (PK). PK digestion was then stopped by the addition of 2 mm PMSF. In some experiments, samples were incubated on ice for 2 h with the hyperactive TEV protease (2 μg/ml) after the addition of 1 mm DTT. To analyze the structural integrity of passenger domain molecules released into the culture medium, pulse-chase labeling was performed as described above, and radiolabeled cells were removed by centrifugation (3,000 × g, 4 °C, 10 min). The supernatant was then treated with PK as described above. In all experiments, proteins were precipitated by the addition of cold 10% TCA. Immunoprecipitations were then performed as described previously (32), and proteins were resolved by SDS-PAGE on 8–16% mini gels (Invitrogen). Radioactive proteins were visualized using a Fujifilm FLA-9000 phosphoimager. For quantitative analysis, cell surface exposure was defined as 1 − (pro-EspP(+PK)/(pro-EspP(−PK) + β-domain(−PK))). Passenger domain cleavage was defined as β-domain(−PK)/(β-domain(−PK) + pro-EspP(−PK)). The signal from each band was normalized to account for differences in the number of radioactive amino acids. The data were graphed using OriginPro 8.5 software (OriginLab Corp.).
Analysis of EspP Passenger Domain Stability at Steady State
AD202 cells transformed with an appropriate plasmid were grown overnight in LB medium. Overnight cultures were washed, diluted, and grown in fresh medium to OD550 ∼ 0.4. espP expression was then induced by the addition of 10 μm IPTG. After 30 min, cultures were chilled on ice for 15 min. Cells were removed by centrifugation (twice at 3,000 × g for 10 min at 4 °C) and discarded. Equal fractions of each supernatant were then subjected to protease digestion or left untreated. PK digestions were performed as described above. In some experiments, samples were incubated with trypsin or chymotrypsin at the indicated concentrations for 30 min at 0 °C. Following protease digestion, proteins were TCA-precipitated, resolved by SDS-PAGE, and transferred to nitrocellulose filters. Western blotting was conducted by incubating the filters with the anti-EspP passenger domain antiserum. Antibody-antigen complexes were detected using HRP-linked protein A (GE Healthcare) with a SuperSignal West Pico chemiluminescence kit (Thermo Scientific).
RESULTS
N-terminal Mutations in the EspP β-Helix Reduce the Structural Integrity of the Passenger Domain and Impede Translocation
Previous work has shown that the mutation of conserved large hydrophobic amino acids that reside near the C terminus of the EspP passenger domain (as well as the passenger domain of another SPATE protein) significantly reduces the efficiency of secretion (19, 33–35). Pulse-chase analysis showed that although the F963A and W990A mutations have no effect on the initiation of translocation (i.e. the exposure of the passenger domain on the cell surface), they both strongly delay the completion of the translocation reaction. In a typical experiment ∼80% of the wild-type passenger domains were completely secreted within 2 min, but only ∼15% of the mutant passenger domains were secreted within this period. Furthermore, secretion of ∼80% of the mutant passenger domains was observed only after 10 min (19). The mutations were also shown to impair the refolding of the purified passenger domain in vitro and to abolish the formation of an ∼17-kDa PK-resistant C-terminal fragment. Presumably, the mutations hamper folding by eliminating a key side chain that is required to stabilize the formation of a hydrophobic core. Indeed, the elimination of buried hydrophobic side chains has been shown to strongly impair the folding of an unrelated β-helical protein (36, 37). In any case, the observation that mutations that disrupt the folding of the C terminus of the passenger domain drastically reduce secretion efficiency strongly suggests that the folding of at least one segment of the polypeptide plays an important role in driving the translocation reaction.
To determine whether these results could be generalized to support the idea that passenger domain secretion is driven by a stepwise folding process, we introduced similar point mutations into medial and N-terminal segments of the EspP β-helix. We individually converted 15 large hydrophobic residues located in the N-terminal 15 rungs of the β-helix to alanine (Fig. 1B). Based on the crystal structure of the EspP passenger domain (28), the side chains of all of these residues (except isoleucine 348) point toward the interior of the β-helix. Furthermore, all of these residues are conserved in that a large hydrophobic residue is found at the equivalent position in most or all other SPATE proteins. To test the effect of these mutations on passenger domain secretion, AD202 cells transformed with a plasmid encoding either wild-type or mutant EspP under the control of the trc promoter were grown in minimal medium. Cells were subjected to pulse-chase labeling after the addition of IPTG, and PK was added to half of each sample. EspP polypeptides were then immunoprecipitated using antisera directed against both the passenger domain and a peptide derived from the C terminus of the β-domain. In these experiments, the exposure of the passenger domain on the cell surface was monitored by measuring the fraction of proEspP (the form of the protein in which passenger and β-domains are covalently linked) that was sensitive to PK digestion. Because cleavage of the passenger domain is strictly dependent on the secretion of the entire passenger domain (10), the completion of the translocation reaction was assessed by monitoring the cleavage of proEspP into discrete passenger and β-domain fragments. Finally, passenger domain folding was assessed by measuring the level of a stable ∼80-kDa N-terminal fragment that is derived from PK treatment of the passenger domain when it is correctly folded (10).
