

Background: Myelin transcription factor 1 (MyT1) contains seven similar zinc finger domains that bind DNA specifically.
Results: A three-dimensional structural model explains how a double zinc finger unit is able to recognize DNA.
Conclusion: DNA-binding residues are conserved among all MyT1 zinc fingers, suggesting an identical DNA binding mode.
Significance: Determination of the molecular details of DNA interaction will be crucial in understanding MyT1 function.
Keywords: Computer Modeling, Myelin, Nuclear Magnetic Resonance, Protein Structure, Zinc Finger
Abstract
Myelin transcription factor 1 (MyT1/NZF2), a member of the neural zinc-finger (NZF) protein family, is a transcription factor that plays a central role in the developing central nervous system. It has also recently been shown that, in combination with two other transcription factors, the highly similar paralog MyT1L is able to direct the differentiation of murine and human stem cells into functional neurons. MyT1 contains seven zinc fingers (ZFs) that are highly conserved throughout the protein and throughout the NZF family. We recently presented a model for the interaction of the fifth ZF of MyT1 with a DNA sequence derived from the promoter of the retinoic acid receptor (RARE) gene. Here, we have used NMR spectroscopy, in combination with surface plasmon resonance and data-driven molecular docking, to delineate the mechanism of DNA binding for double ZF polypeptides derived from MyT1. Our data indicate that a two-ZF unit interacts with the major groove of the entire RARE motif and that both fingers bind in an identical manner and with overall two-fold rotational symmetry, consistent with the palindromic nature of the target DNA. Several key residues located in one of the irregular loops of the ZFs are utilized to achieve specific binding. Analysis of the human and mouse genomes based on our structural data reveals three putative MyT1 target genes involved in neuronal development.
Introduction
Myelin transcription factor 1 (MyT1,5 or neural zinc finger 2 (NZF2)) is a transcription factor that contains seven zinc finger (ZF) modules. These ZFs all contain a C2HC arrangement of zinc ligands and are located in the protein in a 1 + 2 + 4 topology (see Fig. 1), although a second isoform exists that lacks finger 1 (F1) (1). MyT1 was first discovered through its ability to bind to sites in the proteolipid protein promoter (2). This protein (3) plays a major role in the structure and compaction of the myelin sheath that is located around the axons of the central nervous system. MyT1 has also been shown to promote commitment to a neuronal fate in Xenopus laevis (4) and, more recently, has been found to interact with Sin3B, a transcriptional coregulator that mediates transcriptional repression by recruiting histone deacetylases (5).
FIGURE 1.

MyT1 sequence information. A, schematic of the mouse MyT1 protein. ZFs 1–7 are indicated, as are the limits of the double ZF constructs used in this study. B, sequence alignment of MyT1 ZFs as well as the domains from NZF1 and NZF3 for which structures are available (rat NZF1 (rNZF1), PDB ID 1PXE; human NZF3 (hNZF3), PDB ID 2CS8). Arrows and asterisks indicate DNA-binding residues and zinc-coordinating residues, respectively. mMyT1, mouse MyT1. C, RARE DNA sequence with numbering used in this work. Box indicates the protein-binding motif.
In humans, there are two paralogs of MyT1: MyT1-like (MyT1L/NZF1) (6) and suppressor of tumorigenicity 18 (ST18/NZF3) (7). NZF3 has been shown to be a breast cancer tumor suppressor gene (8) and has also been implicated in the regulation of mRNA levels of proapoptotic and proinflammatory genes in fibroblasts (9). The other paralog, MyT1L, has recently attracted substantial interest due to its ability to act in concert with two other transcription factors (Ascl1 and Brnd2) to transform mouse as well as human stem cells directly into functional neurons (10–12). The resulting neuronal cells displayed functional properties such as the generation of trains of action potentials and synapse formation, properties that might enable them to be used for applications in neurological disease modeling or regenerative medicine. However, the molecular mechanisms through which these three transcription factors, in particular MyT1L, act are not well understood.
Several studies have shown that MyT1-type ZFs (from MyT1, MyT1L, and ST18) are able to recognize DNA in a sequence-specific manner (4, 7, 13). The motif AAGTT (the retinoic acid receptor element, or RARE), which is found in the human proteolipid protein promoter and is the core sequence in the cis-regulatory element of the retinoic acid receptor gene, has been identified as the consensus-binding sequence for MyT1.
We recently assessed the binding of MyT1 to DNA and showed that the fifth zinc finger can fit into the DNA major groove and make contacts with the central AGT of the consensus sequence (14). Our data also indicated that double-finger constructs of both the two-ZF and the four-ZF clusters (F1F2, F4F5, F5F6, and F6F7) interact with the full AAAGTT consensus site with a higher affinity than does a single finger.
In this study, we have examined the DNA binding properties of a double-finger polypeptide consisting of MyT1 fingers 4 and 5 and calculated a data-driven structural model using a combination of NMR and SPR affinity data. We reveal that the full AAGTT site can accommodate both fingers and that finger 4 contacts the DNA sequence in an orientation that differs by 180° from that observed for finger 5, consistent with the partially palindromic nature of the DNA site. Our NMR data also show that this binding mode is conserved among other MyT1 double ZF constructs, indicating that the full-length protein might be able to recognize two or three distinct AAGTT sites in vivo.
EXPERIMENTAL PROCEDURES
Subcloning, Expression, and Purification of MyT1 Constructs
The original plasmid encoding mouse 6-ZF myelin transcription factor 1 (mMyT1) was a gift of Dr Lynn Hudson (National Institutes of Health). Both F4F5 and F5F6 constructs of MyT1 (see Fig. 1) were cloned from the original plasmid (residues 18–904), and mutants were constructed using either overlap extension PCR or site-directed mutagenesis. All constructs were cloned into the pGEX-6P vector and overexpressed as GST fusions at 37 °C under standard conditions; isotopically labeled proteins were overexpressed using the protocol described previously (15). Proteins were purified using GSH affinity chromatography, HRV-3C cleavage, and gel filtration (Superdex-75 in SPR buffer: 50 mm NaCl, 10 mm HEPES, 1 mm DTT, pH 7.2). Protein concentrations were determined by absorbance at 215, 225, and 280 nm (16). Fractions were stored in the presence of protease inhibitors at −20 °C until required.
