

Background: 3-Hydroxybenzoate 6-hydroxylase (3HB6H) is a flavoprotein hydroxylase catalyzing the para-hydroxylation of 3-hydroxybenzoate.
Results: The oxidative half-reaction was studied using transient kinetics. The enzyme reaction mechanism was elucidated.
Conclusion: The product release and the hydroxylation limit the turnovers and the enzyme shows characteristics different from the ortho-hydroxylation enzymes.
Significance: This is the first report on the oxygenation mechanism of a para-hydroxylating flavoenzyme.
Keywords: Enzyme Kinetics, Enzyme Mechanisms, Flavin, Flavoproteins, Hydroxylase
Abstract
3-Hydroxybenzoate 6-hydroxylase (3HB6H) from Rhodococcus jostii RHA1 is an NADH-specific flavoprotein monooxygenase that catalyzes the para-hydroxylation of 3-hydroxybenzoate (3HB) to form 2,5-dihydroxybenzoate (2,5-DHB). Based on results from stopped-flow spectrophotometry, the reduced enzyme-3HB complex reacts with oxygen to form a C4a-peroxy flavin with a rate constant of 1.13 ± 0.01 × 106 m−1 s−1 (pH 8.0, 4 °C). This intermediate is subsequently protonated to form a C4a-hydroperoxyflavin with a rate constant of 96 ± 3 s−1. This step shows a solvent kinetic isotope effect of 1.7. Based on rapid-quench measurements, the hydroxylation occurs with a rate constant of 36 ± 2 s−1. 3HB6H does not exhibit substrate inhibition on the flavin oxidation step, a common characteristic found in most ortho-hydroxylation enzymes. The apparent kcat at saturating concentrations of 3HB, NADH, and oxygen is 6.49 ± 0.02 s−1. Pre-steady state and steady-state kinetic data were used to construct the catalytic cycle of the reaction. The data indicate that the steps of product release (11.7 s−1) and hydroxylation (36 ± 2 s−1) partially control the overall turnover.
Introduction
Monooxygenases or hydroxylases are useful biocatalysts for catalyzing regio-specific oxygenation by molecular oxygen under mild conditions. Many hydroxylation reactions of aromatic compounds found in nature are carried out by flavin-dependent enzymes that belong to the class of external flavoprotein monooxygenases that require NADH or NADPH as an external reductant (1–3). The overall reaction of these enzymes can be divided into a reductive half-reaction, in which two electrons are transferred from NAD(P)H to the enzyme-bound flavin, and an oxidative half-reaction in which the hydroxylation of aromatic substrate takes place. The overall reaction can be carried out within a single polypeptide (designated as Class A monooxygenases in Ref. 1) or by two-protein components (designated as Class D monooxygenases in Ref. 1). The current knowledge obtained from studying monooxygenases that catalyze ortho-hydroxylation reactions support a model in which a flavin adduct intermediate (C4a-hydroperoxyflavin) participates as an electrophile in the electrophilic aromatic substitution reaction to give a C4a-hydroxyflavin and a hydroxylated product (4–9). The best understood enzyme of this class is p-hydroxybenzoate 3-hydroxylase (PHBH),2 for which detailed studies on the enzyme structure and kinetics have revealed a great deal of complexity in the protein dynamics (3).
3-Hydroxybenzoate 6-hydroxylase (3HB6H) is one of the flavin-dependent enzymes that have been reported to catalyze para-hydroxylation of aromatic compounds (10). These para-hydroxylation enzymes are involved in the degradation pathways of phenolic compounds by bacteria (1, 11) or the biosynthetic pathways of antibiotics such as angucyclines and rhodomycin (12, 13). 3HB6H from Rhodococcus jostii RHA1 catalyzes the para-hydroxylation of 3-hydroxybenzoate to yield 2,5-dihydroxybenzoate (Fig. 1). The enzyme was cloned and expressed in Escherichia coli, and was shown to be a dimeric protein containing one FAD per 47-kDa subunit (10). The catalytic reaction of 3HB6H consists of two half-reactions similar to other single-component flavoprotein hydroxylases. Investigation on the kinetics of the reductive half-reaction has demonstrated that when bound to the enzyme, substrate and product act as effectors to increase the rate of flavin reduction by 2 orders of magnitude (14). It was also shown that 3HB6H catalyzes the regio-specific para-hydroxylation of a number of 3-hydroxybenzoate analogs (10). Therefore, 3HB6H is potentially useful for synthesizing gentisate compounds that are drugs or antioxidants (15, 16). Recently, the crystal structures of the wild-type and mutant 3HB6H enzymes were solved (17). Although the overall structure of 3HB6H is similar to other single-component flavoprotein aromatic hydroxylases, the arrangement of the active site residues is quite different from ortho-hydroxylation enzymes such as PHBH. The difference in the active site environment is presumably important for the regio-specific para-hydroxylation process (17).
FIGURE 1.
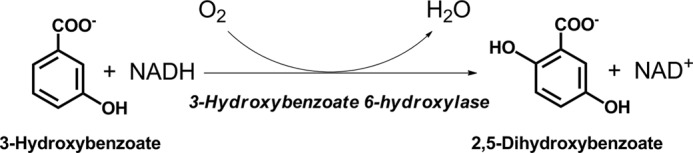
Catalytic reaction of 3HB6H.
Up to now, there have been no reports on the kinetic mechanisms related to the overall catalytic cycle of a para-hydroxylation flavoenzyme. Therefore, we set out to investigate the kinetics of the oxidative half-reaction of 3HB6H from R. jostii RHA1 using stopped-flow and rapid-quench flow techniques. Solvent kinetic isotope effects were used to identify the steps that involved proton transfer. The results provide an understanding of the oxygenation mechanism of 3HB6H and reveal different catalytic features between para- and ortho-hydroxylating flavoenzymes.
EXPERIMENTAL PROCEDURES
Reagents
NADH (purity ≥95%) and FAD (purity ≥95%) were purchased from Sigma. 3-Hydroxybenzoic acid (3HB) and 2,5-dihydroxybenzoic acid (2,5-DHB) were purchased from Merck. The concentrations of compounds were determined using the following absorption coefficients at pH 8.0; NADH, ϵ340 = 6.22 × 103 m−1 cm−1; FAD, ϵ450 = 11.3 × 103 m−1 cm−1; 3HB, ϵ288 = 2.0 × 103 m−1 cm−1; and 2,5-DHB, ϵ320 = 4.1 × 103 m−1 cm−1. All of the absorption coefficient values are for the compounds in solution at pH 8.0. 3HB6H was purified according to the protocol previously described (14). Concentrations of the enzyme were determined using the known absorption coefficient ϵ452 = 11.00 ± 0.03 × 103 m−1 cm−1 (one FAD per subunit) (14).
Spectroscopic Studies
UV-visible absorbance spectra were recorded using a Hewlett-Packard diode array spectrophotometer (HP8453), a Shimadzu 2501PC spectrophotometer, or a Cary 300Bio double-beam spectrophotometer at 25 °C. All spectrophotometers were equipped with thermostated cell compartments. Typical assays contained 10 mm 3HB, 2 μm FAD, and 1 mm NADH. As this concentration NADH would give absorbance of ∼6 at 340 nm, we thus measured the decrease in absorbance at 395 nm (ϵ395 of 0.22 × 103 m−1 cm−1), which still gives good linearity for enzyme assays.
Steady-state Kinetics
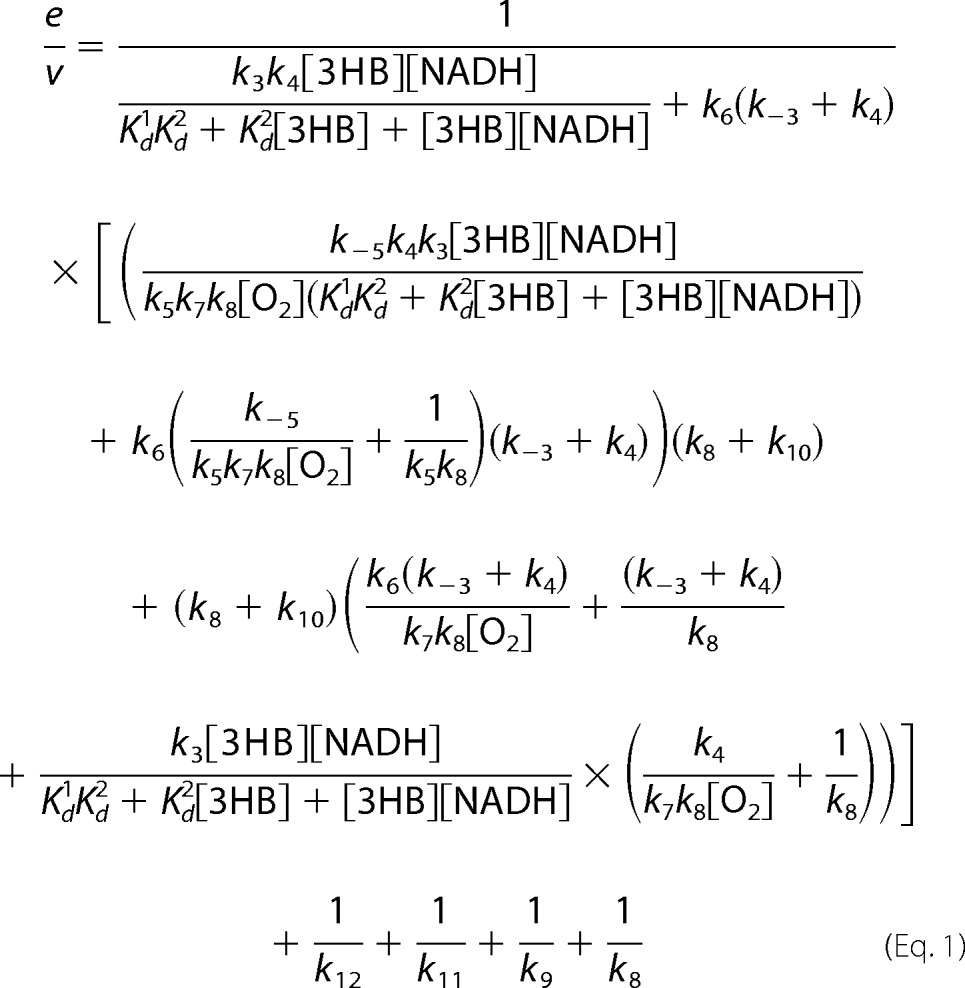
To determine an apparent catalytic constant (kcat) at 4 °C, a solution of the enzyme at 1 μm in the presence of 10 mm 3HB and 2 μm FAD in air saturation (0.26 mm oxygen) was mixed with an oxygenated buffer (1.92 mm oxygen) plus 10 mm 3HB, 10 mm NADH, and 2 μm FAD using a stopped-flow spectrophotometer. Final concentrations of the reagents after mixing were 0.5 μm enzyme, 10 mm 3HB, 5 mm NADH, and 1.09 mm oxygen. The apparent kcat from steady-state experiments was compared with the value calculated based on Equation 1 and individual rate constants obtained from rapid-kinetics experiments. Derivations of Equation 1 were carried out according to the method described by Cha (18) and Cramer's rule (19) (see derivations in supplemental Schemes S1 and S2).