One of the point mutations, F349A, appeared to moderately impair passenger domain secretion. Although the mutant passenger domain was exposed on the cell surface as rapidly as its wild-type counterpart, the cleavage of the pro form of the mutant into discrete passenger and β-domain fragments (which are recognized by anti-passenger domain and anti-C-terminal peptide antisera, respectively) was noticeably delayed (Fig. 2, A, B, F and G). Because the mutation is located far away from the passenger domain cleavage site and because only the last few residues of the passenger domain appear to influence the cleavage reaction per se (27, 38), the defect in proteolytic maturation is almost certainly an indirect effect of a delay in the completion of translocation. Consistent with this conclusion, the F349A mutation had no effect on the folding of the C terminus of the passenger domain. In the presence of PK, an ∼47-kDa C-terminal fragment that contains the ∼30-kDa β-domain plus a stable ∼17-kDa segment of the passenger domain was detected in cells that produced both wild-type and mutant EspP (Fig. 2A, lanes 6–10 and 16–20). This fragment results from the partial digestion of passenger domains that are exposed but not yet released from the cell surface and is observed only when the ∼17-kDa C-terminal segment folds correctly (19). In contrast, the F349A mutation moderately reduced the accumulation of a stable ∼80-kDa N-terminal fragment and therefore moderately impaired the folding of a more proximal segment of the passenger domain (Fig. 2B, lanes 8–10 and 18–20). Taken together, the results show that this mutation had a much more modest effect on both secretion and folding than similar alanine substitutions introduced into the C terminus of the EspP passenger domain (see Ref. 19). Perhaps more surprisingly, none of the other 15 point mutations significantly affected EspP biogenesis, and none of the mutations that were tested (L418A, F646A, L641A, and L639A) produced folding defects (data not shown). In addition, the triple mutation L639A/L641A/F646A did not affect either passenger domain secretion or folding. These results suggest that most of the EspP β-helix is much more refractory to structural distortions than the C terminus.
FIGURE 2.

Mutations at the N terminus of the β-helix impede EspP biogenesis and passenger domain folding. A and B, AD202 cells transformed with a plasmid encoding wild-type EspP under the control of the trc promoter (pRLS5) or a derivative encoding the F349A mutant were subjected to pulse-chase labeling following the addition of IPTG. Half of each culture sample was treated with PK, and immunoprecipitations were conducted using antiserum generated against an EspP C-terminal peptide (A) or the EspP passenger domain (B). C, close-up view of the N terminus of the β-helix showing Phe-349 and other hydrophobic residues that were mutagenized to create double and triple mutations. The illustration was generated using PyMOL. D and E, AD202 cells transformed with a pRLS5 derivative encoding the specified EspP mutant were subjected to pulse-chase labeling following the addition of IPTG. Half of each culture sample was treated with PK, and immunoprecipitations were conducted using antisera generated against an EspP C-terminal peptide (D) or the EspP passenger domain (E). F and G, the fraction of the passenger domain that was surface-exposed and released by proteolytic cleavage at each time point in A and D is shown.
Although most of the point mutations did not affect EspP biogenesis, we hypothesized that combining the F349A mutation with other mutations that eliminate large hydrophobic side chains would further destabilize the hydrophobic core and lead to greater defects in EspP assembly and passenger domain folding. To test this idea, we created double and triple mutants in which residues in the same rung of the β-helix were converted to alanine (F349A/I355A and L347A/I348A/F349A) (Fig. 2C). We also created two double mutants in which residues in adjacent rungs were converted to alanine (L332A/F349A and F349A/F370A) (Fig. 2C). In addition to potentially destabilizing the two rungs independently, the latter mutation appears to abrogate a stacking interaction between the two rungs. AD202 cells transformed with a plasmid encoding one of the EspP mutants were subjected to pulse-chase labeling, and half of the cells were treated with PK as described above. Immunoprecipitations with the anti-EspP C-terminal peptide antiserum showed that unlike the F349A mutation, several of the double and triple mutations noticeably delayed the cell surface exposure of the passenger domain (Fig. 2, D and F). The immunoprecipitations also showed that the double and triple mutations slightly exacerbated the passenger domain cleavage defect caused by the single mutation and concomitantly prolonged the lifetime of the ∼47-kDa C-terminal fragment (Fig. 2, D and G). Given that these mutations tended to slightly impair the initiation of translocation, it is unclear that they exerted a significantly greater effect on the completion of translocation than the F349A mutation alone. They did, however, exert a much more profound effect on passenger domain stability. Immunoprecipitations with the anti-passenger domain antiserum showed that the mutations greatly reduced (and, in the case of the triple mutation, nearly eliminated) the accumulation of the ∼80-kDa PK-resistant passenger domain fragment (Fig. 2E). These results demonstrate that the removal of large hydrophobic side chains can strongly impair the folding of at least one segment of the passenger domain while producing only moderate effects on passenger domain secretion.