β-RARE DNA and Mutant Oligonucleotides
Single-stranded β-RARE DNA (5′-ACCGAAAGTTCAC and 5′-GTGAACTTTCGGT), mutant oligonucleotides and biotinylated DNA for SPR experiments were obtained from Sigma-Aldrich, annealed in SPR buffer without DTT (heated to 95 °C for 5 min and then cooled to room temperature over the course of 1–2 h), and purified using gel filtration (Superdex-75). Concentrations were calculated from absorbance at 260 nm.
Surface Plasmon Resonance
All experiments were performed on a Biacore 3000 system (Biacore AB) at flow rates of 20 μl/min in SPR buffer to which was added 0.01% polysorbate 20 (P20) detergent. Biotinylated DNA (∼10–100 nm) was immobilized on streptavidin-coated Biacore SA chips (50–100 resonance units). MyT1 and MyT1 mutants (0.2–10 μm) were injected in SPR buffer, and binding was monitored. The system was washed with 1 m NaCl (1 min) after each experiment. For kinetics studies, the Biacore BiaEvaluation software was utilized to calculate affinity constants using global fitting algorithms. In the competition experiments, F5F6 (5 μm) was added to prebound DNA in the presence of 5 molar eq of competitor DNA oligonucleotides.
NMR Spectroscopy
F4F5 or F5F6 (unlabeled, 15N-labeled, or 15N/13C-labeled) were exchanged into NMR buffer (50 mm NaCl, 10 mm phosphate, 1 mm DTT, pH 7.2) with 1 mm DTT and concentrated in Microsep 3K cutoff filters to 200–1000 μm. Resonance assignments were made from standard triple-resonance experiments that were acquired at 25 °C on Bruker Avance 600 and 800 NMR spectrometers equipped with cryoprobes. 15N HSQC titrations as well as two-dimensional NOESY experiments of proteins with β-RARE DNA were carried out in NMR buffer at 25 °C. Chemical shift changes were calculated as a weighted average of HN, N, and Cα changes, using a previously reported equation (17, 18). Assignments of the DNA alone were obtained from our previous study (14). One-bond HN residual dipolar couplings (RDCs) were recorded for the F4F5-DNA complex in NMR buffer containing 22.2 mg/liter Pf1 phage (ASLA Biotech), using the in-phase/anti-phase pulse sequence (19). Alignment was assessed by measuring the D2O splitting (19 Hz). The program PALES (20) was used for the calculation of the magnitude and orientation of the sterically induced alignment tensor (see below for details). NMR data were processed using Topspin (Bruker, Karlsruhe) and analyzed with SPARKY 3 (37).
HADDOCK Docking
F4F5 was docked to the DNA using the program HADDOCK 1.3 (21–23). The starting structure for the DNA was a B-form model of the double helix DNA fragment (5′-ACCGAAAGTTCAC) constructed with the Nucleic Acid Builder package (24). Based on our NMR data (see Figs. 2 and 3), a starting structure of F4F5 was made in silico by fusing two individual ZF domains and the native linker sequence (see Fig. 1) together using the calculated NMR structure of F5 (14) (Protein Data Bank (PDB) ID 2JYD) as a template. A total of 10 different starting orientations between F4F5 and the DNA were chosen as starting structures for the docking. Sequences shown to be disordered in our previous NMR analysis, namely residues 799–800 (N-terminal) and 872–873 (C-terminal), were defined as fully flexible during the calculations, as was the internal linker (828–845) (14). Ambiguous interaction restraints for both the protein and the DNA were chosen based both on our NMR data from Figs. 2 and 3 and on solvent accessibility (>30%, determined by the program MOLMOL) and were fixed at 2 Å. For the DNA fragment, ambiguous interaction restraints were defined solely from the unique base atoms of bases Ade6, Ade7, Thy20, and Thy21, whereas for F5, DNA bases Thy9, Thy10, Ade17, and Ade18 were selected. For the protein, restraints between unique side-chain atoms of F4 (residues His-812, Tyr-817, Ser-819, Arg-821, Ser-822, Leu-823, Ser-824) as well as corresponding residues in F5 (His-856, Tyr-861, Ser-863, Arg-865, Ser-866, Leu-867, Ser-868) were chosen. A total of 48 ambiguous interaction restraints resulted from these definitions and were used as input into HADDOCK for all 10 different F4F5-DNA starting configurations. Additional restraints to maintain base planarity and Watson-Crick bonds for the DNA, intramolecular noncrystallographic symmetry restraints between F4F5 and DNA (F4+Ade6/7 = F5+Ade17/18 and F4+Thy20/21 = F5+Thy9/10), and zinc-coordinating restraints for F4F5 were introduced. During the rigid body energy minimization, 1000 structures were calculated, and the 200 best solutions based on the intermolecular energy were used for the semiflexible, simulated annealing. 10 different runs were carried out with the 10 F4F5-DNA starting orientations, respectively. The best 10 structures of each run were part of the lowest energy cluster (cut-off of 0.5 Å root mean square deviation (RMSD) based on the pairwise backbone RMSD matrix).
FIGURE 2.

Both F4 and F5 bind DNA using the same subset of residues. A, portion of the 15N HSQC of 15N-labeled double ZF construct F4F5 in the absence (gray) and presence (black) of 1 molar eq of RARE DNA. Assignments are indicated. B, graph showing magnitude (black dots, left and bottom axes) and direction (gray boxes, right and top axes) of chemical shift changes (measured from 15N HSQC spectra as indicated in the inset) that occur for residues in F4 as compared with the corresponding residues in F5 upon the addition of 1 molar eq of RARE DNA. Only residues 802–826 in F4 and 846–870 in F5 are shown. The high correlation between F4 and F5 changes for a given residue strongly suggests the same mode of binding for both ZFs. C, summary of chemical shift changes for HN, N, and Cα nuclei. Total chemical shift changes, weighted according to Ayed et al. (36), are also shown.
FIGURE 3.

Both strands of the RARE site bind MyT1. A, imino region of a one-dimensional 1H NMR spectrum of the RARE oligonucleotide in the absence (above) and presence (below) of 1 molar eq of F4F5. Base numbers (numbering from Fig. 1C) are indicated. B, summary of chemical shift changes for DNA protons following binding of F4F5. Black, gray, and white bars represent imino (thymines and guanines), amino (cytosines and adenines), and methyl (thymine) protons, respectively. Significant changes (more than one standard deviation greater than the mean change for all base protons) are indicated by an arrowhead, and the corresponding DNA bases are marked in gray in the sequence above each graph. The two graphs show changes for the two strands of the DNA. C and D, two portions of the imino region of a two-dimensional NOESY spectrum (temperature = 298 K) of an F4F5-DNA (1:1) complex. No intermolecular NOEs are observed.