 |
Rapid Reaction Experiments
Reactions were carried out in 100 mm Tris-H2SO4 (pH 8.0), 4 °C, unless otherwise specified. The measurements were performed using a TgK Scientific model SF-61DX or a TgK Scientific model SHU-61SX2 stopped-flow spectrophotometer in single-mixing mode. The stopped-flow apparatus was made anaerobic by flushing the flow system with an anaerobic buffer solution containing 0.5 mg/ml of dithionite in 100 mm sodium phosphate (pH 7.0), and equilibrated in the dithionite solution overnight. The flow system of the stopped-flow instrument was washed with anaerobic 100 mm Tris-H2SO4 (pH 8.0) three times before starting the experiments.
For preparation of a reduced enzyme solution, an anaerobic oxidized enzyme solution was placed in a tonometer and reduced with an equivalent amount of dithionite (0.2 mg/ml in 100 mm Tris-H2SO4, pH 8.0), which was delivered from a gas-tight syringe with a microtitrator attached to the tonometer. The enzyme reduction was monitored through a cuvette attached to the tonometer to ensure a stoichiometric reduction process (20). The reduced enzyme solution was mixed with buffers containing various oxygen concentrations in the stopped-flow spectrophotometer. All oxygen concentrations used are at more than a 5-fold excess of the enzyme concentration to ensure pseudo-first order conditions.
Apparent rate constants (kobs) were calculated from the kinetic traces using exponential fits and the software packages Kinetic Studio (Hi-Tech Scientific, Salisbury, UK) and Program A (written at the University of Michigan by Rong Chang, Jung-yen Chiu, Joel Dinverno, and David P. Ballou). Rate constants were obtained from oxygen using Marquardt-Levenberg nonlinear fit algorithms included in KaleidaGraph (Synergy Software). Simulations were performed by numerical methods using Runge-Kutta algorithms implemented in Berkeley Madonna 8.3 and a time step of 2 × 10−4 s for simulations of the oxidative half-reaction of the enzyme-3HB complex. A five-step irreversible consecutive reaction model was used for simulations of the oxidative half-reaction.
Global analyses of absorption spectra acquired during oxidative half-reactions were performed using ReactLabTM KINETICS (Jplus Consulting Ltd.), a software package used for calculation of intermediate spectra using multistep irreversible consecutive reactions. The rate constants of each step were obtained from kinetic analysis of fits of single-wavelength kinetic traces.
To study solvent kinetic isotope effects (SKIE) on the oxidative half-reaction of 3HB6H, all enzyme solutions were exchanged into a Tris buffer made with deuterium oxide. In brief, Tris(hydroxymethyl)aminomethane (24.2 g) was dissolved in ∼30 ml of 99.9% deuterium oxide, and the resultant solution was equilibrated for 13–15 h (overnight) inside an anaerobic glove box (Belle Technology). The equilibrated solution was then evaporated at 60 °C for 2 h using a rotary evaporator to obtain H2O-free Tris(hydroxymethyl)aminomethane powder. The resultant powder was dissolved in 99.9% deuterium oxide and the same process was repeated to ensure that the buffer contained at least 99.9% D2O. The dried powder was then redissolved in ∼195 ml of 99.9% D2O. The buffer pD was adjusted by adding 1 m D2SO4 into the solution while monitoring the pD using a pH meter (pD = pH measured + 0.4) (21). The volume of the resulting buffer was adjusted to 200 ml with D2O to obtain 100 mm Tris-D2SO4 (pD 8.0). 3-Hydroxybenzoaic acid (3HB) (0.173 g) was dissolved in ∼20 ml of 99.9% D2O. The pD of the 3HB solution was adjusted to 8.0 by adding 0.1 m sodium deuterium oxide (NaOD). The solutions were dried twice as described above using a rotary evaporator. The dried powder of 3HB was re-dissolved in 25 ml of D2O to obtain 50 mm 3HB in 100 mm Tris-D2SO4 (pD 8.0). To avoid an exchange of deuterium with protium from air moisture, all processes were performed inside an anaerobic glove box. To prepare enzyme in D2O buffer, the concentrated enzyme solution (700 μl, A452 ∼ 5.1) was equilibrated inside the anaerobic glove box for 30 min to remove oxygen. The solution was loaded onto a PD-10 column equilibrated with 100 mm Tris-D2SO4 (pD 8.0) and the enzyme was eluted with D2O buffer. The eluted enzyme solution was mixed with a solution of 3HB prepared in the same D2O buffer and the volume was adjusted to ∼6 ml to obtain a final absorbance at 452 nm of ∼0.6 and a final concentration of 3HB ∼10 mm. The enzyme-3HB complex in D2O buffer was reduced by adding a solution of 0.5 mg/ml of dithionite solution, which was dissolved in D2O buffer. Enzyme reduction was monitored using a spectrophotometer inside the anaerobic glove box to ensure stoichiometric reduction. The solution of reduced enzyme-3HB complex was transferred into a tonometer and left overnight (∼18 h) at 4 °C prior to the stopped-flow experiment. This preparation process was to assure that the enzyme was fully equilibrated in D2O and that all of the exchangeable sites on the reduced enzyme had incorporated deuterium.
Analysis of the Hydroxylation Reaction Using Rapid-quench Flow Techniques
The experiments were performed using a TgK Scientific model RQF-63, DimentionTM D1 rapid quench-flow system in an anaerobic glove box. The rapid-quench flow system consisted of three syringes. Syringe A contained an anaerobic solution of 25 μm reduced enzyme plus 10 mm 3HB. Syringe B contained a solution of quencher, 0.15 m HCl. Syringe C contained an air-saturated buffer (0.26 mm oxygen) plus 10 mm 3HB. The solution of reduced enzyme-3HB complex was mixed with the air-saturated buffer and the reaction mixture was allowed to age for various periods of time: 0.012, 0.015, 0.019, 0.024, 0.030, 0.037, 0.040, 0.046, 0.058, 0.062, 0.068, 0.090, 0.093, 0.134, 0.150, 0.230, 0.330, 0.430, or 0.530 s, before being quenched with 0.15 m HCl solution. Quenched samples were collected from the sample loop, and the enzyme was separated using a Microcon unit (Amicon YM-10). A solution of 1 m HCl was added to the filtrates to give a final concentration of 0.5 m HCl and the samples were analyzed for the amount of 2,5-DHB produced from the reaction using an HPLC (Agilent 1100 Series) with a 3.9 × 150-mm Nova-pak C18 reverse-phase column (Waters) and detected by a photodiode array detector. The column was equilibrated with 5% methanol and 0.1% formic acid in H2O as a mobile phase (flow rate of 0.5 ml/min) before sample injection. A gradient of methanol (increased from 5 to 40% in 20 min) and 0.1% formic acid was used to separate 3HB and 2,5-DHB. 2,5-DHB was identified by the absorbance at 330 nm and eluted at the retention time of 14.8 min after injection. The concentrations of product formed were estimated based on a standard curve in the range of 2–40 μm 2,5-DHB.
RESULTS
Reaction of Free Reduced Enzyme with O2 and the Effects of NAD+
A reduced enzyme solution was mixed with buffers containing various oxygen concentrations. Flavin oxidation was monitored at 10-nm intervals from 300 to 600 nm. No signs of formation of the intermediate C4a-hydroperoxyflavin were detected, as an increase of absorbance at all wavelengths was a single exponential (Fig. 2A). A plot of the observed rate constants versus oxygen concentrations is linear without a significant intercept value, corresponding with a second-order rate constant of 4.96 ± 0.4 × 103 m−1 s−1 (Fig. 2A, inset).
FIGURE 2.