Further analysis of one of the double mutants (L332A/F349A) provided evidence that the mutations exerted a local effect on passenger domain folding. In one set of experiments, radiolabeled cells that produced either wild-type EspP or the L332A/F349A mutant were pelleted, and half of the culture medium was treated with PK. As expected, immunoprecipitations using the anti-passenger domain antiserum showed that PK converted most of the wild-type passenger domain that was released into the medium into an ∼80-kDa stable fragment (Fig. 3A, lanes 8–10). In contrast, PK treatment of the mutant passenger domain that was released into the medium yielded a much smaller amount of the ∼80-kDa fragment. Instead, the protease converted a significant fraction of the mutant protein into a novel ∼50-kDa fragment (Fig. 3A, lanes 19–20). Presumably this fragment was not clearly detected in the experiments described above because the cleaved passenger domain was not as highly enriched in samples that included both the cell and culture medium fractions. In any case, the size of the fragment suggests that it results from cleavage of the ∼80-kDa fragment near the site of the mutations (which would release an ∼30-kDa fragment). To confirm that the ∼50-kDa polypeptide represented an internal fragment of the passenger domain, immunoprecipitations were performed using an antiserum raised against an EspP N-terminal peptide. As predicted, the anti-peptide antiserum recognized the ∼80-kDa fragment, but not the ∼50-kDa fragment (Fig. 3B, lanes 9–10 and 19–20). These results imply that the double mutation perturbs the folding of the β-helix around residues ∼330–350, but not segments of the passenger domains that are closer to the C terminus.
FIGURE 3.

EspP double mutation L332A/F349A disrupts the structure of the passenger domain locally. AD202 cells transformed with pRLS5 or a derivative encoding the L332A/F349A mutant were subjected to pulse-chase labeling following the addition of IPTG. Radiolabeled cells were removed by centrifugation, and half of the culture medium was treated with PK. Immunoprecipitations were then performed using antisera generated against the EspP passenger domain (A) or an EspP N-terminal peptide (B).
Finally, we wished to confirm that the passenger domain cleavage defects associated with the mutations were secondary consequences of defects in the completion of passenger domain translocation. To this end, we introduced a TEV protease recognition site into an exposed loop in the N-terminal globular domain (at Asn-214) of wild-type EspP and one of the double mutants (F349A/I355A) to create plasmids pWK15 and pWK16 (see Fig. 1A). We expected that if the translocation of the polypeptide segment N-terminal to the mutations was slowed, then the accessibility of the pro form of the mutant protein to TEV digestion should likewise be delayed. AD202 cells transformed with pWK15 or pWK16 were subjected to pulse-chase labeling and TEV digestion, and immunoprecipitations were performed using the anti-passenger domain antiserum. As expected, the appearance of an ∼120-kDa TEV fragment (a fragment that is ∼15 kDa smaller than proEspP) preceded the cleavage of the wild-type passenger domain (Fig. 4, lanes 1–5). This observation is consistent with previous results showing that the N terminus of the passenger domain is exposed on the cell surface prior to proteolytic maturation (10). An ∼90-kDa TEV fragment that corresponds to the bulk of the passenger domain was also observed around the time that the free passenger domain appeared, but it disappeared at later time points presumably because the TEV site became inaccessible. As predicted, the delay in the cleavage of the mutant passenger domain correlated with a later appearance of both the ∼120- and ∼90-kDa TEV fragments (Fig. 4, lanes 6–10). These results demonstrate that the mutations affect the exposure of the globular domain on the cell surface and rule out the possibility that they affect the passenger domain cleavage reaction per se.
FIGURE 4.