The 10 best structures from run I were subjected to a second round of semiflexible annealing following the inclusion of 43 HN RDCs as additional direct restraints (using the SANI statement); axial and rhombic components of the alignment tensor (Da and Dr) were calculated using the 10 run I structures and the software PALES (20). The alignment tensor was then recalculated based on the resulting best 10 of a total of 200 calculated structures (lowest SANI energies), and HADDOCK was run again (see above) using these new values. After this protocol, the final 10 structures were not significantly different from the ones calculated without the RDCs (RMSD over all atoms of the lowest energy structure = 0.3 Å). These structures were analyzed using standard HADDOCK protocols.
PRE Measurements
To attach a paramagnetic moiety to the RARE oligonucleotide, lyophilized modified RARE DNA containing a phosphorothioate linkage at Thy15 (which is located next to the DNA-binding site) was resuspended in 100 mm phosphate buffer (pH 7) to a concentration of ∼400 μm. This solution was then used to dissolve the complementary strand, and the DNA was annealed by heating to 95 °C for 5 min and then cooling to room temperature over a period of 1 h. A 100 molar excess of 3-(2-iodoacetamido-)proxyl radical (in 100% ethanol) was added to the annealed oligonucleotide and incubated in the dark for 20 h under shaking. To remove excess single-stranded DNA and unreacted 3-(2-iodoacetamido-)proxyl, the reaction mixture was subjected to size exclusion chromatography using NMR buffer. The progress of the reaction was monitored using UV spectroscopy and mass spectroscopy. The final product (containing about 50% labeled and 50% unlabeled double-stranded DNA) was used to carry out a semiquantitative paramagnetic resonance enhancement (PRE) analysis.
To acquire PRE data, two-time point HSQC experiments as described in Ref. 25 were performed (Ta = 0 ms; Tb = 14 ms) on a 1 mm F5-DNA complex. Spectra were first recorded for the paramagnetic (oxidized) sample and then for the diamagnetic (reduced) sample following the addition of a 50 molar excess of sodium dithionate. After normalizing all four 15N HSQC spectra, peak intensities were obtained for the assigned residues and PRE rates (25) were calculated. Distances shown in Fig. 8B are calculated from the phosphate of Thy15 to the HN backbone atom of the corresponding residue.
FIGURE 8.

Comparison to the single-finger F5-DNA interaction. A, portion of the 15N HSQC of 15N-labeled double ZF protein F4F5 (upper) and single ZF F5 (14) (lower), respectively, in the absence (gray) and presence (black) of 1 molar eq of RARE DNA. Note the similarity in both the magnitude and the direction of the chemical shift changes. B, semiquantitative PRE analysis of F5 in the presence of nitroxide spin-labeled RARE DNA. The PRE rate calculated from a two time point 15N HSQC experiment (25) is plotted as a function of the amino acid (upper part) and compared with the actual distance between the phosphorus atom of Thy15 (on which the PRE label is located) and the corresponding backbone HN proton for the two different F5-DNA models, respectively (middle and lower parts). The PRE rate profile agrees better with the distance profile of the new F5-DNA model than with that of our previous model (14). Residues colored in green are those with distances to the Thy15 phosphorus (in green as well) that are below the average distance minus one standard deviation. These residues are indicated on the corresponding model (right part of figure).
RESULTS
Identification of the DNA-binding Interface of the MyT1 Double-finger F4F5
In our recent study (14), we tested different combinations of MyT1 ZFs and their ability to bind a 13-bp oligonucleotide containing the RARE sequence (Fig. 1C). Our data showed that F2 and F5 bind with similar affinities (Ka ∼1 × 106 m−1 under the conditions of our binding experiments), whereas F3, F4, and F6 bind at least 10-fold less tightly (precise affinities were not measured). In contrast, the affinity of polypeptides comprising two ZF (F2F3, F4F5, F5F6, and F6F7) is significantly higher and displays less variation. Using isothermal titration calorimetry, we also showed that the F4F5-RARE complex forms with a 1:1 stoichiometry. Taken together, these data suggest that both fingers in the two-ZF polypeptides contribute to DNA binding.
Previously, we experienced problems with degradation and intermediate chemical exchange during NMR studies of the double-finger proteins (14). We therefore optimized our experimental conditions (e.g. the addition of protease inhibitor and change in buffer components) to circumvent these issues and have consequently been able to record backbone triple resonance spectra for 15N MyT1 F4F5 and assign the 15N HSQC spectrum before and after the addition of 1 molar eq of RARE DNA (Fig. 2A). Comparison of the F4 sequence with F5 (Fig. 1) reveals that all residues between the first and last zinc-coordinating cysteines are conserved between the two domains. Consistent with this conservation, most signals occur in closely spaced pairs or as two superimposed peaks in 15N HSQC spectra (Fig. 2A), indicating that the fold of the two domains is essentially identical. Moreover, the direction and magnitude of chemical shift changes that occur for residues in F4 as compared with their counterparts in F5 upon the addition of RARE DNA are very similar (Fig. 2B). Comparison of all chemical shift changes for HN, N, and Cα atoms in F4 with those in F5 (Fig. 2C) shows clearly that the DNA-binding surface is conserved between the two domains.
We used our assignments of the RARE oligonucleotide (14) to assess which part of the DNA was involved in the interaction with F4F5 (Fig. 3). Analysis of amino-, imino-, and methyl- proton chemical shift changes (Fig. 3, A and B) reveals the sequence AAAGTTCA (palindromic sequence is underlined) as the F4F5 interaction surface. These data are in good agreement with our SPR and NMR analysis of the F5-DNA interaction (14), which showed that F5 has a footprint on the DNA that comprises the smaller AGT motif.
