

Background: Overexpressed UCP2 and ERBB2 are oncogenic, but their functions at low expression are unclear.
Results: ERBB2 regulates UCP2 levels at low and overexpression settings. The rapid regulation of mitochondria by ERBB2 requires UCP2, but not its transporter activity.
Conclusion: At low levels, UCP2 integrates mitochondria into the acute (2 h) signaling response.
Significance: This study links growth factor signaling, mitochondrial adaptability, and oncogenic deregulation.
Keywords: Glucose Metabolism, Mitochondria, Receptor Tyrosine Kinase, Signal Transduction, Uncoupling Proteins
Abstract
Overexpression of the ERBB2 receptor tyrosine kinase and the mitochondrial inner membrane protein UCP2 occurs frequently in aggressive cancers with dysfunctional mitochondria. Overexpressed ERBB2 signals constitutively and elevated UCP2 can uncouple mitochondria and alleviate oxidative stress. However, the physiological contributions of UCP2 and ERBB2 at the low expression levels that are typical of most tissues, as well as the path to oncogenic deregulation, are poorly understood. We now show that ERBB2 directly controls UCP2 levels, both at low physiological levels and oncogenic overexpression. At low levels of receptor and UCP2, ligand stimulation creates a distinct temporal response pattern driven by the opposing forces of translational suppression of the exceptionally short lived UCP2 protein and a time delayed transcriptional up-regulation. The latter becomes dominant through constitutive signaling by overexpressed ERBB2, resulting in high levels of UCP2 that contribute mitochondrial uncoupling. By contrast, ligand stimulation of non-overexpressed ERBB2 transiently removes UCP2 and paradoxically reduces the mitochondrial membrane potential, oxygen consumption, and OXPHOS on a signaling time scale. However, neither the transporter activity nor down-regulation of already low UCP2 levels drive this reduction in mitochondrial activity. Instead, UCP2 is required to establish mitochondria that are capable of responding to ligand. UCP2 knockdown impairs proliferation at high glucose but its absence specifically impairs ligand-induced growth when glucose levels fluctuate. These findings demonstrate the ability of growth factor signaling to control oxidative phosphorylation on a signaling time scale and point toward a non-transporter role for low levels of UCP2 in establishing dynamic response capability.
Introduction
The decision of the cell to commit to a proliferative state is closely linked to the availability of energy sources as well as external stimuli, such as growth factors. By contrast, relative homeostasis is a defining feature of mitochondrial activity on an intermediate time scale. The assumption of relative homeostasis and stable numbers of mitochondria per cell forms the foundation of widely used formazan dye-based proliferation assays. Provided experimental conditions do not introduce uncoupling or other pharmacological perturbances of mitochondrial function, the conversion of these dyes is a reflection of total mitochondrial activity that in turn is taken to be proportional to the number of cells. This raises the question whether mitochondrial oxidative phosphorylation (OXPHOS)2 has adaptive capability on the more rapid time scale of external nutrient fluctuations or proliferative signaling cascades. In addition, progressive metabolic and mitochondrial deregulations are hallmarks of cancer cells. The driving forces behind these transitions and how they relate to the “normal” regulation of mitochondrial function are still poorly understood.
Uncoupling proteins (UCPs) can regulate mitochondrial OXPHOS efficiency by multiple mechanisms. For UCP1, found first in the inner mitochondrial membrane of brown adipocytes, this is known to involve direct proton leakage from the mitochondrial matrix and an uncoupling of the proton gradient from ATP synthesis to generate heat (1–5). The function of other UCP family members, especially at their physiological expression levels that are very low in all but few tissues, has remained much more controversial. Among the three closely related homologues in humans, UCP2 also stands out through generally very low and variable protein expression across tissues despite relatively uniform mRNA levels and an exceptionally short half-life of 20–30 min. This compares to more than 10 h for UCP1 or 3–4 h for the closely related UCP3 (6–9). In most tissues UCP2 protein levels can pose a challenge for detection. Because the most convincing demonstrations of an uncoupling contribution by UCP2 are based either on conditions of experimental or pathological overexpression, or select tissue with naturally elevated levels (see below), this has resulted in two questions. Beyond the scope of our current study, it is being debated whether the physiological uncoupling by elevated levels of UCP2 is indeed the result of direct proton leakage, as shown for UCP1, or alternatively the secondary consequence of transporting charged metabolites. Second, the exceptionally short half-life and very low protein levels fall short of expectations based on mRNA levels. This is difficult to reconcile with a primary role as an uncoupler in a manner that resembles UCP1. This has raised the question whether UCP2 at these low protein levels could serve an additional purpose.