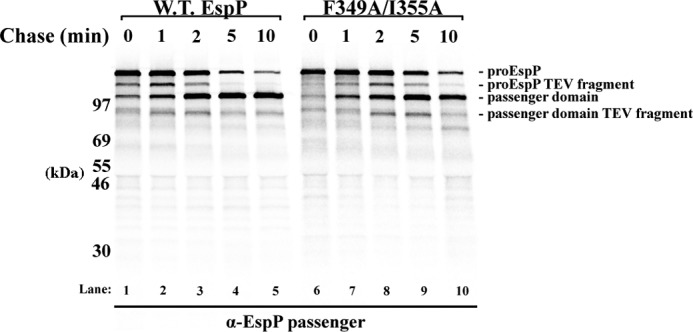
EspP mutation F349A/I355A delays the surface exposure of the globular domain. AD202 cells transformed with a plasmid encoding wild-type EspP or the F349A/I355A mutant with a TEV insertion at residue 214 (pWK15 or pWK16) were subjected to pulse-chase labeling after the addition of IPTG, and culture samples were treated with TEV protease. Immunoprecipitations were then performed using the anti-EspP passenger domain antiserum.
Mutations That Abolish Stable Folding of the EspP β-Helix Only Partially Impair Passenger Domain Translocation
The results described above indicate that the elimination of buried large hydrophobic side chains around residue 350 creates a local folding defect that reduces the efficiency with which the adjacent N-terminal globular domain is secreted. The observation that none of the mutations completely abolished the formation of a stable ∼80-kDa N-terminal PK fragment suggested, however, that they perturbed the stability of the β-helix only moderately. Thus, it is conceivable that although many of the mutant passenger domain molecules do not fold perfectly into compact structures, they often fold sufficiently well to drive the completion of the translocation reaction. It is also possible that the mutations exert differential effects on the folding of individual passenger domain molecules. In that case, the molecules that are not secreted may represent the population of molecules that are the most severely destabilized by the mutations. Furthermore, the finding that only alanine substitutions near the N terminus of the β-helix impaired protein folding in the aforementioned experiments raised the possibility that this segment has unique properties and that the folding of much of the passenger domain cannot be affected by the removal of hydrophobic residues.
To address these issues, we mutagenized up to seven buried hydrophobic residues located near the N terminus and the middle of the EspP β-helix. In one mutant (mutant S), Val-317, Met-319, Leu-332, Phe-349, Ile-355, Phe-370, and Tyr-376 were replaced with alanine. Because all of the side chains are located within ∼5 Å of Phe-349 (Fig. 5A), we predicted that the mutations would remove a large hydrophobic patch from a single region of the protein. In a second mutant (mutant Q), the tightly clustered Leu-546, Val-550, Met-565, Phe-634, and Val-654 residues were changed to alanine (Fig. 5B). The insertion of a peptide linker into a disordered loop that projects from this region of the β-helix has been previously shown to impair passenger domain folding (10). To examine the effect of the mutations on the stability of the secreted passenger domain, AD202 cells transformed with a plasmid encoding wild-type or mutant EspP were grown in LB medium, and the culture medium was incubated with PK. Western blots probed with the anti-passenger domain antiserum showed that the protease converted most of the wild-type passenger domain to an ∼80-kDa fragment but completely degraded the mutant passenger domains (Fig. 5C). To confirm that the mutations severely destabilized the β-helix, we compared the susceptibility of the secreted wild-type and mutant passenger domains to digestion by two other proteases. In the presence of 10 μg/ml chymotrypsin, the wild-type passenger domain remained intact, but both mutant polypeptides were completely degraded (Fig. 5D). Even at a much higher concentration (100 μg/ml), chymotrypsin digested only about half of the wild-type passenger domain (Fig. 5E). Because chymotrypsin selectively cleaves peptide bonds formed by large hydrophobic residues, the results strongly suggest that the mutations increase the accessibility of amino acids that are normally situated inside the hydrophobic core of the passenger domain. In addition, most of the mutant passenger domain molecules were degraded by low concentrations of trypsin (1–2 μg/ml), whereas the wild-type protein was degraded only by considerably higher concentrations of the protease (Fig. 5, F and G). Taken together, the results provide strong evidence that the mutations abolish the folding of at least the segments of the passenger domain into which they were introduced into a native state.
FIGURE 5.

Multiple mutations strongly destabilize the EspP β-helix. A and B, close-up view of two regions of the β-helix showing the residues that were converted to alanine to create mutants S and Q. The illustration was generated using PyMOL. C, AD202 cells transformed with pRLS5 or a plasmid encoding mutant S or Q were grown in LB medium, and expression of the plasmid-borne gene was induced by the addition of IPTG. Cells were removed by centrifugation, and half of the culture medium was treated with PK. Western blot analysis was then conducted using the anti-EspP passenger domain antiserum. D–G, the experiment described for C was repeated except that chymotrypsin or trypsin was added to equal portions of the culture medium at the indicated concentrations. In E and G, only the culture medium of cells transformed with pRLS5 was analyzed.