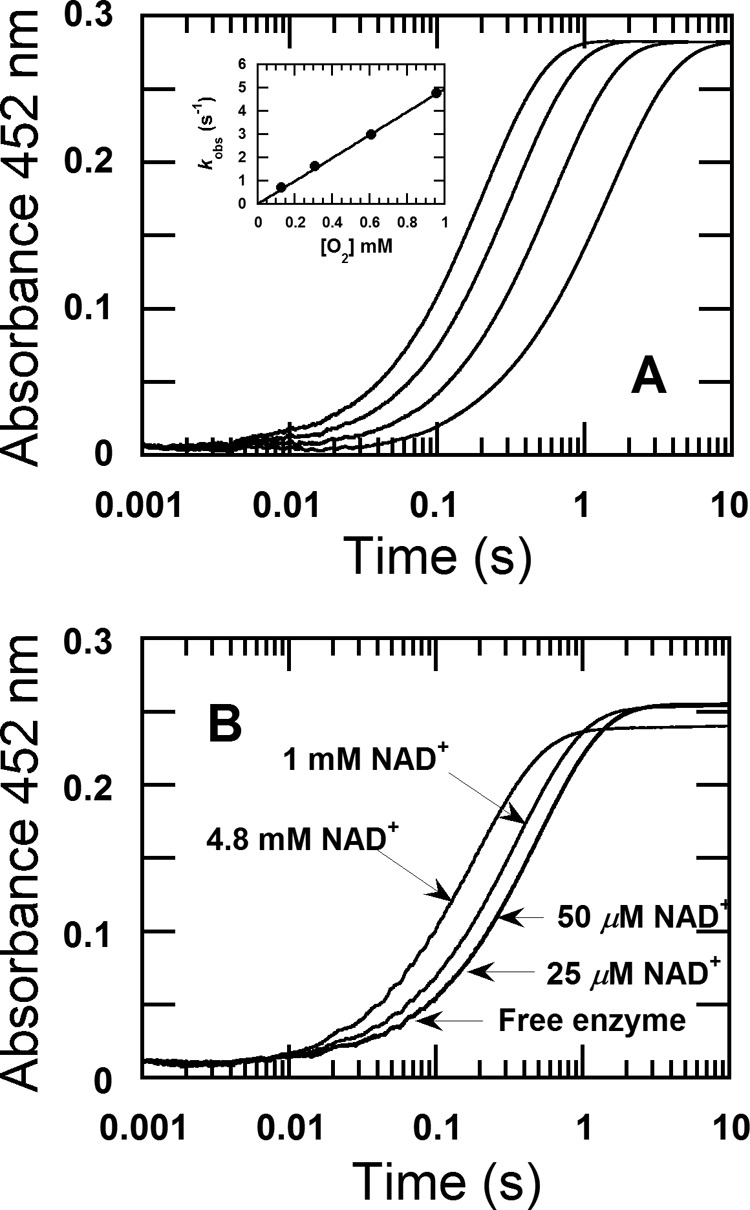
Reaction of free reduced 3HB6H enzyme with oxygen and the effect of NAD+ on the re-oxidation. A, a solution of the reduced enzyme (26 μm) was mixed with buffer containing various oxygen concentrations, 0.13, 0.31, 0.61, and 0.96 mm in 100 mm Tris-H2SO4 (pH 8.0) at 4 °C. All given concentrations are after mixing. The reaction was monitored at a wavelength of 452 nm with a stopped-flow spectrophotometer. The kinetic traces from left to right correspond to decreasing oxygen concentrations. Inset shows a plot of the observed rate constants versus oxygen concentrations. B, a solution of the reduced enzyme (25 μm) was mixed with different NAD+ concentrations of 25 μm, 50 μm, 1 mm, and 4.8 mm (indicated by arrows) under anaerobic conditions in the first mixing. All concentrations are given as after mixing performed in 100 mm Tris-H2SO4 (pH 8.0) at 4 °C. The mixture solution of reduced enzyme and NAD+ was aged for 200 s and then the reaction was mixed with 0.61 mm oxygen in the second mixing. The oxidation reactions were monitored at 452 nm.
The effect of NAD+ on the above reaction was investigated by mixing a solution of the reduced enzyme plus NAD+ with oxygen-containing buffer using the double-mixing mode of the stopped-flow spectrophotometer. For the first mixing, the reduced enzyme (100 μm before mixing) was mixed with various concentrations of NAD+ of 100 μm, 200 μm, 4 mm, and 19.2 mm (concentrations before mixing) under anaerobic conditions. The resulting mixture was aged for 200 s to allow any binding of NAD+ to take place before being mixed with buffer containing 1.92 mm oxygen (concentration before mixing) at the second mixing step. The final concentration of the enzyme was 25 μm, and those of NAD+ were 25 μm, 50 μm, 1 mm, and 4.8 mm. The reactions were monitored at 452 nm. The control reaction in which the reduced enzyme was mixed with anaerobic buffer without NAD+ in the first mixing was also carried out. The results showed that the kinetic traces of the reaction of free enzyme without NAD+, the reaction containing 25 μm NAD+, and the reaction containing 50 μm NAD+ were nearly the same, with observed rate constants of 1.98 ± 0.006, 2.11 ± 0.03, and 2.20 ± 0.07 s−1, respectively (as indicated by the arrow in Fig. 2B). These data indicated that low concentrations of NAD+ have no significant effect on the rate of enzyme oxidation. However, when NAD+ concentrations were increased to 1 and 4.8 mm, the rate of enzyme oxidation was affected as the observed rate constants were 2.77 ± 0.07 and 5.23 ± 0.12 s−1, respectively. These results imply that NAD+ shows weak or nonspecific binding to the reduced enzyme. However, as the reductive half-reaction only produces 1 eq of NAD+, the data suggest that under turnover, NAD+ hardly binds to reduced enzyme and likely leaves prior to the oxygen reaction.
Reaction of Reduced Enzyme-3HB Complex with Oxygen Detected by Stopped-flow Spectrophotometry
A solution of the reduced enzyme plus 10 mm 3HB was mixed with buffers containing 10 mm 3HB and various oxygen concentrations in the stopped-flow spectrophotometer. Flavin oxidation was monitored at 10-nm intervals from 300 to 600 nm. In general, C4a-adduct intermediates (C4a-hydroperoxyflavin or C4a-hydroxyflavin) have their absorbance peaks around 360–410 nm with little absorption around 450 nm region. Therefore, formation and decay of C4a-hydroperoxyflavin and C4a-hydroxyflavin were detected at 400 nm, whereas flavin oxidation was monitored at 452 nm (Fig. 3, A and B). At the initial time of measurement (0.002 s), the absorbance detected was different from absorbance of the reduced enzyme, indicating that part of the reaction of the reduced enzyme-3HB complex and oxygen was rapid and occurred during the dead time of the stopped-flow mixing. The kinetics showed five exponential phases. At the highest oxygen concentration of 0.96 mm, where separation of each kinetic phase was the clearest, the first phase (0.002–0.003 s) was characterized by an increase in absorbance at 400 nm (Fig. 3A) without an absorbance change at 452 nm. The plot of kobs versus oxygen concentration was linear, yielding a second-order rate constant of 1.13 ± 0.01 × 106 m−1 s−1 (Fig. 3B, inset). The second phase (0.003–0.017 s) was characterized by a decrease in absorbance at 400 nm (Fig. 3A). The observed rate constant of this phase was independent of oxygen concentration with an observed rate constant of 96 ± 3 s−1. The kinetics of the decrease in absorbance at 400 nm of this phase was concurrent with an increase in absorbance at 452 nm (∼16% change in the total amplitude) (Fig. 3B). The third phase (0.017–0.060 s) showed an increase in absorbance at 400 nm independent of the oxygen concentration, and was consistent with an observed rate constant of 36 ± 2 s−1 (Fig. 3A). This phase was also characterized by a major concomitant increase in absorbance at 452 nm (∼82% change in the total amplitude).
FIGURE 3.

Reaction of reduced 3HB6H enzyme in the presence of 3HB with oxygen. A solution of the reduced enzyme (30 μm) plus 10 mm 3HB was mixed with buffer containing various oxygen concentrations of 0.13, 0.31, 0.61, and 0.96 mm, plus 10 mm 3HB in the stopped-flow spectrophotometer. All concentrations as described were after mixing. The reaction was performed in 100 mm Tris-H2SO4 (pH 8.0) at 4 °C. A, the reaction was monitored by absorbance change at 400 nm to detect formation of C4a-flavin adduct intermediates and B, at 452 nm for detecting flavin oxidation. The lower to upper kinetic traces at 400 nm correspond to increasing oxygen concentrations. The dotted lines are from simulations using a four-step consecutive reaction according to a → b → c → d → e → f. The rate constants of each step used for simulations are according to those indicated in Table 1, k7 = 1.6 × 106 m−1 s−1, k8 = 83 s−1, k9 = 36 s−1, k10 = 13 s−1, k12 = 11.7 s−1, k13 = 0.92 s−1, and the molar absorption coefficients used for the simulations were ϵ400 of a (Ered) = 3,300 m−1 cm−1, ϵ400 of b (E-C4a-peroxyflavin) = 7,500 m−1 cm−1, ϵ400 of c (E-C4a-hydroperoxyflavin) = 6,300 m−1 cm−1, ϵ400 of d (Eox-2,5-DHB) = 6,800 m−1 cm−1, ϵ400 of e (E′ox) = 6,900 m−1 cm−1, ϵ400 of (Eox) = 6,910 m−1 cm−1 and at 452 nm, ϵ452 of a = 1,200 m−1 cm−1, ϵ452 of f b = 1,300 m−1 cm−1, ϵ452 of c = 1,400 m−1 cm−1, ϵ452 of d = 10,300 m−1 cm−1, ϵ452 of e = 10,600 m−1 cm−1, ϵ452 of f = 11,000 m−1 cm−1. Inset in A shows a plot of product formed versus time obtained from rapid quench-flow experiments (filled circles) under the same conditions as those in the stopped-flow experiment. Simulations of product formation according to the model used in the main figures but with the observed hydroxylation rate constant of 36 s−1 at the second step or 96 s−1 at the third step are shown in solid and dotted lines, respectively. Inset in B shows a plot of the observed rate constants versus oxygen concentrations.