In an attempt to obtain intermolecular NOEs, we recorded two-dimensional NOESY spectra of F4F5 in the presence of RARE DNA (Fig. 3, C and D) at both 25 °C and 4 °C. Although F4F5 binds DNA with higher affinity than the single finger F5, no intermolecular NOEs could be unambiguously identified under these conditions. Fig. 3, C and D, show two different portions of the DNA imino-proton region of the 25 °C NOESY spectrum; no cross-peaks to any protein resonances are observed. Notably, resonances from a number of DNA base protons as well as F4F5 side-chain protons that are likely to form part of the protein-DNA interface could not be located in the spectra of the complex. These signals were most likely experiencing intermediate exchange, preventing the observation of NOEs. In summary, our HSQC data indicate that the structures of both F4 and F5 are largely conserved and that both fingers use the same surface to bind the same target sequence on the DNA, explaining the partly palindromic nature of the RARE DNA motif.
Data-driven HADDOCK Docking Yields a Structural Model of the F4F5-DNA Complex
Due to the lack of any intermolecular NOEs, structure determination using the standard NOE-based approach was not possible. Thus, we used our NMR data to calculate a structural model of an F4F5-DNA complex (Figs. 4 and 6) using HADDOCK (23, 26). To achieve this, we first created an F4F5 polypeptide by linking two individual ZF domains and the native linker sequence (Fig. 1) in silico, using the NMR structure of F5 (14) as a template. We defined ambiguous restraints between DNA atoms and specific side-chain atoms of DNA-interacting residues of each ZF, based on our NMR data (Figs. 2 and 3). Restraints to keep the structure of the DNA in a standard B-form geometry were also included. Consistent with our chemical shift data, which indicate that both fingers interact in an identical manner, we additionally introduced intramolecular noncrystallographic symmetry constraints between F4F5 and the palindromic part of the DNA (AAGTT). During the docking calculations, active residues were defined as semiflexible (side-chain atoms were allowed to move freely), whereas the linker region and the last two residues at both the N-terminal and the C-terminal ends were defined as fully flexible. We carried out 10 runs with different starting orientations of the F4F5 construct relative to the DNA in the presence and absence of noncrystallographic symmetry restraints; all other restraints were kept constant for all runs. In 2 of the 10 docking calculations, we observed significant convergence as seen from HADDOCK energy versus RMSD graphs (Fig. 4A, runs I and II, black and blue) following two-stage docking and simulated annealing. Both total HADDOCK energy (Fig. 4A) and intermolecular HADDOCK energy (Fig. 4B) are significantly higher (less favorable) for run II as compared with run I. Based on these data, the lowest energy cluster of structures from run I was used to represent a model of the complex. Inspection of the structures with the lowest energies of each run (Fig. 4C) reveals that run II differs from run I by a rotation of each individual zinc finger of 90° around an axis running perpendicular to the major groove of the DNA.
FIGURE 4.
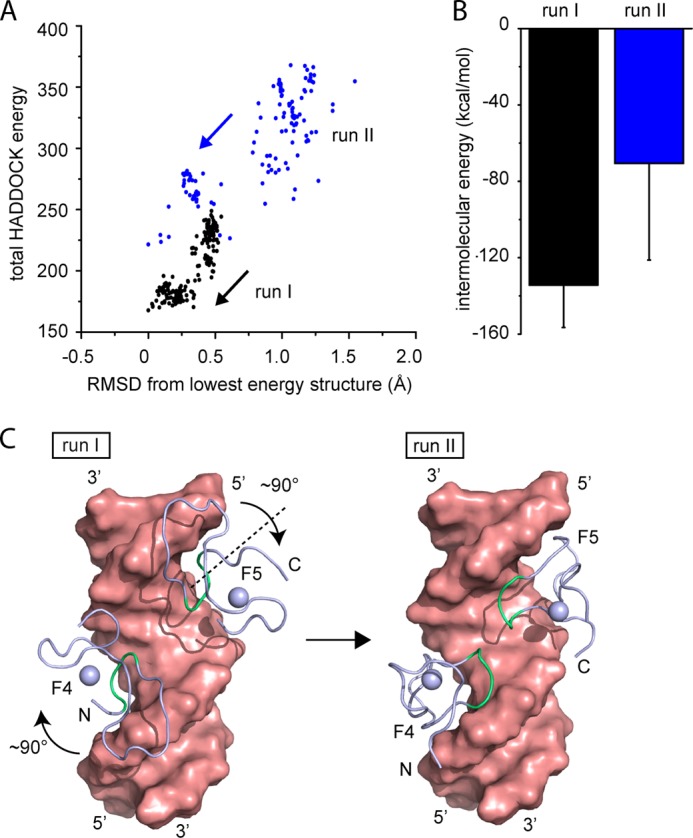
Data-driven structural modeling of F4F5-DNA interaction. A, graph showing total HADDOCK energy as a function of the RMSD to the lowest energy structure of two runs (run I and II) from HADDOCK docking of an F4F5 polypeptide to double-stranded RARE DNA (different starting orientations were used for each run). Significant convergence is observed in both runs. B, calculated intermolecular energy (using standard HADDOCK protocols) of the 10 best structures of each run. Error bars indicate ± S.E. C, space-filling representation of lowest energy structures from run I and II without the linker region between the ZFs. Residues colored in green are the DNA-binding residues.
FIGURE 6.

A structural model of an F4F5-DNA complex. A, overlay of the lowest energy MyT1-F4F5 complex HADDOCK model before (green) and after (blue, PDB ID 2MF8) the introduction of RDCs for refinement. F4F5 and DNA (salmon) are shown as graphics. B, surface representation of the DNA from the lowest energy model and ribbon diagram of F4F5. The side chains of several interacting residues are shown as sticks. The zinc ions are shown as gray spheres. C, roll and twist angles as well as the width of the major groove of the DNA in the complex structure. Expected values for B-form DNA are shown as dotted lines. Error bars represent S.D. from the 10 best (SANI energy) HADDOCK structures.
Mutational Analysis and RDCs Were Used to Evaluate and Refine the Structural Model
Based on our 15N HSQC titration data and a visual examination of the structural model, we made a series of point mutations in both ZFs in an effort to assess the validity of our HADDOCK model. Residues were mutated in pairs (the two corresponding residues in each ZF), and seven double mutants were made, namely I813A/I857A, N816A/N860A, Y817F/Y861F, S819D/S863D, R821K/R865K, S822D/S866D, and S824D/S868D. Note that mutations of serine to alanine were avoided as such mutations had been previously demonstrated to disrupt the fold of these domains (14). All seven mutants were correctly folded as judged by their 15N HSQC spectra. The ability of each mutant to bind RARE DNA was then assessed by recording 15N HSQC spectra. Fig. 5A depicts weighted chemical shift changes of the backbone HN and N atoms of Tyr-817/861 (residues that underwent large chemical shift changes in the wild-type protein following the addition of DNA) upon the addition of 1 molar eq of RARE DNA for each mutant. Changes in the 15N HSQC spectra for three of the seven mutants (I813A/I857A, N816A/N860A, Y817F/Y861F) following the addition of DNA closely resembled the changes observed for the wild-type protein, whereas the remainder exhibited substantially smaller chemical shift changes (see Fig. 5D for representative 15N HSQC spectra for mutants Y817F/Y861F and S824D/S868D).