We next examined the effect of the mutations on EspP assembly by subjecting cells transformed with a plasmid encoding either mutant S or mutant Q to pulse-chase analysis and PK treatment. Immunoprecipitations with the anti-EspP C-terminal peptide antiserum showed that the mutations did not significantly affect the exposure of the passenger domain on the cell surface (Fig. 6, A and Fig. C). A moderate reduction in the efficiency of passenger domain cleavage (presumably due to a defect in the completion of translocation) and a concomitant persistence of the ∼47-kDa C-terminal fragment were observed in cells that produced mutant S (Fig. 6A, lanes 1–10, and Fig. 6D). The magnitude of the effect was, however, similar to that produced by the L332/F349A double mutant (see Fig. 2E). Mutant Q produced an even milder secretion defect (Fig. 6A, lanes 11–20, and Fig. 6D). Interestingly, immunoprecipitations with the anti-passenger domain antiserum confirmed that the mutations abolished the formation of an ∼80-kDa PK-resistant passenger domain fragment (Fig. 6B). Taken together, these results show that even severe folding defects caused by the elimination of multiple buried hydrophobic residues from either N-terminal or medial segment of the β-helix only partially impair the completion of the secretion reaction.
FIGURE 6.

EspP mutants S and Q display only moderate passenger domain translocation defects. A and B, AD202 cells transformed with pRLS5 or a derivative encoding mutant S or Q were subjected to pulse-chase labeling following the addition of IPTG. Half of each culture sample was treated with PK, and immunoprecipitations were conducted using antisera generated against an EspP C-terminal peptide (A) or the EspP passenger domain (B). C and D, the fraction of the passenger domain that was surface-exposed and released by proteolytic cleavage at each time point in A is shown. For comparison, the results obtained in the experiment shown in Fig. 2 in which we analyzed the assembly of wild-type EspP are also plotted.
DISCUSSION
In this study, we obtained evidence that the folding of autotransporter passenger domains in the extracellular space plays only a modest role in driving their translocation across the OM. Based on the finding that the mutation of conserved hydrophobic residues located near the C terminus of the EspP passenger domain to alanine strongly disrupts both folding and the completion of translocation (19), we introduced similar mutations at the medial and N-terminal portions of the β-helix. We found, however, that most single alanine substitutions in this region did not significantly affect either the structural integrity of the passenger domain or the efficiency of translocation. The results suggest that many internal segments of the β-helix are stabilized by multiple redundant interactions. One mutation, F349A, slightly reduced the stability of the passenger domain and the efficiency of translocation, and multiple mutations in the same region of the β-helix led to progressively greater defects in both the structural integrity and secretion of the passenger domain. This region may be especially sensitive to perturbation because it is located near the N-terminal cap of the β-helix, where mutations may increase the accessibility of the hydrophobic core to water molecules most substantially. Although double and triple mutations that include F646A did not affect passenger domain stability, a quintuple mutation in this region abolished the resistance of the passenger domain to protease treatment and partially impaired translocation. The relative sensitivity of this region to mutation may likewise reflect its position adjacent to a disordered loop that projects from the β-helix to form a small globular subdomain and that forms an irregularity in the β-helix. In any case, the finding that multiple mutations that effectively destabilize the passenger domain only moderately impair translocation demonstrates that secretion cannot be completely explained by a simple vectorial folding model.
It is important to note that the mutations described here significantly impaired the completion of translocation but only minimally affected the initiation of translocation. Furthermore, the appearance of a stable ∼47-kDa C-terminal PK fragment immediately following the initiation of translocation indicates that the mutations did not affect the folding of the ∼17-kDa C-terminal passenger domain segment, which is the first segment that emerges on the cell surface. Taken together, these observations are consistent with a sequential folding model in which the mutations act locally and do not affect either the folding or translocation of segments that lie closer to the C terminus. It was therefore surprising that unlike C-terminal passenger domain mutations such as F963A, N-terminal and medial mutations did not appear to stall translocation at a unique location and trap a discrete N-terminal fragment inside the periplasm (19). One explanation of the data is that the N-terminal and medial mutations exerted more heterogeneous effects on the folding of a relatively large segment of the passenger domain. In any case, the finding that C-terminal mutations impaired the secretion reaction more dramatically and more specifically than N-terminal and medial mutations strongly suggests that the C terminus of the passenger domain plays an especially important role in facilitating translocation. Indeed, the relatively high sequence conservation of the C terminus of the passenger domain (21, 39) may reflect a unique function. As suggested previously (5, 21), the C-terminal region may promote efficient secretion at least in part by nucleating the folding of the β-helix and thereby catalyzing the rate-limiting step in the sequential folding reaction.