The fourth phase (0.13–0.28 s) is a small amplitude change at 452 nm (∼3% of total amplitude change at 452 nm), which can be fitted with a rate consent of 8–12 s−1 (Fig. 3B). This phase is proposed to be a product release from the oxidized enzyme (this conclusion was later confirmed under “Discussion”). The trend of absorbance increase upon product leaving is also consistent with absorbance characteristics of the Eox-product complex and free Eox previously observed in Ref. 14. The fifth phase was a slow phase (∼0.92 s−1) with a small absorbance increase at 452 nm (∼2% of the total amplitude change) (Fig. 3B). This phase might result from a small fraction of inactive enzyme because the rate constant of this step is much slower than the overall turnover number of 6.49 ± 0.02 s−1 (Table 1). Therefore, the observed fourth phase was not included in the interpretation of the reaction mechanism. When the reduced enzyme-3HB complex was prepared using NADH as the reductant (stoichiometric reduction), and used in the same experiment as described above, the reduced enzyme-3HB complex in the presence of NAD+ showed the same results (data not shown). These data again confirm that NAD+ does not bind to the reduced enzyme-3HB complex.
TABLE 1.
Values of rate constants described in Figs. 4 and 9
The values were obtained from experimental data performed in 100 mm Tris-H2SO4 (pH 8.0) at 4 °C using the stopped-flow spectrophotometer or kinetic simulations.
| Observed rate constants from experimental data | Rate constants from simulations |
|---|---|
| k1 = 2.6 ± 0.03 × 105 m−1s−1a | k1 = 4 × 105 m−1s−1a |
| k−1 = 65 ± 3 s−1a | k−1 = 64 ± 3 s−1a |
| k2 = 4.4 × 106 m−1s−1a | |
| k−2 = 7600 s−1a | |
| k3 = 377 ± 8 s−1a | k3 = 340 s−1a |
| k−3 = 12 s−1a | |
| k4 = 48 ± 2 s−1a | k4 = 51 s−1a |
| k5 = 7.3 ± 0.5 × 102 m−1s−1b | |
| k−5 = 0.056 s−1 | |
| k6 = 43 ± 2 m−1s−1a | |
| k7 = 1.13 ± 0.01 × 106 m−1s−1 | k7 = 1.6 × 106 m−1s−1c |
| k8 + k10 = 96 ± 3 s−1 | |
| k8 = 83 s−1d | k8 = 83 s−1c |
| k9 = 36 ± 2 s−1 | k9 = 36c |
| k10 = 13 s−1d | k10 = 13 s−1 |
| k11 (−NaN3) = NDe | |
| k11 (+NaN3) = 9.9 s−1 | |
| k12 = 11.7 s−1f | |
| k13 (−NaN3) = 0.92 s−1g | |
| k13 (+NaN3) = 0.35 s−1g | |
| kH2Ocat(app)= 6.49 ± 0.02 s−1 | |
| kD20cat(app)> = 5.43 ± 0.04 s−1 |
a Rate constants from simulations and experimental data were according to the previous report (14).
b The apparent bimolecular rate constant.
c The values from simulations according to “Experimental Procedures” and Fig. 3.
d The values calculate from percent coupling (86%).
e ND, not determined.
f The value was calculated from kcat and Equation 2.
g The rate constant of fifth phase in Fig. 3, which is not in the catalytic cycle but only used for simulation with the experimental traces.
Reaction of Reduced Enzyme-3HB Complex with Oxygen Detected by Rapid-quench Techniques
Based on the stopped-flow data alone, the interpretation of each kinetic phase could not be assigned. Therefore, rapid-quench flow techniques (“Experimental Procedures”) were used to identify the hydroxylation rate constant. The results of the rapid-quench experiments gave a kinetic trace for product formation (Fig. 3A, inset, filled circle). For the stopped-flow experiment in which the oxygen concentration was the same as the rapid-quench experiment (0.13 mm after mixing), the observed rate constant of the first step (formation of C4a-hydroperoxyflavin) was 161 s−1 (kobs1), that of the second step was 96 ± 3 s−1, and that of the third step was 36 ± 2 s−1. Therefore, simulations of product formation using a four-step irreversible consecutive reaction model with the hydroxylation rate constant of 96 s−1 (the second phase as the hydroxylation step, Fig. 3A, inset, dotted line) or 36 s−1 (the third phase as the hydroxylation step, Fig. 3A, inset, solid line) were carried out to identify the correlation between the rate constants measured from the stopped-flow and rapid-quench experiments. The comparison of rapid-quench data and simulations show that the simulation trace using the third step as the hydroxylation step (solid line) fits well with the experimental data, whereas the other trace does not. Therefore, the third kinetic phase of the stopped-flow data reflects the step involved with product formation. As this phase is concurrent with flavin oxidation, it implies that the rate of C4a-hydroxyflavin decay is much faster than the rate of C4a-hydroxyflavin formation, thus preventing accumulation and detection of this intermediate. As a result, the overall kinetic mechanism of the oxidative half-reaction of 3HB6H can be summarized according to Fig. 4.
FIGURE 4.

The oxygenation reaction of 3HB6H. The first step is formation of C4a-peroxy anion. The second step is a parallel reaction of protonation of a peroxy group to form C4a-hydroperoxyflavin and an uncoupling path for hydrogen peroxide elimination. The third step is a reaction of hydroxylation. The fourth step is dehydration of water from C4a-hydroxyflavin to return to the oxidized flavin, of which the reaction is very fast, preventing the detection of C4a-hydroxyflavin.
According to the model in Fig. 4, two forms of C4a-flavin adduct exist prior to the hydroxylation step. The first step is proposed to be formation of the C4a-peroxyflavin anion with an observed rate constant of 161 s−1 (k1). The second step is bifurcation of the pathways of coupling (hydroxylation) and uncoupling (non-hydroxylation) paths. For the coupling path (82%), the proton transfer occurs to form the C4a-hydroperoxyflavin-3HB complex, whereas for the uncoupling path (16%), the H2O2 elimination takes place rapidly after the proton transfer to form the oxidized enzyme. The existence of the uncoupling pathway is also supported by results of product analysis and diode array detection (shown later). The observed rate constant of this phase (96 ± 3 s−1 (k2)) is combined rate constants from both pathways and the spectrum c in Fig. 6 is combined characteristics of C4a-hydroperoxyflavin-3HB complex and oxidized flavin. The third step is the hydroxylation step with a rate constant of 36 ± 2 s−1 (Table 1). The fourth step is the dehydration of C4a-hydroxyflavin to form oxidized flavin and H2O. Because the hydroxylation step was observed at the same time as the majority of the flavin oxidation (∼82% of the total amplitude change, Fig. 3, A and B), the data imply that the dehydration step (step 4, Fig. 4) is rapid and could not be detected as a separate kinetic step.
FIGURE 6.

Spectra of intermediates in the reaction of reduced enzyme-3HB with oxygen. Reactions similar to those in Fig. 3 were monitored using the stopped-flow diode-array spectrometer. All reactions were performed in 100 mm Tris-H2SO4 (pH 8.0) at 4 °C. A, a solution of the reduced enzyme (32 μm) plus 10 mm 3HB was mixed with 0.96 mm oxygen plus 10 mm 3HB. All concentrations as described were after mixing. The solid line spectrum represents the reduced enzyme-3HB complex (a). Filled circle and open circle lines are spectra of C4a-peroxyflavin anion (b) and C4a-hydroperoxyflavin (c), respectively. Spectrum d represents the oxidized enzyme-2,5-DHB complex. Spectra e and f represent different forms of the oxidized enzyme-3HB complex.
Reaction of Reduced Enzyme-3HB Complex with Oxygen in the Presence of Sodium Azide
It is known that the presence of inhibitory anions such as azide can slow down the dehydration step of C4a-hydroxyflavin in single-component flavoprotein hydroxylases (22–25). Therefore, we performed the experiment shown in Fig. 3 in the presence of sodium azide (NaN3) so that the identity of the C4a-hydroxyflavin could be resolved. At a concentration of 0.96 mm oxygen, the reaction containing 50 mm NaN3 showed a kinetic trace with five exponential phases. For the kinetic traces detected at 400 nm (Fig. 5A, open circle line), the first phase (0.002–0.003), the second phase (0.003–0.017 s), and the third phase (0.017–0.060 s) gave observed rate constants of 1500, 96, and 36 s−1 (the rate constant for hydroxylation), respectively, which are the same as for the reaction in the absence of sodium azide (Fig. 3, Table 1). For the traces detected at 452 nm, the data showed multiphasic kinetics similar to those observed at 400 nm. A notable effect of sodium azide addition was the presence of another intermediate that formed with the same rate constant as the hydroxylation step and decayed with a rate constant of 9.9 ± 0.2 s−1. The decay step was concurrent with ∼64% of the total absorbance increase at 452 nm. Therefore, this intermediate (Fig. 5B, open circle line) was assigned as C4a-hydroxyflavin that dehydrates to form oxidized flavin. The fifth phase is a slow phase with a small absorbance increase at 452 (∼2% of amplitude change) with a rate constant of 0.35 s−1, which is probably due to a small inactive enzyme fraction similar to that observed in the reaction in the absence of azide. Altogether, the data clearly support the kinetic model proposed in Fig. 4. The same experiments as in Figs. 3 and 5 but with fluorescence detection were also carried out. However, the data were not useful for kinetic interpretation because only oxidized enzyme is fluorescent, whereas none of the flavin intermediates give fluorescence signals.
FIGURE 5.