FIGURE 5.

Mutational and RDC analysis to evaluate the MyT1-DNA interaction. A, weighted chemical shift changes for backbone H and N from Tyr-817/Tyr-861 in the absence and presence of 1 molar eq of RARE DNA. The number on top of the bars (average from n = 3–4 ± S.E.) indicates binding association constants from SPR experiments carried out with immobilized biotinylated double-stranded RARE DNA and mutant F4F5 constructs in solution. Green bars indicate equal or higher affinity constants as compared with wild-type F4F5. Red bars indicate mutations that result in significantly lower association constants and are therefore important for the interaction. B, graphic (left) and space-filling representation (middle and right) of F4 with mutations that result in equal/stronger and weaker DNA binding colored in green and red (as in panel A), respectively. The second ZF domain (F5, not shown) shows an identical pattern (with corresponding mutations). C, space-filling representation of F4 from run I and run II in the same orientation as in the middle of panel B with residues that make specific contacts to the DNA colored in dark gray. Note that the DNA-binding residues in run I match red residues (important for binding) in B (in contrast to run II), in agreement with our structural model. D, portion of the 15N HSQC of 15N-labeled double ZF mutant Y817F/Y861F (top) and S824D/S868D (bottom) in the absence (gray) and presence (black) of 1 molar eq of RARE DNA (as in Fig. 2A). Note the significant difference between the two mutants. E, representative SPR binding curves for the interaction of the Y817F/Y861F double mutant (0.2–6 μm) in solution with immobilized biotinylated double-stranded RARE DNA. F, plot of calculated (from refined structures) versus measured one-bond HN-N RDCs (average from the 10 lowest SANI energy HADDOCK structures).
To corroborate these data, biotin-tagged DNA was immobilized on streptavidin-coated chips and treated with mutant proteins in SPR experiments. Association constants were calculated from equilibrium binding data (Fig. 5A, top of bars; see Fig. 5E for a representative SPR sensorgram of mutant Y817F/Y861F). Notably, the double mutations at Ile-813/Ile-857, Asn-816/Asn-860, and Tyr-817/Tyr-861 did not decrease the binding affinity (Fig. 5A, green bars). Interestingly, the asparagine double mutant exhibits stronger binding than the wild-type protein. In contrast, the F4F5 double mutants S819A/S863A, R821K/R821K, S822D/S866D, and S824D/S824D (Fig. 5A, red bars) exhibit significantly smaller association constants, underscoring the importance of these residues for DNA binding. To visualize the results of this analysis, all mutated residues were mapped onto the structure of F4 in Fig. 5B. Green- and red-colored spheres indicate mutants that bind with equal/stronger or weaker affinity, respectively. Fig. 5C shows in dark green the residues that make specific contacts with DNA in each of our two converged HADDOCK runs (runs I and II). Comparison of these inferred DNA-binding surfaces reveals that all mutations that reduce DNA binding (red color in the figure) are also part of the predicted interaction surface from run I, whereas two of three residues that form part of the DNA-binding interface in the alternative structure from run II (Ile-813 and Tyr-817) do not lead to a reduction in binding when mutated (Fig. 5, B and C). Overall, these mutational data strongly support the conclusion that the model derived from run I is the best representation of the F4F5-DNA structure.
To further confirm and refine our run I-derived model, we recorded one-bond HN RDCs for the F4F5-DNA complex in Pf1 phage using in-phase/anti-phase 15N HSQC experiments (19). A total of 43 RDCs were measured and then introduced as additional restraints for a further two-stage HADDOCK docking analysis (see ”Experimental Procedures“ for more details). For the final 10 structures, the correlation between the predicted and observed HN RDCs is good (r = 0.90; Fig. 5F). Fig. 6A shows the structural model calculated with (blue) and without (light green) RDCs, revealing no significant difference (RMSD over all atoms = 0.3 Å), supporting overall the validity of our structural model.
Polar Interactions Are Mostly Responsible for Specific DNA Binding
Both F4 and F5 contact the major groove of the DNA, together making contacts over the entire AAGTT motif (Fig. 6B). Base-specific hydrogen bonds observed in >50% of the structures were identified between base protons of Ade7/18 (that is, the symmetry-related bases from the AAGTT motif on the two DNA strands) and Arg-821/865 (from F4/F5), Ade6/17, and Ser-824/868 as well as Thy9/20 and Ser-822/866, whereas Ser-819/Ser-863 contacts the DNA backbone. Multiple hydrophobic contacts made by residues Leu-823 and Leu-867 to methyl groups of Thy20 and Thy9, respectively, suggest that these residues might be important for binding. Taken together, these observations indicate that polar interactions are a strong determinant of binding affinity to MyT1 F4F5 and that several serines and arginines, which are conserved in any of the MyT1 ZF domains apart from finger F1 (Fig. 1), play an important role in recognition of the DNA. We have also analyzed the conformation of the DNA after the docking process using the software 3DNA (27) (Fig. 6C). Consistent with the restraints applied during the calculation, the geometry of the DNA is conserved, and only minor deviations from the original B-form conformation are observed.
DNA Binding Properties of an F5F6 Construct
To determine whether DNA binding of other MyT1 double ZF domains is similar to F4F5, we expressed a 15N-labeled F5F6 polypeptide and recorded NMR 15N HSQC spectra. A portion of the spectrum in the presence and absence of 1 molar eq of RARE DNA is shown in Fig. 7A. As seen in the F4F5 construct, resonances from the corresponding residues in each ZF module occur in pairs, and furthermore, the direction and magnitude of the chemical shift changes upon binding of DNA are conserved, indicating that F6 has a similar fold and DNA-binding interface to F5. To determine the importance of each DNA base for the interaction, we carried out SPR competition experiments as described earlier (14) (Fig. 7B). Single base changes across the whole GAAAGTT motif reduced the ability of a RARE-based oligonucleotide to compete with WT RARE DNA for binding to F5F6; the profile obtained closely resembled that measured previously for F4F5 (14). This similarity in DNA binding was further confirmed by one-dimensional 1H NMR spectroscopy, which showed that the chemical shifts of nitrogen-bound guanine and thymine protons of RARE DNA in the presence of F5F6 are almost identical to corresponding protons in a complex formed with F4F5 (Fig. 7C).