It is conceivable that the mutations we analyzed only moderately impaired passenger domain secretion because the repetitive nature and/or fundamental robustness of the β-helical structure tends to compensate for local folding defects. In one scenario, residues N-terminal to a destabilized region of the passenger domain occasionally diffuse across the OM and fold, thereby bypassing the structural defect. The existence of this type of phenomenon would explain why point mutations that prevent the folding of the C terminus of the EspP passenger domain only delay but do not block secretion (19) and why C-terminal deletions in the passenger domains of other autotransporters do not appear to significantly impair secretion (39, 40). Alternatively, the structural plasticity of the β-helix might allow sufficient packing of the hydrophobic core to drive secretion despite the presence of multiple disruptive substitutions. This hypothesis is consistent with the observation that the complete removal of the C-terminal segment of the EspP passenger domain has a much more drastic effect on secretion than single point mutations (19).
Our results also raise the possibility that only a portion of the energy that is required to drive passenger domain secretion is derived from protein folding. Most notably, we found that multiple point mutations that potentially affect the structural integrity of a sizeable portion of the β-helix reduced secretion efficiency by <50%. Indeed several lines of evidence strongly suggest that external sources of energy can play a key role in driving secretion. First, chimeric passenger domains that contain modules that fold in the periplasm and many other non-native polypeptides whose conformation in the periplasm and ability to fold in the extracellular space presumably vary considerably have been reported to be secreted efficiently (17, 31, 41–43). In addition, at least a few native passenger domains are globular rather than β-helical (8). Consistent with the hypothesis that passenger domain folding does not fully account for the energetics of secretion, we recently found that a chimeric EspP passenger domain that contains a large intrinsically disordered segment is secreted as efficiently as the native passenger domain (31). Interestingly, our results indicate that the strongly acidic character of the disordered polypeptide is important for efficient translocation. This observation suggests that charge repulsion between the polypeptide chain and acidic groups in phospholipids or LPS molecules or dielectric properties of the OM itself may help to drive the secretion reaction. It is also possible that the secretion of both native and chimeric passenger domains is driven through an interaction with an unidentified inner membrane protein that hydrolyzes ATP or draws energy from the membrane potential. Although an inner membrane protein called TamB was recently reported to be required for efficient passenger domain secretion (44), we have found that it is not required for EspP assembly.3 In any case, the available evidence suggests that the β-helical structure of the passenger domain may not have been conserved simply to drive secretion. As suggested previously, β-helical structures tend to fold slowly and therefore may confer an advantage by helping to prevent aggregation (5, 22).
Acknowledgment
We thank Susan Buchanan for generously providing recombinant TEV protease.
This work was supported, in whole or in part, by the NIDDK Intramural Research Program of the National Institutes of Health.
W. Kang'ethe and H. D. Bernstein, unpublished data.
- OM
- outer membrane
- TEV
- tobacco etch virus
- IPTG
- isopropyl β-d-thiogalactopyranoside
- PK
- proteinase K.
REFERENCES
- 1. Leyton D. L., Rossiter A. E., Henderson I. R. (2012) From self sufficiency to dependence: mechanisms and factors important for autotransporter biogenesis. Nat. Rev. Microbiol. 10, 213–225 [DOI] [PubMed] [Google Scholar]
- 2. Emsley P., Charles I. G., Fairweather N. F., Isaacs N. W. (1996) Structure of Bordetella pertussis virulence factor P.69 pertactin. Nature 381, 90–92 [DOI] [PubMed] [Google Scholar]
- 3. Otto B. R., Sijbrandi R., Luirink J., Oudega B., Heddle J. G., Mizutani K., Park S. Y., Tame J. R. (2005) Crystal structure of hemoglobin protease, a heme binding autotransporter protein from pathogenic Escherichia coli. J. Biol. Chem. 280, 17339–17345 [DOI] [PubMed] [Google Scholar]