Reaction of reduced enzyme-3HB complex with oxygen in the presence of sodium azide. A solution of the reduced enzyme (23 μm) containing 10 mm 3HB and 50 mm sodium azide was mixed with buffer containing 10 mm 3HB, 50 mm sodium azide, and 0.96 mm oxygen. All concentrations described were after mixing. The reactions were performed in 100 mm Tris-H2SO4 (pH 8.0) at 4 °C. A, the reactions were monitored by absorbance change at 400 nm to monitor C4a-flavin adduct species. The open circle line was the kinetic trace in the presence of 50 mm sodium azide. Sodium azide slows down the rate of C4a-hydroxyflavin decay (from >36 s−1 to 9.9 ± 0.2 s−1) so that the existence of this intermediate can be identified (at 0.05 s) when compared with the same reaction without sodium azide (solid line trace). Simulations of the reaction in the presence of sodium azide are shown in dotted line, which is superimposed to the open circle line. The simulations used a five-step consecutive reaction according to a model, a → b → c → d → e → f. Rate constants for each step used for simulations are according to those indicated in Table 1, k7 = 1.6 × 106 m−1 s−1, k8 = 83 s−1, k9 = 36 s−1, k10 = 13 s−1, k11 (+NaN3) = 10 s−1, k13 = 0.35 s−1 and the molar absorption coefficients used for simulations were ϵ400 of a (Ered) = 4,100 m−1 cm−1, ϵ400 of b (E-C4a-peroxyflavin) = 8,100 m−1 cm−1, ϵ400 of c (E-C4a-hydroperoxyflavin) = 7,500 m−1 cm−1, ϵ400 of d (E-C4a-hydroxyflavin) = 8,500 m−1 cm−1, ϵ400 of e (E′ox) = 6,880 m−1 cm−1, ϵ400 of f (Eox) = 6,887 m−1 cm−1 and at 452 nm, ϵ452 of a = 1,300 m−1 cm−1, ϵ452 of b = 1,700 m−1 cm−1, ϵ452 of c = 2,100 m−1 cm−1, ϵ452 of d = 5,900 m−1 cm−1, ϵ452 of e = 10,700 m−1 cm−1, and ϵ452 of f = 11,000 m−1 cm−1. B, the reactions were monitored by absorbance change at 452 nm to detect flavin oxidation. The open circle line and the solid line were kinetic traces for the reaction with and without 50 mm sodium azide, respectively.
Product Formation and Hydroxylation Ratio
The model in Fig. 4 indicates that a fraction of C4a-hydroperoxyflavin bifurcates to form H2O2 without hydroxylating the substrate. Therefore, the percentage of 2,5-DHB formed per NADH consumed (coupling ratio) at pH 8.0 under the same conditions as the stopped-flow experiment was determined using an HPLC method (“Experimental Procedures”). The reaction was set with a limiting concentration of NADH (60 μm 3HB6H, 50 μm NADH, 10 mm 3HB) such that the reaction only occurs under single turnover. The results gave a coupling ratio of 86%, which is consistent with the uncoupling ratio of ∼16% observed in Figs. 3 and 4. As the rate constant observed for decay of flavin hydroperoxide is the combined rate constant of the parallel reactions of H2O2 elimination. It is assumed that the ∼86% fraction of total enzyme carried out with hydroxylation, whereas the ∼14% fraction was eliminated as hydrogen peroxide. Therefore, the rate constant for hydrogen peroxide elimination (kH2O2) at the second step was ∼13 s−1 (k10 in Fig. 9). Simulations according to the model in Fig. 4 agree well with the experimental data (Fig. 3, dashed lines), validating the model in Fig. 4 and the rate constants in Table 1.
FIGURE 9.

The kinetic mechanism for the overall reaction of 3HB6H. Reaction of 3HB and NADH to the oxidized enzyme can be described as a branched bimolecular mechanism. However, the binding of 3HB to the oxidized enzyme is more preferred (major path). This model was confirmed by kinetic simulations (shown in Fig. 3).
Spectra of Flavin Intermediates Involved in the Reoxidation of 3HB6H
A similar experiment as those shown in Fig. 3, A and B, in the presence of 3HB substrate was carried out, but with diode-array detection (“Experimental Procedures”). Global analysis of the data according to the model: a → b → c → d → e → f, and with rate constants from the analysis of Fig. 3, A and B, and from the simulations in Table 1 were used to identify the spectra of flavin intermediates involved in the reaction. Species a and e are the reduced enzyme-3HB complex and the oxidized flavin, respectively (Fig. 6). The analysis yielded the filled circle line, which exhibits a maximum absorbance at 388 nm corresponding to the first intermediate (spectrum b in Fig. 6). Spectrum c (open circle), which corresponds to the second intermediate, shows a peak absorbance at a shorter wavelength (378 nm) with an increase in absorbance in the 440–500 nm range of oxidized enzyme (∼14%). According to the model in Fig. 4, spectrum b represents the characteristic of C4a-peroxyflavin, whereas spectrum c represents the C4a-hydroperoxyflavin. Spectrum d, a third intermediate, shows characteristics of the oxidized enzyme that slowly converts to the final species, spectrums e (E′ox:3HB) and f (Eox:3HB) (as indicated by the arrow).
SKIE
Fig. 4 proposes that the first two forms of oxygenated flavin adducts are C4a-peroxyflavin, resulting from the reaction of the reduced enzyme-3HB complex with oxygen, and the C4a-hydroperoxyflavin that results from protonation of the first species. Therefore, the SKIE was used to verify whether the second step that is supposed to involve proton transfer was affected when the reaction was carried out in D2O buffer (“Experimental Procedures”). The kinetic traces at 400 nm shown by the open circle line and the solid line are those from experiments in D2O and H2O buffer, respectively (Fig. 7A). When compared, both reactions showed that the first intermediate (which is supposed to be the C4a-peroxyflavin) formed maximally at 0.003 s and that no SKIE was detected for this step. The second phase (0.003–0.04 s) of the reaction in D2O buffer (Fig. 7A, open circle line) showed a lower rate constant of 56 ± 1 s−1 when compared with the reaction in H2O buffer (96 s−1), corresponding to a SKIE of 1.7. These results indicate that the second step involved proton transfer, supporting the reaction mechanism proposed in Fig. 4. The third phase (0.04–0.2 s), which is supposed to reflect hydroxylation, also shows a lower rate constant of 19.9 ± 0.4 s−1 in D2O buffer when compared with the data obtained for the H2O experiment (36 ± 2 s−1, Table 1), corresponding to a SKIE of 1.8. Although this phase is a combination of hydroxylation and dehydration, the SKIE value is contributed mainly by the hydroxylation step because the dehydration step is much faster than the hydroxylation. Altogether, the SKIE data support the reaction mechanism proposed in Fig. 4.
FIGURE 7.
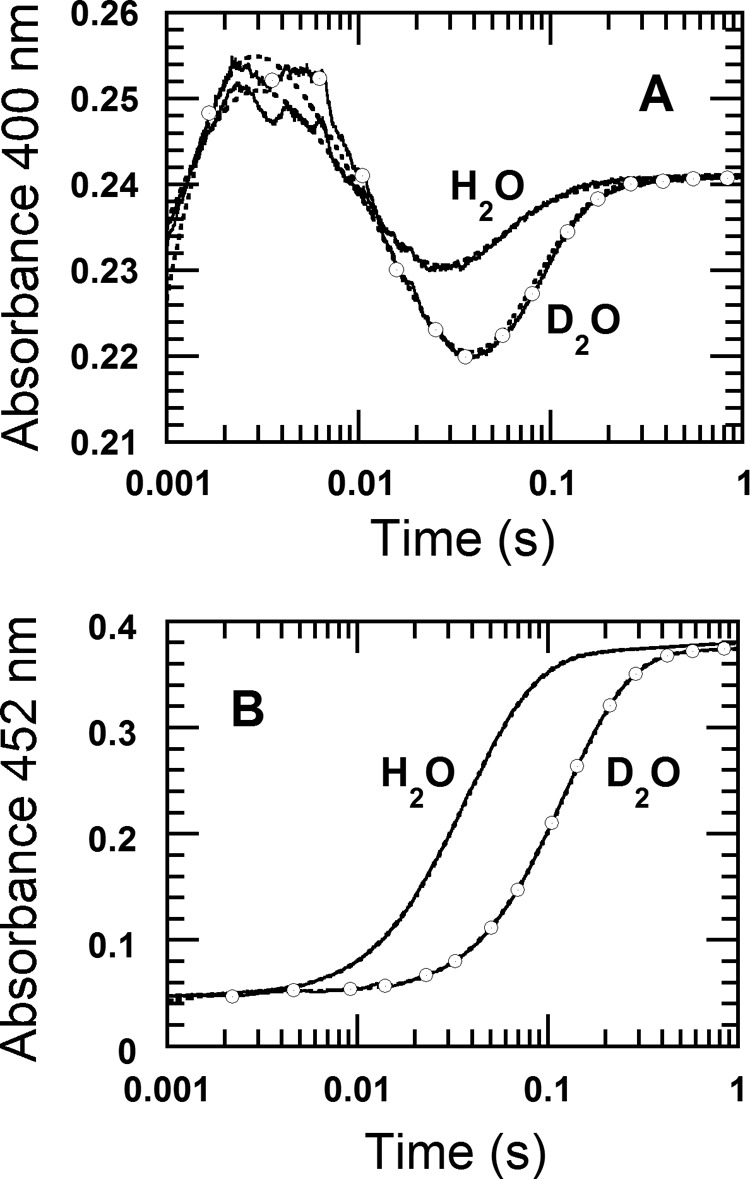
Reactions of reduced enzyme-3HB complex with oxygen in H2O and D2O buffers. A solution of the reduced enzyme (23 μm) containing 10 mm 3HB and 50 mm sodium azide was mixed with buffer containing 10 mm 3HB, 50 mm sodium azide, and 0.96 mm oxygen. All concentrations described were after mixing. The reactions were performed in 100 mm Tris-H2SO4 (pH(D) 8.0) at 4 °C. A, the reactions were monitored by absorbance change at 400 nm to detect formation of C4a-flavin adducts. Solid and open circle lines were the reactions in H2O and D2O buffer, respectively. B, the reactions were monitored by absorbance change at 452 nm to detect flavin oxidation. Solid and open circle lines were the reactions in H2O and D2O buffer, respectively. Dotted lines in A and B are kinetic traces obtained from simulations.