FIGURE 7.

NMR analysis of the interaction of F5F6 with DNA. A, portion of the 15N HSQC of 15N-labeled F5F6 (right) in the absence (gray) and presence (black) of 1 molar eq of RARE DNA and comparison with the same data for F4F5 (left). B, SPR data (average from n = 3 replicates) showing the ability of mutant oligonucleotides to compete with chip-bound RARE DNA for the binding of F5F6 in the wild-type complex. All mutants (black bars) except C11G (right gray bar) compete significantly less well than wild-type RARE (left gray bar), indicating that these bases most likely are contacted by F5F6. C, imino region of a one-dimensional 1H NMR spectrum of RARE DNA in the presence of F5F6 (solid line) and comparison with the same region of a spectrum of the F4F5-RARE complex (dotted line).
All DNA-binding residues identified within the F4 and F5 are conserved in F6, as well as in all other MyT1 zinc fingers (with the exception of the first finger) and a single alanine residue in place of a leucine in F6 (Fig. 1). These observations strongly suggest that the DNA binding mode is conserved among all MyT1 double ZF modules.
DISCUSSION
Differences in the DNA Binding Mode between MyT1 Single and Double Fingers
In this study, we have used a combination of NMR and SPR data to calculate a structural model of a MyT1 double ZF domain bound to DNA. We have shown that these domains interact with the major groove of the entire RARE motif and that both fingers bind in an identical manner with two-fold symmetry. Specificity is achieved through several key residues, namely Arg-821/865, Ser-822/866, and Ser-824/868, which make hydrogen bonds to base protons of adenines and thymines, whereas the two leucines Leu-823 and Leu-867 make hydrophobic interactions with the methyl groups of the thymines in the consensus sequence.
To compare our double ZF F4F5-RARE model with our previous single ZF F5-RARE model (14), we compared the 15N HSQC spectra in the absence (gray) and presence (black) of RARE DNA (Fig. 8A). Both the magnitude and the direction of the chemical shift changes are very similar, suggesting that the DNA binding mode is conserved. However, comparison of our published F5-DNA model with the F4F5-DNA model reveals that the orientations of the ZF and the surface used for DNA recognition are different in the two cases. The high degree of similarity of the NMR spectra argues that the difference observed in the models does not reflect a real difference in solution binding. Indeed, a semiquantitative PRE analysis (Fig. 8B) carried out using F5 in the presence of a paramagnetic nitroxide spin-labeled RARE DNA (see ”Experimental Procedures“ for more details) further confirmed that the DNA binding orientation of F5 is equivalent to that proposed in our new F4F5-DNA model.
Substantially more data were used to derive and validate the model in the F4F5 case; in particular, the NMR measurements that were made on the F4F5 mutants clearly showed in significant detail which mutations did not appreciably affect the interaction. The close agreement of these mutational data with our F4F5-DNA model as well as the good correlation of the measured with the calculated RDCs leads us to conclude that our current model is more likely to represent the true binding mode for these ZFs. It is worth emphasizing the value of recording 15N HSQC spectra of complexes made with point mutants when validating docking models; the high information content of such experiments (compared for example with simple affinity measurements) can be very valuable in distinguishing different possible models (e.g. in our case between run I and II) of the same complex.
Comparison with Members of the NZF Protein Family
MyT1 (NZF2) belongs to the class of ZF transcription factors that is characterized by the presence of multiple Cys-X4-Cys-X4-His-X7-His-X5-Cys sequences. So far, apart from our solution structure of the fifth finger of MyT1, only two other structures have been determined: the first ZF domain of NZF1 (28) (PDB ID 1PXE) and the fifth and sixth ZF of NZF3 (or ST18) (PDB ID 2CS8), which were solved by the RIKEN structural genomics initiative. Both NZF1 and NZF3 double ZF modules have been shown to be able to recognize the RARE DNA motif (7, 13, 29).
The structures of F5 and F6 of NZF3 are highly similar to our MyT1 F5 structure (RMSD < 1.5 Å over folded regions), indicating that the ZF fold is conserved within the whole protein family. This is also in good agreement with our NMR data on the F5F6 construct (Fig. 7). As a consequence, the same loop that binds the DNA specifically in the MyT1 F4F5 construct is likely to be utilized in NZF3. Indeed, sequence alignment of these proteins (Fig. 1B) confirms that the subsets of residues that make specific contacts with the DNA (Fig. 1B, arrows) are either conserved or very similar (e.g. threonine instead of serine).
The DNA binding affinities of constructs containing MyT1-like zinc fingers appear to vary over a wide range depending on the number of individual ZF modules present. For example, although the affinity of all six ZF domains of NZF3 for a double RARE site was estimated to be around 2 nm by EMSA experiments (7), a double ZF polypeptide consisting of the second and third finger of NZF1 binds with ∼10 nm as determined by fluorescence methods (30). In contrast, our isothermal titration calorimetry (14) and SPR data (Fig. 5) revealed a RARE DNA binding affinity of between 0.3 and 1 μm for the F4F5 construct. However, in all cases, a double ZF module was found to be required and sufficient to recognize one single palindromic AANTT site, indicating that binding of four or more zinc fingers most likely occurs via interaction of multiples of these units with DNA.
Other Putative Target Sites of MyT1
To search for other putative MyT1 DNA-binding sites, we used our structural and biophysical data to define a minimal DNA-binding site. The majority of the specific contacts are made to the two bases flanking the central guanine of the AAGTT site (underlined), in agreement with our SPR competition data (14). We therefore initially searched the GenBankTM database for a triple RANTR (R = G/A, N = any base) site that would allow both the MyT1 F2F3 and the F4F5F6F7 ZF clusters to bind simultaneously. Due to the importance of MyT1 in neuronal development, we limited our search to neuronally expressed genes with promoters of mice and humans available in GenBank. No hits were found with this search sequence, but a double RANTR motif was located at three sites (Fig. 9).