- 4. Gangwer K. A., Mushrush D. J., Stauff D. L., Spiller B., McClain M. S., Cover T. L., Lacy D. B. (2007) Crystal structure of the Helicobacter pylori vacuolating toxin p55 domain. Proc. Natl. Acad. Sci. U.S.A. 104, 16293–16298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Junker M., Schuster C. C., McDonnell A. V., Sorg K. A., Finn M. C., Berger B., Clark P. L. (2006) Pertactin β-helix folding mechanism suggests common themes for the secretion and folding of autotransporter proteins. Proc. Natl. Acad. Sci. U.S.A. 103, 4918–4923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Oomen C. J., van Ulsen P., van Gelder P., Feijen M., Tommassen J., Gros P. (2004) Structure of the translocator domain of a bacterial autotransporter. EMBO J. 23, 1257–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Barnard T. J., Dautin N., Lukacik P., Bernstein H. D., Buchanan S. K. (2007) Autotransporter structure reveals intra-barrel cleavage followed by conformational changes. Nat. Struct. Mol. Biol. 14, 1214–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. van den Berg B. (2010) Crystal structure of a full-length autotransporter. J. Mol. Biol. 396, 627–633 [DOI] [PubMed] [Google Scholar]
- 9. Ruiz-Perez F., Henderson I. R., Leyton D. L., Rossiter A. E., Zhang Y., Nataro J. P. (2009) Roles of periplasmic chaperone proteins in the biogenesis of serine protease autotransporters of Enterobacteriaceae. J. Bacteriol. 191, 6571–6583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ieva R., Bernstein H. D. (2009) Interaction of an autotransporter passenger domain with BamA during its translocation across the bacterial outer membrane. Proc. Natl. Acad. Sci. U.S.A. 106, 19120–19125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ieva R., Tian P., Peterson J. H., Bernstein H. D. (2011) Sequential and spatially restricted interactions of assembly factors with an autotransporter β-domain. Proc. Natl. Acad. Sci. U.S.A. 108, E383–E391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sauri A., Soprova Z., Wickström D., de Gier J. W., Van der Schors R. C., Smit A. B., Jong W. S., Luirink J. (2009) The Bam (Omp85) complex is involved in secretion of the autotransporter haemoglobin protease. Microbiology 155, 3982–3991 [DOI] [PubMed] [Google Scholar]
- 13. Voulhoux R., Bos M. P., Geurtsen J., Mols M., Tommassen J. (2003) Role of a highly conserved bacterial protein in outer membrane protein assembly. Science 299, 262–265 [DOI] [PubMed] [Google Scholar]
- 14. Wu T., Malinverni J., Ruiz N., Kim S., Silhavy T. J., Kahne D. (2005) Identification of a multicomponent complex required for outer membrane biogenesis in Escherichia coli. Cell 121, 235–245 [DOI] [PubMed] [Google Scholar]
- 15. Hagan C. L., Kim S., Kahne D. (2010) Reconstitution of outer membrane protein assembly from purified components. Science 328, 890–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pohlner J., Halter R., Beyreuther K., Meyer T. F. (1987) Gene structure and extracellular secretion of Neisseria gonorrhoeae IgA protease. Nature 325, 458–462 [DOI] [PubMed] [Google Scholar]
- 17. Skillman K. M., Barnard T. J., Peterson J. H., Ghirlando R., Bernstein H. D. (2005) Efficient secretion of a folded protein domain by a monomeric bacterial autotransporter. Mol. Microbiol. 58, 945–958 [DOI] [PubMed] [Google Scholar]
- 18. Jong W. S., ten Hagen-Jongman C. M., den Blaauwen T., Slotboom D. J., Tame J. R., Wickström D., de Gier J. W., Otto B. R., Luirink J. (2007) Limited tolerance towards folded elements during secretion of the autotransporter Hbp. Mol. Microbiol. 63, 1524–1536 [DOI] [PubMed] [Google Scholar]
- 19. Peterson J. H., Tian P., Ieva R., Dautin N., Bernstein H. D. (2010) Secretion of a bacterial virulence factor is driven by the folding of a C-terminal segment. Proc. Natl. Acad. Sci. U.S.A. 107, 17739–17744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Klauser T., Pohlner J., Meyer T. F. (1992) Selective extracellular release of cholera toxin B subunit by Escherichia coli: dissection of Neisseria IgAβ-mediated outer membrane transport. EMBO J. 11, 2327–2335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Renn J. P., Clark P. L. (2008) A conserved stable core structure in the passenger domain β-helix of autotransporter virulence proteins. Biopolymers 89, 420–427 [DOI] [PubMed] [Google Scholar]
- 22. Junker M., Clark P. L. (2010) Slow formation of aggregation-resistant β-sheet folding intermediates. Proteins 78, 812–824 [DOI] [PubMed] [Google Scholar]