Binding of 3HB to the Reduced Enzyme
The previous study has shown that 3HB binds to the oxidized enzyme via a single-step reaction with a kon of 4 × 105 m−1 s−1 (k1) and a koff of 4600 s−1 (k−1) (14). In this experiment, we investigated whether 3HB can also bind to the reduced enzyme if the enzyme was first reduced by NADH or dithionite. The experiment was performed using the double-mixing mode of the stopped-flow spectrophotometer. In the first mixing, a solution of the reduced enzyme was mixed with various concentrations of 3HB under anaerobic conditions and incubated for 50 s. This incubation period should be long enough to allow complete binding if a complex of the reduced enzyme and 3HB can form. In the second mixing, the solution from the first mixing was mixed with buffer containing 1.92 mm oxygen (before mixing concentration). The reaction was monitored at 400 nm to detect formation of C4a-hydroperoxyflavin (Fig. 8A). The kinetic data in Figs. 2 and 3 could be used to distinguish the characteristics of the reaction in the absence or presence of substrate because in the absence of 3HB binding, the reaction would not form any C4a-hydroperoxyflavin (Fig. 2A). Results in Fig. 8A showed that the amplitude of the absorbance at 400 nm (characteristic of flavin C4a-adduct) increased with increased 3HB concentration, indicating that 3HB can bind to the reduced enzyme. The control experiment in which the reduced enzyme was mixed with anaerobic buffer at the first mixing step did not result in any increase in absorbance at 400 nm at 0.004 s (Fig. 8A, dashed line). Therefore, the increase in 400 nm absorbance at 0.004 s was plotted against the concentration of 3HB to calculate the dissociation constant (Kd) of the reduced enzyme-3HB complex, giving a value of 0.077 ± 0.005 mm (Fig. 8A, inset). For the third kinetic phase (0.017–0.060 s, Fig. 8A) that corresponds to hydroxylation and flavin oxidation (Fig. 4), the observed rate constants of this phase increased from 2.2 s−1 to 36 s−1 (lower to upper traces) when the substrate concentration was increased. This rate increment was due to the change in the rate of flavin oxidation as the enzyme was converted from the substrate-free to the substrate-bound form. The data also did not indicate any sign of substrate inhibition on the flavin oxidation step generally found in many ortho-hydroxylation flavoenzymes.
FIGURE 8.

Binding of 3HB to the reduced enzyme. Experiments were performed using the double-mixing mode of the stopped-flow spectrophotometer. The reactions were performed in 100 mm Tris-H2SO4 (pH 8.0) at 4 °C. A, in the first mixing, a solution of the reduced enzyme at 127 μm (concentration before mixing) was mixed with buffer containing various 3HB concentrations, 0.1, 0.2, 0.4, 0.8, 2, 8, and 20 mm (concentrations before mixing) under anaerobic conditions. The mixture of reduced enzyme and 3HB was incubated for 50 s before it was mixed with buffers containing 0.96 mm oxygen and various 3HB concentrations of 0.05, 0.1, 0.2, 0.4, 1, 4, and 10 mm. All concentrations were indicated according to before mixing conditions and the traces of low to high 3HB concentrations correspond to lower to upper traces (indicated by the arrow). The reactions were monitored at 400 nm to detect formation of C4a-hydroperoxyflavin. The increase of amplitude at 400 nm due to formation of C4a-hydroperoxyflavin represents the amount of the reduced enzyme-3HB complex. The dotted line trace is the control reaction without 3HB. Inset in A shows a plot of the absorbance change at 0.003 s versus the concentration of 3HB. B, a solution of the reduced enzyme, 120 μm (concentration before mixing), was mixed with 20 mm 3HB in the first mixing under anaerobic conditions. The reaction was incubated at various age times of 0.05, 0.1, 0.2, 0.5, 1, 5, 10, and 20 s (indicated by arrow) before the mixture was mixed with buffer containing 1.92 mm oxygen and 10 mm 3HB (concentration as before mixing) in the second mixture. The dotted line trace was a control reaction without 3HB. Inset in B shows a plot of the absorbance change at the time of 0.003 s versus age time.
The kinetics for binding of 3HB to the reduced enzyme was also investigated using the double-mixing mode. In the first mixing, the reduced enzyme was mixed with 20 mm 3HB (before mixing concentration) and incubated for various age times. The mixture from the first mixture was added to an oxygenated buffer containing 10 mm 3HB in the second mixture and the reaction was monitored for absorbance changes at 400 nm. An increase in absorbance at 400 nm due to the formation of C4a-hydroperoxyflavin was used as an indication of the amount of reduced enzyme-3HB complex (Fig. 8B), and this signal was plotted against the age time to yield the kinetics of complex formation. The analysis gave an observed rate constant of 7.3 ± 0.5 s−1 (Fig. 8B, inset). A biomolecular rate constant of 3HB binding was calculated by dividing the observed rate constant with 10 mm 3HB as 7.3 ± 0.5 × 102 m−1 s−1 (k5 in Table 1, Fig. 9). This calculation is valid because the dissociation rate constant (koff) is small compared with the observed pseudo first-order rate constant. Based on the dissociation constant (Kd) of the reduced enzyme-3HB complex of 0.077 ± 0.005 mm (Fig. 8A), the rate constant for dissociation of 3HB from the reduced enzyme-3HB complex (koff) is equivalent to 0.056 s−1 (k−5 in Table 1, Fig. 9).
DISCUSSION
This work reports on the kinetics of the oxidative half-reaction of 3HB6H from R. jostii RHA1 using various transient kinetic and mechanistic tools. The results reveal for the first time the catalytic features of a flavin-dependent para-hydroxylation reaction that are different as well as similar in many ways to the known reactions of ortho-hydroxylation. The data reported here are also useful for understanding factors that control the overall catalysis. Although the overall structure of 3HB6H is similar to PHBH and other Class A flavoprotein monooxygenases, the arrangement of active site residues is rather different. In 3HB6H, the hydroxyl group of 3HB is proposed to make an H-bond interaction with a His-213 residue, whereas the carboxylate group interacts with Gln-49 and Tyr-105 (17) (Fig. 10). In PHBH, the hydroxyl group of PHB makes an H-bond interaction with Tyr-201, whereas the carboxylate group of the substrate interacts with Arg-214 (3) (Fig. 10). The difference in active site structures presumably governs the opposite regio-specific hydroxylation between ortho- and para-hydroxylation enzymes and also contributes to the observed characteristics of 3HB6H that are different from PHBH, such as the presence of discrete steps in the formation of C4a-peroxyflavin and C4a-hydroperoxyflavin, and the absence of substrate inhibition on the flavin oxidation.
FIGURE 10.
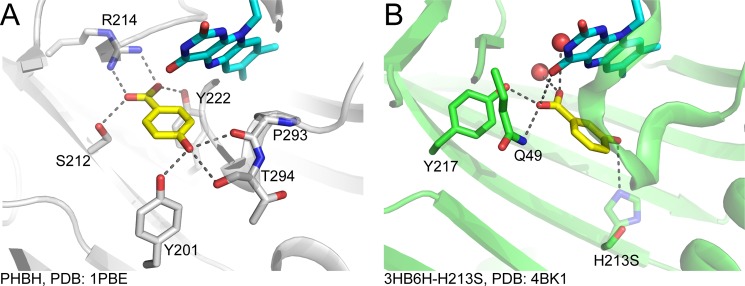
Comparison of active site structures of PHBH (ortho-hydroxylation) and 3HB6H (para-hydroxylation). A, active site of PHBH (1PBE) (41); B, active site of 3HB6H (4BK1) (17). The crystal structure of 3HB6H was obtained from co-crystallization of 3HB and the H213S variant of 3HB6H (17). A residue His-213 in the wild-type enzyme was modeled by replacing Ser-213 in the variant structure with His. The modeled His-213 is shown in light color. The 3-OH group of 3HB may interact with His-213.
3HB6H is different from most of ortho-hydroxylation enzymes in that 3HB6H forms the C4a-peroxyflavin prior to protonation to form C4a-hydroperoxyflavin. This conclusion is drawn from the solvent kinetic isotope effect of 1.7 on the second step following the formation of the first intermediate, and from rapid-quench experiments that identified the hydroxylation step to be after formation of the second intermediate (Figs. 3 and 7). For other enzymes such as PHBH (26), MHPCO (24), and the oxygenase component of p-hydroxyphenylacetate hydroxylase (20, 27), the reaction with oxygen and proton transfer coincide because only the C4a-hydroperoxyflavin (not C4a-peroxyflavin) was detected as an intermediate prior to hydroxylation. An exception to this was found in the reaction of phenol hydroxylase (27) in which the enzyme showed formation of C4a-peroxyflavin anion before protonation to C4a-hydroperoxyflavin. The latter species is required in these aromatic hydroxylases because their reactions are involved in electrophilic aromatic substitution in which the C4a-hydroperoxyflavin acts as an electrophile (4, 7, 9). Although the advantage of having discrete steps of C4a-peroxyflavin and C4a-hydroperoxyflavin formation in 3HB6H and phenol hydroxylase is not clear, it highlights the differences in the proton transfer pathways among flavin-dependent hydroxylases.