FIGURE 9.

Putative MyT1 target sites. Sequence alignment of portions of three human and mouse genes (International Nucleotide Sequence Database Collaboration (INSDC) database accession numbers in parentheses) that contain potential target sites for MyT1 double or quadruple ZF clusters are shown.
The first putative site is located around 350 bp upstream of the promoter of NeuroD1, a basic helix-loop-helix transcription factor that when combined with Ascl1, Brnd2, and MyT1L can convert fetal and postnatal human fibroblasts into functional neurons (11). This site consists of two separate RANTR sites that are separated by 18 nucleotides and could accommodate F2F3 and two zinc fingers of the F4F5F6F7 cluster.
The second potential binding site is part of the promoter of SLC1A3 (GLAST/EAAT-1), a glial glutamate transporter found to be important in brain function (31, 32). This site is made up of two AANTT motifs separated by 5 bp that are almost identical to the RARE sequence (AAGTT) and thus highly likely to be able to bind either F2F3 or two of the four fingers of the F4F5F6F7 cluster.
Similarly, the third site contains two MyT1 double ZF-binding sites (AANTT and AANTR, 4 bp apart) and is located around 140 nucleotides upstream of the start of the transcription site of neurogenin1, a transcription factor essential for neuronal differentiation and subtype specification during embryogenesis (33, 34). Interestingly, when NZF3, a member of the same protein family to which MyT1 belongs, and Neurog1 are co-expressed in cells, the rate of neuronal differentiation is significantly increased, suggesting that these proteins act in concert to stimulate these processes (35).
In conclusion, we have used a combination of structural and biophysical methods to determine the molecular basis of the interaction of double ZF domains of myelin transcription factor 1 with DNA. Our work has revealed that the entire RARE motif, which has been identified previously as a relevant binding site, can accommodate a two-ZF module that occupies the major grove of the DNA (14). The orientation of the two fingers relative to each other differs by 180°, and both domains contact the same bases using the same set of residues, consistent with the partly palindromic nature of the DNA. Using our structural data, we have identified three potential MyT1 target sites proximal to genes that are important in neuronal development.
This work was supported by the National Health and Medical Council of Australia (NHMRC) and the Australian Research Council (ARC).
The atomic coordinates and structure factors (code 2MF8) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- MyT1
- myelin transcription factor 1
- NZF
- neural zinc-finger
- ZF
- zinc finger
- RARE
- retinoic acid receptor
- RDC
- residual dipolar coupling
- RMSD
- root mean square deviation
- PRE
- paramagnetic resonance enhancement
- HSQC
- heteronuclear single quantum correlation.
REFERENCES
- 1. Nielsen J. A., Berndt J. A., Hudson L. D., Armstrong R. C. (2004) Myelin transcription factor 1 (Myt1) modulates the proliferation and differentiation of oligodendrocyte lineage cells. Mol. Cell. Neurosci. 25, 111–123 [DOI] [PubMed] [Google Scholar]
- 2. Kim J. G., Hudson L. D. (1992) Novel member of the zinc finger superfamily: A C2-HC finger that recognizes a glia-specific gene. Mol. Cell. Biol. 12, 5632–5639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Macklin W. B., Campagnoni C. W., Deininger P. L., Gardinier M. V. (1987) Structure and expression of the mouse myelin proteolipid protein gene. J. Neurosci. Res. 18, 383–394 [DOI] [PubMed] [Google Scholar]
- 4. Bellefroid E. J., Bourguignon C., Hollemann T., Ma Q., Anderson D. J., Kintner C., Pieler T. (1996) X-MyT1, a Xenopus C2HC-type zinc finger protein with a regulatory function in neuronal differentiation. Cell 87, 1191–1202 [DOI] [PubMed] [Google Scholar]
- 5. Romm E., Nielsen J. A., Kim J. G., Hudson L. D. (2005) Myt1 family recruits histone deacetylase to regulate neural transcription. J. Neurochem. 93, 1444–1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kim J. G., Armstrong R. C., v Agoston D., Robinsky A., Wiese C., Nagle J., Hudson L. D. (1997) Myelin transcription factor 1 (Myt1) of the oligodendrocyte lineage, along with a closely related CCHC zinc finger, is expressed in developing neurons in the mammalian central nervous system. J. Neurosci. Res. 50, 272–290 [DOI] [PubMed] [Google Scholar]
- 7. Yee K. S., Yu V. C. (1998) Isolation and characterization of a novel member of the neural zinc finger factor/myelin transcription factor family with transcriptional repression activity. J. Biol. Chem. 273, 5366–5374 [DOI] [PubMed] [Google Scholar]
- 8. Jandrig B., Seitz S., Hinzmann B., Arnold W., Micheel B., Koelble K., Siebert R., Schwartz A., Ruecker K., Schlag P. M., Scherneck S., Rosenthal A. (2004) ST18 is a breast cancer tumor suppressor gene at human chromosome 8q11.2. Oncogene 23, 9295–9302 [DOI] [PubMed] [Google Scholar]
- 9. Yang J., Siqueira M. F., Behl Y., Alikhani M., Graves D. T. (2008) The transcription factor ST18 regulates proapoptotic and proinflammatory gene expression in fibroblasts. FASEB J. 22, 3956–3967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lujan E., Chanda S., Ahlenius H., Südhof T. C., Wernig M. (2012) Direct conversion of mouse fibroblasts to self-renewing, tripotent neural precursor cells. Proc. Natl. Acad. Sci. U.S.A. 109, 2527–2532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pang Z. P., Yang N., Vierbuchen T., Ostermeier A., Fuentes D. R., Yang T. Q., Citri A., Sebastiano V., Marro S., Südhof T. C., Wernig M. (2011) Induction of human neuronal cells by defined transcription factors. Nature 476, 220–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vierbuchen T., Ostermeier A., Pang Z. P., Kokubu Y., Südhof T. C., Wernig M. (2010) Direct conversion of fibroblasts to functional neurons by defined factors. Nature 463, 1035–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jiang Y., Yu V. C., Buchholz F., O'Connell S., Rhodes S. J., Candeloro C., Xia Y. R., Lusis A. J., Rosenfeld M. G. (1996) A novel family of Cys-Cys, His-Cys zinc finger transcription factors expressed in developing nervous system and pituitary gland. J. Biol. Chem. 271, 10723–10730 [DOI] [PubMed] [Google Scholar]