- 23. Renn J. P., Junker M., Besingi R. N., Braselmann E., Clark P. L. (2012) ATP-independent control of autotransporter virulence protein transport via the folding properties of the secreted protein. Chem. Biol. 19, 287–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Driessen A. J. (1992) Precursor protein translocation by the Escherichia coli translocase is directed by the protonmotive force. EMBO J. 11, 847–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shi L. X., Theg S. M. (2013) Energetic cost of protein import across the envelope membranes of chloroplasts. Proc. Natl. Acad. Sci. U.S.A. 110, 930–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lattman E. E., Rose G. D. (1993) Protein folding–what's the question? Proc. Natl. Acad. Sci. U.S.A. 90, 439–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dautin N., Barnard T. J., Anderson D. E., Bernstein H. D. (2007) Cleavage of a bacterial autotransporter by an evolutionarily convergent autocatalytic mechanism. EMBO J. 26, 1942–1952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Khan S., Mian H. S., Sandercock L. E., Chirgadze N. Y., Pai E. F. (2011) Crystal structure of the passenger domain of the Escherichia coli autotransporter EspP. J. Mol. Biol. 413, 985–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Akiyama Y., Ito K. (1990) SecY protein, a membrane-embedded secretion factor of E. coli, is cleaved by the OmpT protease in vitro. Biochem. Biophys. Res. Commun. 167, 711–715 [DOI] [PubMed] [Google Scholar]
- 30. Szabady R. L., Peterson J. H., Skillman K. M., Bernstein H. D. (2005) An unusual signal peptide facilitates late steps in the biogenesis of a bacterial autotransporter. Proc. Natl. Acad. Sci. U.S.A. 102, 221–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kang'ethe W., Bernstein H. D. (2013) Charge-dependent secretion of an intrinsically disordered protein via the autotransporter pathway. Proc. Natl. Acad. Sci. U.S.A. 110, E4246–E4255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ulbrandt N. D., Newitt J. A., Bernstein H. D. (1997) The E. coli signal recognition particle is required for the insertion of a subset of inner membrane proteins. Cell 88, 187–196 [DOI] [PubMed] [Google Scholar]
- 33. Velarde J. J., Nataro J. P. (2004) Hydrophobic residues of the autotransporter EspP linker domain are important for outer membrane translocation of its passenger. J. Biol. Chem. 279, 31495–31504 [DOI] [PubMed] [Google Scholar]
- 34. Brockmeyer J., Spelten S., Kuczius T., Bielaszewska M., Karch H. (2009) Structure and function relationship of the autotransport and proteolytic activity of EspP from Shiga toxin-producing Escherichia coli. PLoS ONE 4, e6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Soprova Z., Sauri A., van Ulsen P., Tame J. R., den Blaauwen T., Jong W. S., Luirink J. (2010) A conserved aromatic residue in the autochaperone domain of the autotransporter Hbp is critical for initiation of outer membrane translocation. J. Biol. Chem. 285, 38224–38233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Betts S., Haase-Pettingell C., Cook K., King J. (2004) Buried hydrophobic side-chains essential for the folding of the parallel β-helix domains of the P22 tailspike. Protein Sci. 13, 2291–2303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Simkovsky R., King J. (2006) An elongated spine of buried core residues necessary for in vivo folding of the parallel β-helix of P22 tailspike adhesin. Proc. Natl. Acad. Sci. U.S.A. 103, 3575–3580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dautin N., Bernstein H. D. (2011) Residues in a conserved α-helical segment are required for cleavage but not secretion of an Escherichia coli serine protease autotransporter passenger domain. J. Bacteriol. 193, 3748–3756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Oliver D. C., Huang G., Nodel E., Pleasance S., Fernandez R. C. (2003) A conserved region within the Bordetella pertussis autotransporter BrkA is necessary for folding of its passenger domain. Mol. Microbiol. 47, 1367–1383 [DOI] [PubMed] [Google Scholar]
- 40. Ohnishi Y., Nishiyama M., Horinouchi S., Beppu T. (1994) Involvement of the COOH-terminal pro-sequence of Serratia marcescens serine protease in the folding of the mature enzyme. J. Biol. Chem. 269, 32800–32806 [PubMed] [Google Scholar]
- 41. Jose J., Meyer T. F. (2007) The autodisplay story, from discovery to biotechnical and biomedical applications. Microbiol. Mol. Biol. Rev. 71, 600–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jong W. S., Saurí A., Luirink J. (2010) Extracellular production of recombinant proteins using bacterial autotransporters. Curr. Opin. Biotechnol. 21, 646–652 [DOI] [PubMed] [Google Scholar]
- 43. Sevastsyanovich Y. R., Leyton D. L., Wells T. J., Wardius C. A., Tveen-Jensen K., Morris F. C., Knowles T. J., Cunningham A. F., Cole J. A., Henderson I. R. (2012) A generalised module for the selective extracellular accumulation of recombinant proteins. Microb. Cell Fact. 11, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Selkrig J., Mosbahi K., Webb C. T., Belousoff M. J., Perry A. J., Wells T. J., Morris F., Leyton D. L., Totsika M., Phan M.-D., Celik N., Kelly M., Oates C., Hartland E. L., Robins-Browne R. M., Ramarathinam S. H., Purcell A. W., Schembri M. A., Strugnell R. A., Henderson I. R., Walker D., Lithgow T. (2012) Discovery of an archetypal protein transport system in bacterial outer membranes. Nat. Struct. Mol. Biol. 19, 506–510 [DOI] [PubMed] [Google Scholar]