In addition, 3HB6H does not exhibit substrate inhibition on the flavin oxidation step, a common characteristic found in ortho-hydroxylation enzymes. Substrate inhibition typically results in enzyme trapping in the form of C4a-hydroxyflavin, which prevents regeneration of the enzyme to the starting oxidized species. Many ortho-hydroxylation enzymes including PHBH (3, 5), phenol hydroxylase (28), 2-methyl-3-hydroxypyridine-5-carboxylic acid oxygenase (24), and the oxygenase component of p-hydroxyphenylacetate hydroxylase (20, 27) are inhibited by excess substrate. The difference of 3HB6H from ortho-hydroxylation enzymes in this respect is probably attributable to the difference in substrate binding mode and the dynamics of ligand exchange. It remains to be seen in future investigations if the absence of substrate inhibition is also common in other para-hydroxylation enzymes.
The overall reaction of 3HB6H can be described according to a branched bimolecular mechanism model (29) in which an aromatic compound is the first substrate to bind to the enzyme to form the enzyme-substrate complex prior to NADH binding. The results in Fig. 8 and from a previous study (14) indicate that another pathway in which NADH binds first and reduces the enzyme-bound FAD prior to aromatic substrate binding also exists (minor path in Fig. 9). The data suggest that the path in which 3HB binds first is a preferred path because the binding occurs with a rate constant (kon) of 4 × 105 m−1 s−1 compared with a bimolecular rate constant of 43 m−1 s−1 for the other path (14). For the reaction of PHBH, the binding of p-hydroxybenzoate and NADPH to the oxidized enzyme could be described as a random-order type (3, 23, 30). For other Class A flavoprotein monooxygenases, the sequence of substrate binding has not been fully explored as in the case of 3HB6H; however, it can be assumed that the flavin reduction by NAD(P)H is very slow unless a substrate is bound. This catalytic feature of Class A enzymes is advantageous for preventing NADH consumption and H2O2 production in the absence of aromatic substrate (physiological demand for hydroxylation reaction).
3HB6H shows a typical characteristic of Class A enzymes in that formation of C4a-adduct intermediates can only be detected in the presence of aromatic substrates (Fig. 3A). The results in Fig. 2B suggest that after the reduction of the enzyme-bound FAD by NADH is completed, NAD+ is released prior to the oxygen reaction because there was no evidence showing any significant effect of NAD+ on the oxidative half-reaction. The oxidation reaction of reduced enzyme-3HB, prepared either by NADH or dithionite reduction, showed similar kinetics. For Class B flavoprotein monooxygenases such as phenylacetone monooxygenase (31), cyclohexanone monooxgenase (32), and l-ornithine monooxygenase (33–35), the binding of NAD(P)+ is required to stabilize the C4a-peroxyflavin intermediate (36, 37). For Class C enzymes such as bacterial luciferase (38, 39) and Class D enzymes such as the oxygenase component of p-hydroxyphenylacetate hydroxylase (27, 40), C4a-hydroperoxyflavin can be detected in the absence of any bound ligand.
Based on pre-steady state and steady state kinetics, the rate-limiting step for the overall reaction of 3HB6H is product release. Results from stopped-flow and rapid-quench studies in previous and current reports unambiguously assign rate constants associated with individual steps. Under a similar condition to the pre-steady state kinetics studies, the apparent kcat was measured as 6.49 ± 0.02 s−1 (“Experimental Procedures,” Table 1). Because there is no step with a rate constant in this range, the data suggest that product release (k12 in Fig. 9) likely controls the overall rate of the reaction. Calculation of kcat according to Equation 1 at saturation concentrations of all substrates according to the mechanism in Fig. 9 can be described as in Equation 2 (derivations in supplemental Schemes S1 and S2).
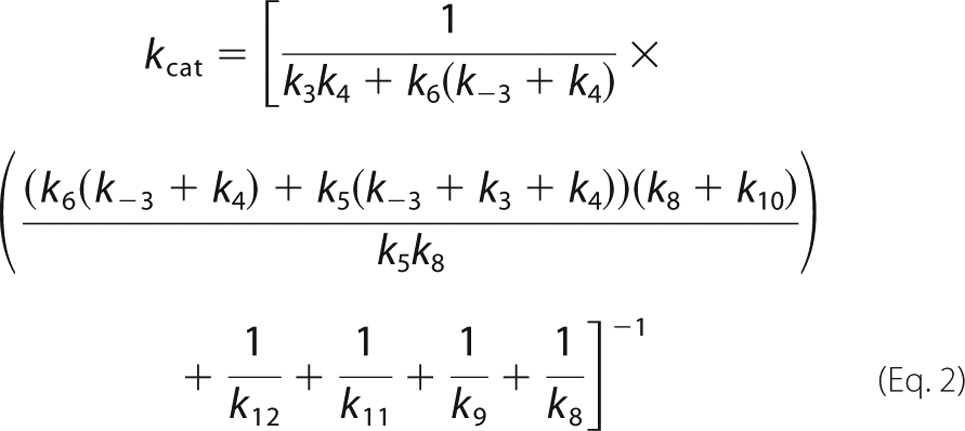 |
Using the individual rate constants and dissociation constants obtained from experimental data and simulations (Table 1), the value of k12 was calculated to be ∼11.7 s−1. This number also agrees well with the fourth phase of the stopped-flow data that is proposed to be the product release step (8–12 s−1).
The kinetic mechanism of the reaction in Fig. 9 indicates that both product release (k12 = 11.7 s−1) and hydroxylation (k9 = 36 s−1) partially control the overall catalytic turnover. To confirm this conclusion, we measured the kcat under the same condition in D2O buffer (100 mm Tris-D2SO4, pD 8.0). The kcat in D2O was 5.43 ± 0.04 s−1 (Table 1), which corresponds to a SKIE of 1.2. These results support the conclusion above that the step predominantly controlling the overall reaction (k12 = 11.7 s−1) is insensitive to D2O, whereas the hydroxylation (k9 = 36 s−1), which has the intrinsic SKIE of 1.7 (Fig. 7), partially controls the overall catalytic turnover. Therefore, the data in this report are useful in identifying that the hydroxylation and product release control the overall catalysis of 3HB6H. Although several flavoenzymes catalyzing para-hydroxylation of aromatic compounds have been reported (11), no detailed kinetic and mechanistic investigations of these enzymes have been carried out.
In conclusion, this study has elucidated the reaction mechanism of 3HB6H from R. jostii RHA1. The results clearly show that two forms of flavin C4a-adduct, C4a-peroxyflavin and C4a-hydroperoxyflavin, form prior to the hydroxylation step. The kinetic mechanism of 3HB6H can be described as a branched bimolecular mechanism in which 3HB is preferred to bind as the first substrate, followed by NADH that leaves prior to the reaction with oxygen. The overall reaction of 3HB6H is controlled by hydroxylation and product release steps. These results represent the first report of the oxygenation mechanism of a para-hydroxylating flavoenzyme and serve as the grounds for future exploration of factors that govern the regio-specific hydroxylation at the para-position.
Acknowledgment
We thank Barrie Entsch for critical reading of the manuscript.
This work was supported by Thailand Research Fund Grants RSA5580050 (to J. S.), RTA5680001 (to P. C.), and MRG5680043 (to D. P.), and grants from the National Research University Project of CHE (to D. P.), and Chulalongkorn University through the Ratchadaphiseksomphot Endowment Fund, the Faculty of Dentistry, Chulalongkorn University through the Developing Research Unit: Cell Signaling and Protein Functions (to J. S.), the Faculty of Science, Mahidol University (to P. C.), and the Netherlands Organization for Scientific Research (NWO) through the Integrated Biosynthesis Organic Synthesis (IBOS) programme (to W. v. B.).

This article contains supplemental Schemes S1 and S2.
- PHBH
- p-hydroxybenzoate 3-hydroxylase
- 3HB6H; PHB
- p-hydoxybenzoate 3-hydroxybenzoate 6-hydroxylase
- 3HB
- 3-hydroxybenzoic acid
- 2,5-DHB
- 2,5-dihydroxybenzoic acid
- SKIE
- solvent kinetic isotope effect.