- 14. Gamsjaeger R., Swanton M. K., Kobus F. J., Lehtomaki E., Lowry J. A., Kwan A. H., Matthews J. M., Mackay J. P. (2008) Structural and biophysical analysis of the DNA binding properties of myelin transcription factor 1. J. Biol. Chem. 283, 5158–5167 [DOI] [PubMed] [Google Scholar]
- 15. Cai M., Huang Y., Sakaguchi K., Clore G. M., Gronenborn A. M., Craigie R. (1998) An efficient and cost-effective isotope labeling protocol for proteins expressed in Escherichia coli. J. Biomol. NMR 11, 97–102 [DOI] [PubMed] [Google Scholar]
- 16. Wolf P. (1983) A critical reappraisal of Wadell's technique for ultraviolet spectrophotometric protein estimation. Anal. Biochem. 129, 145–155 [DOI] [PubMed] [Google Scholar]
- 17. Seavey B. R., Farr E. A., Westler W. M., Markley J. L. (1991) A relational database for sequence-specific protein NMR data. J. Biomol. NMR 1, 217–236 [DOI] [PubMed] [Google Scholar]
- 18. Ko L. J., Prives C. (1996) p53: puzzle and paradigm. Genes Dev. 10, 1054–1072 [DOI] [PubMed] [Google Scholar]
- 19. Cordier F., Dingley A. J., Grzesiek S. (1999) A doublet-separated sensitivity-enhanced HSQC for the determination of scalar and dipolar one-bond J-couplings. J. Biomol. NMR 13, 175–180 [DOI] [PubMed] [Google Scholar]
- 20. Zweckstetter M., Hummer G., Bax A. (2004) Prediction of charge-induced molecular alignment of biomolecules dissolved in dilute liquid-crystalline phases. Biophys. J. 86, 3444–3460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. van Dijk A. D., Bonvin A. M. (2006) Solvated docking: introducing water into the modelling of biomolecular complexes. Bioinformatics 22, 2340–2347 [DOI] [PubMed] [Google Scholar]
- 22. van Dijk M., van Dijk A. D., Hsu V., Boelens R., Bonvin A. M. (2006) Information-driven protein-DNA docking using HADDOCK: it is a matter of flexibility. Nucleic Acids Res. 34, 3317–3325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dominguez C., Boelens R., Bonvin A. M. (2003) HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 125, 1731–1737 [DOI] [PubMed] [Google Scholar]
- 24. Macke T., Case D. A. (1998) Modeling unusual nucleic acid structures in Molecular Modeling of Nucleic Acids (Leontis N. B., Santa Lucia J., Jr., eds), pp. 379–393, American Chemical Society, Washington [Google Scholar]
- 25. Iwahara J., Tang C., Marius Clore G. (2007) Practical aspects of 1H transverse paramagnetic relaxation enhancement measurements on macromolecules. J. Magn. Reson. 184, 185–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. de Vries S. J., van Dijk A. D., Krzeminski M., van Dijk M., Thureau A., Hsu V., Wassenaar T., Bonvin A. M. (2007) HADDOCK versus HADDOCK: new features and performance of HADDOCK2.0 on the CAPRI targets. Proteins 69, 726–733 [DOI] [PubMed] [Google Scholar]
- 27. Lu X. J., Olson W. K. (2003) 3DNA: a software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res. 31, 5108–5121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Berkovits-Cymet H. J., Amann B. T., Berg J. M. (2004) Solution structure of a CCHHC domain of neural zinc finger factor-1 and its implications for DNA binding. Biochemistry 43, 898–903 [DOI] [PubMed] [Google Scholar]
- 29. Berkovits H. J., Berg J. M. (1999) Metal and DNA binding properties of a two-domain fragment of neural zinc finger factor 1, a CCHC-type zinc binding protein. Biochemistry 38, 16826–16830 [DOI] [PubMed] [Google Scholar]
- 30. Besold A. N., Lee S. J., Michel S. L., Sue N. L., Cymet H. J. (2010) Functional characterization of iron-substituted neural zinc finger factor 1: metal and DNA binding. J. Biol. Inorg. Chem. 15, 583–590 [DOI] [PubMed] [Google Scholar]
- 31. Hagiwara T., Tanaka K., Takai S., Maeno-Hikichi Y., Mukainaka Y., Wada K. (1996) Genomic organization, promoter analysis, and chromosomal localization of the gene for the mouse glial high-affinity glutamate transporter Slc1a3. Genomics 33, 508–515 [DOI] [PubMed] [Google Scholar]
- 32. Wilmsdorff M. V., Blaich C., Zink M., Treutlein J., Bauer M., Schulze T., Schneider-Axmann T., Gruber O., Rietschel M., Schmitt A., Falkai P. (2013) Gene expression of glutamate transporters SLC1A1, SLC1A3 and SLC1A6 in the cerebellar subregions of elderly schizophrenia patients and effects of antipsychotic treatment. World J. Biol. Psychiatry 14, 490–499 [DOI] [PubMed] [Google Scholar]
- 33. Kim E. J., Hori K., Wyckoff A., Dickel L. K., Koundakjian E. J., Goodrich L. V., Johnson J. E. (2011) Spatiotemporal fate map of neurogenin1 (Neurog1) lineages in the mouse central nervous system. J. Comp. Neurol. 519, 1355–1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Murray R. C., Tapscott S. J., Petersen J. W., Calof A. L., McCormick M. B. (2000) A fragment of the Neurogenin1 gene confers regulated expression of a reporter gene in vitro and in vivo. Dev. Dyn. 218, 189–194 [DOI] [PubMed] [Google Scholar]
- 35. Kameyama T., Matsushita F., Kadokawa Y., Marunouchi T. (2011) Myt/NZF family transcription factors regulate neuronal differentiation of P19 cells. Neurosci. Lett. 497, 74–79 [DOI] [PubMed] [Google Scholar]
- 36. Ayed A., Mulder F. A., Yi G. S., Lu Y., Kay L. E., Arrowsmith C. H. (2001) Latent and active p53 are identical in conformation. Nat. Struct. Biol. 8, 756–760 [DOI] [PubMed] [Google Scholar]
- 37. Goddard T. D., Kneller D. G. (2007) SPARKY 3 University of California, San Francisco [Google Scholar]