REFERENCES
- 1. van Berkel W. J., Kamerbeek N. M., Fraaije M. W. (2006) Flavoprotein monooxygenases, a diverse class of oxidative biocatalysts. J. Biotechnol. 124, 670–689 [DOI] [PubMed] [Google Scholar]
- 2. Fagan R. L., Palfey B. A. (2010) Comprehensive Natural Products II Chemistry and Biology: Flavin-dependent Enzymes, Elsevier, Oxford [Google Scholar]
- 3. Entsch B., Ballou D. P. (2013) Handbook of Flavoproteins: The Reaction mechanisms of Groups A and B flavoprotein monooxygenases, Walter de Gruyter GmbH, Berlin/Briston [Google Scholar]
- 4. Ortiz-Maldonado M., Ballou D. P., Massey V. (1999) Use of free energy relationships to probe the individual steps of hydroxylation of p-hydroxybenzoate hydroxylase. Studies with a series of 8-substituted flavins. Biochemistry 38, 8124–8137 [DOI] [PubMed] [Google Scholar]
- 5. Entsch B., Cole L. J., Ballou D. P. (2005) Protein dynamics and electrostatics in the function of p-hydroxybenzoate hydroxylase. Arch. Biochem. Biophys. 433, 297–311 [DOI] [PubMed] [Google Scholar]
- 6. Palfey B. A., McDonald C. A. (2010) Control of catalysis in flavin-dependent monooxygenases. Arch. Biochem. Biophys. 493, 26–36 [DOI] [PubMed] [Google Scholar]
- 7. Chaiyen P., Sucharitakul J., Svasti J., Entsch B., Massey V., Ballou D. P. (2004) Use of 8-substituted-FAD analogues to investigate the hydroxylation mechanism of the flavoprotein 2-methyl-3-hydroxypyridine-5-carboxylic acid oxygenase. Biochemistry 43, 3933–3943 [DOI] [PubMed] [Google Scholar]
- 8. Chaiyen P. (2010) Flavoenzymes catalyzing oxidative aromatic ring-cleavage reactions. Arch. Biochem. Biophys. 493, 62–70 [DOI] [PubMed] [Google Scholar]
- 9. Tongsook C., Sucharitakul J., Thotsaporn K., Chaiyen P. (2011) Interactions with the substrate phenolic group are essential for hydroxylation by the oxygenase component of p-hydroxyphenylacetate 3-hydroxylase. J. Biol. Chem. 286, 44491–44502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Montersino S., van Berkel W. J. (2012) Functional annotation and characterization of 3-hydroxybenzoate 6-hydroxylase from Rhodococcus jostii RHA1. Biochim. Biophys. Acta 1824, 433–442 [DOI] [PubMed] [Google Scholar]
- 11. Montersino S., Tischler D., Gassner G. T., van Berkel W. J. (2011) Catalytic and structural features of flavoprotein hydroxylases and epoxidases. Adv. Synth. Catal. 353, 2301–2319 [Google Scholar]
- 12. Kallio P., Patrikainen P., Suomela J. P., Mäntsälä P., Metsä-Ketelä M., Niemi J. (2011) Flavoprotein hydroxylase PgaE catalyzes two consecutive oxygen-dependent tailoring reactions in angucycline biosynthesis. Biochemistry 50, 5535–5543 [DOI] [PubMed] [Google Scholar]
- 13. Wang Y., Niemi J., Mäntsälä P. (2002) Modification of aklavinone and aclacinomycins in vitro and in vivo by rhodomycin biosynthesis gene products. FEMS Microbiol. Lett. 208, 117–122 [DOI] [PubMed] [Google Scholar]
- 14. Sucharitakul J, Wongnate T, Montersino S, van Berkel WJ, Chaiyen P. (2012) Reduction kinetics of 3-hydroxybenzoate 6-hydroxylase from Rhodococcus jostii RHA1. Biochemistry 51, 4309–4321 [DOI] [PubMed] [Google Scholar]
- 15. Joshi R., Gangabhagirathi R., Venu S., Adhikari S., Mukherjee T. (2012) Antioxidant activity and free radical scavenging reactions of gentisic acid. In vitro and pulse radiolysis studies. Free Radic. Res. 46, 11–20 [DOI] [PubMed] [Google Scholar]
- 16. Ashidate K., Kawamura M., Mimura D., Tohda H., Miyazaki S., Teramoto T., Yamamoto Y., Hirata Y. (2005) Gentisic acid, an aspirin metabolite, inhibits oxidation of low-density lipoprotein and the formation of cholesterol ester hydroperoxides in human plasma. Eur. J. Pharmacol. 513, 173–179 [DOI] [PubMed] [Google Scholar]
- 17. Montersino S., Orru R., Barendregt A., Westphal A. H., van Duijn E., Mattevi A., van Berkel W. J. (2013) Crystal structure of 3-hydroybenzoate 6-hydroxylase uncovers lipid-assisted flavoprotein strategy for regioselective aromatic hydroxylation. J. Biol. Chem. 288, 26235–26245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cha S. (1968) A simple method for derivation of rate equations for enzyme-catalyzed reactions under the rapid equilibrium assumption or combined assumptions of equilibrium and steady state. J. Biol. Chem. 243, 820–825 [PubMed] [Google Scholar]
- 19. Kuby S. A. (2000) A Study of Enzymes: Historical Introduction, Theories of Enzyme Catalysis, and Some Elementary Considerations of Enzyme Kinetics, CRC Press, Boca Raton, FL [Google Scholar]
- 20. Sucharitakul J., Chaiyen P., Entsch B., Ballou D. P. (2006) Kinetic mechanisms of the oxygenase from a two-component enzyme, p-hydroxyphenylacetate 3-hydroxylase from Acinetobacter baumannii. J. Biol. Chem. 281, 17044–17053 [DOI] [PubMed] [Google Scholar]
- 21. Schowen K. B., Schowen R. L. (1982) Solvent isotope effects of enzyme systems. Methods Enzymol. 87, 551–606 [PubMed] [Google Scholar]
- 22. Steennis P. J., Cordes M. M., Hilkens J. H., Müller F. (1973) On the interaction of p-hydroxybenzoate hydroxylase from Pseudomonas fluorescens with halogen ions. FEBS Lett. 36, 177–180 [DOI] [PubMed] [Google Scholar]
- 23. Entsch B., Ballou D. P., Massey V. (1976) Flavin-oxygen derivatives involved in hydroxylation by p-hydroxybenzoate hydroxylase. J. Biol. Chem. 251, 2550–2563 [PubMed] [Google Scholar]
- 24. Chaiyen P., Brissette P., Ballou D. P., Massey V. (1997) Unusual mechanism of oxygen atom transfer and product rearrangement in the catalytic reaction of 2-methyl-3-hydroxypyridine-5-carboxylic acid oxygenase. Biochemistry 36, 8060–8070 [DOI] [PubMed] [Google Scholar]
- 25. Chaiyen P., Brissette P., Ballou D. P., Massey V. (1997) Reaction of 2-methyl-3-hydroxypyridine-5-carboxylic acid (MHPC) oxygenase with N-methyl-5-hydroxynicotinic acid. Studies on the mode of binding, and protonation status of the substrate. Biochemistry 36, 13856–13864 [DOI] [PubMed] [Google Scholar]
- 26. Entsch B., van Berkel W. J. (1995) Structure and mechanism of para-hydroxybenzoate hydroxylase. FASEB J. 9, 476–483 [DOI] [PubMed] [Google Scholar]
- 27. Ruangchan N., Tongsook C., Sucharitakul J., Chaiyen P. (2011) pH-dependent studies reveal an efficient hydroxylation mechanism of the oxygenase component of p-hydroxyphenylacetate 3-hydroxylase. J. Biol. Chem. 286, 223–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Maeda-Yorita K., Massey V. (1993) On the reaction mechanism of phenol hydroxylase. New information obtained by correlation of fluorescence and absorbance stopped flow studies. J. Biol. Chem. 268, 4134–4144 [PubMed] [Google Scholar]
- 29. Keitaro H. (1979) Kinetics of Fast Enzyme Reactions: Analysis of Fast Reactions: Transient Kinetics, Kodansha Ltd., Tokyo [Google Scholar]
- 30. Husain M., Massey V. (1979) Kinetic studies on the reaction of p-hydroxybenzoate hydroxylase. Agreement of steady state and rapid reaction data. J. Biol. Chem. 254, 6657–6666 [PubMed] [Google Scholar]
- 31. Torres Pazmiño D. E., Winkler M., Glieder A., Fraaije M. W. (2010) Monooxygenases as biocatalysts. Classification, mechanistic aspects and biotechnological applications. J. Biotechnol. 146, 9–24 [DOI] [PubMed] [Google Scholar]
- 32. Sheng D., Ballou D. P., Massey V. (2001) Mechanistic studies of cyclohexanone monooxygenase. Chemical properties of intermediates involved in catalysis. Biochemistry 40, 11156–11167 [DOI] [PubMed] [Google Scholar]
- 33. Chocklett S. W., Sobrado P. (2010) Aspergillus fumigatus SidA is a highly specific ornithine hydroxylase with bound flavin cofactor. Biochemistry 49, 6777–6783 [DOI] [PubMed] [Google Scholar]
- 34. Mayfield J. A., Frederick R. E., Streit B. R., Wencewicz T. A., Ballou D. P., DuBois J. L. (2010) Comprehensive spectroscopic, steady state, and transient kinetic studies of a representative siderophore-associated flavin monooxygenase. J. Biol. Chem. 285, 30375–30388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Meneely K. M., Barr E. W., Bollinger J. M., Jr., Lamb A. L. (2009) Kinetic mechanism of ornithine hydroxylase (PvdA) from Pseudomonas aeruginosa. Substrate triggering of O2 addition but not flavin reduction. Biochemistry 48, 4371–4376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Alfieri A., Malito E., Orru R., Fraaije M. W., Mattevi A. (2008) Revealing the moonlighting role of NADP in the structure of a flavin-containing monooxygenase. Proc. Natl. Acad. Sci. U.S.A. 105, 6572–6577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Orru R., Dudek H. M., Martinoli C., Torres Pazmiño D. E., Royant A., Weik M., Fraaije M. W., Mattevi A. (2011) Snapshots of enzymatic Baeyer-Villiger catalysis. Oxygen activation and intermediate stabilization. J. Biol. Chem. 286, 29284–29291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Suadee C., Nijvipakul S., Svasti J., Entsch B., Ballou D. P., Chaiyen P. (2007) Luciferase from Vibrio campbellii is more thermostable and binds reduced FMN better than its homologues. J. Biochem. 142, 539–552 [DOI] [PubMed] [Google Scholar]
- 39. Tinikul R., Thotsaporn K., Thaveekarn W., Jitrapakdee S., Chaiyen P. (2012) The fusion Vibrio campbellii luciferase as a eukaryotic gene reporter. J. Biotechnol. 162, 346–353 [DOI] [PubMed] [Google Scholar]
- 40. Thotsaporn K., Chenprakhon P., Sucharitakul J., Mattevi A., Chaiyen P. (2011) Stabilization of C4a-hydroperoxyflavin in a two-component flavin-dependent monooxygenase is achieved through interactions at flavin N5 and C4a atoms. J. Biol. Chem. 286, 28170–28180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schreuder H. A., Prick P. A., Wierenga R. K., Vriend G., Wilson K. S., Hol W. G., Drenth J. (1989) Crystal structure of the p-hydroxybenzoate hydroxylase-substrate complex refined at 1.9-Å resolution. Analysis of the enzyme-substrate and enzyme-product complexes. J. Mol. Biol. 208, 679–696 [DOI] [PubMed] [Google Scholar]