High levels of UCP2 have been correlated with ROS protection, increased longevity (10–14) and altered viability of cardiomyocytes in the face of ROS stress, depending on the molecular source of ROS (15, 16). Apart from cancer, high expression levels of UCP2 protein are unusual with the exception of pancreatic islet cells and macrophages. Here a reduction in UCP2 increases ROS levels that result in reduced glucose tolerance (17) and constitutively activated macrophages, respectively (18, 19). Insight into the broader function of UCP2 comes from genetic analysis and links it to obesity and type II diabetes (20) as well as cardiovascular risk in men (21). In cancer, substantially elevated UCP2 levels have been observed in various tumors, including those overexpressing ERBB2 (erythroblastic leukemia viral oncogene homolog 2 or human EGF receptor 2 (HER2)). This UCP2 overexpression has been linked to increased resistance to oxidative stress (22). Notably, in MCF7 cells, a non-ERBB2 overexpressing breast cancer line, recombinant overexpression of UCP2 alone can induce aggressive growth with high metastatic potential in a manner that cannot be reproduced by other, uncoupling capable, UCP family members (23). However, this correlation between aggressive cancer cell growth and high levels of UCP2 contrasts studies in fibroblasts from UCP2−/− mice. Here, loss of the otherwise low levels of UCP2 results in accelerated growth (24). This implies a more complex contribution of UCP2 in the regulation of cell proliferation than is apparent from studies of UCP2 at high expression levels.
Alternative models for UCP2 function have identified its contribution to the mitochondrial transport of calcium (25, 26) and the export of pyruvate and unprotonated short chain fatty acids from mitochondria (24, 27). This transport of charged species can equate indirectly to a net-uncoupling (28). Both, glutamine and glucose up-regulate UCP2. Especially glutamine acts rapidly at the level of translation due to a glutamine response element in the UCP2 mRNA (29, 30). In conjunction with its metabolite transport capability, this regulation has been proposed to constitute a “glucose sparing switch” (31, 32). This would make any net contribution of UCP2 to cellular metabolism and mitochondrial membrane potential dependent on metabolic conditions, sufficient transport capacity, and demand.
Beyond the observation of feedback regulation by metabolites and the notable exception of LPS responses in macrophages, little is known about the integration of UCP2 regulation into larger regulatory schemes. The same applies to the forces that drive its overexpression in some cancers. The receptor tyrosine kinase ERBB2 is a well established player in breast cancer and other solid tumor malignancies. Constitutive ERBB2 signaling confers a broad range of systemic changes that generally result in a more aggressive cancer phenotype. In addition, overexpressed ERBB2 (primarily the cleaved cytoplasmic segment) has been found mislocalized to the nucleus. More recently, mtHSP90-dependent localization of full-length ERBB2 to mitochondria has been reported in cells that overexpress the receptor (33). However, the functional consequences of mitochondrial localized ERBB2 are still poorly understood.
Compared with the overall body of work that emphasizes ERBB2 overexpression, we still know little about the role of ERBB2 at low physiological levels. Conditional knockouts of ERBB2 or ERBB4 in mice result in dilated cardiomyopathy (34, 35), and cardiac side effects of ERBB2-targeted cancer therapy in humans are linked to mitochondrial failure (36, 37). Cardiac stress up-regulates ERBB receptors that are critical but barely detectable in cardiomyocytes (38, 39). In mice, the clinical ERBB2 inhibitor lapatinib increases mortality of both pups and mothers because it blocks the pregnancy-induced and protective remodeling of the left ventricle (40). Beyond cardiac functions, ERBB2 suppresses an autophagy dependent cell death in the duct lumen during breast development in a manner that is linked to shifts in metabolite preferences (41). Combined these studies suggest a critical role of low levels of ERBB2 in the control of mitochondrial function, stress response, and metabolic reprogramming. However, it is not clear how this regulation is achieved and which aspects of it are immediate consequences of ERBB2 signaling versus long term adjustments to broader transcriptional changes. In the current study we provide evidence that ligand-regulated and non-overexpressed ERBB2 directly and rapidly controls mitochondrial activity in a manner that requires UCP2. UCP2 is also needed to facilitate ERBB2-mediated and ligand-induced proliferation under conditions of elevated, but especially at fluctuating glucose levels. Although out of balance in cancer cells, the basic components regulating UCP2 protein levels are preserved at low and overexpression levels of ERBB2 and point toward UCP2 as a critical and very dynamic link between growth factor receptor signaling and the regulation of mitochondrial OXPHOS.
EXPERIMENTAL PROCEDURES
Cell Lines and Other Reagents
All cell lines were cultured and maintained in RPMI 1640 medium (Cellgro), 10% fetal bovine serum (Gemini Bioproducts), penicillin (100 units/ml), and streptomycin (0.01%) solution (Cellgro), and humidified with 5% CO2 air gas mixture. Pertuzumab was generously provided by Dr. Mark Sliwkowski (Genentech). Commercial reagents were obtained from the following sources: oligomycin and MAPK inhibitors UO126 and PD98059 (Cell Signaling), Canertinib (LC Laboratories), antimycin, carbonyl cyanide m-chlorophenyl hydrazone (CCCP), dihydroethidium, and the AKT inhibitor LY294002 (Calbiochem), KCN, lipoic acid, and cycloheximide (Sigma), tetramethylrhodamine methyl ester (Invitrogen), Genipin (Wako chemicals), Lipofectamine and Alamar blue reagent (Invitrogen), and control siRNA and UCP2 siRNA (Dharmacon). Recombinant NRGβ1, benchmarked against full-length commercial Ig-NRGβ1, was produced as a thioredoxin fusion protein in Escherichia coli as described previously (42–44). The constitutively active ΔMEK1 construct was provided by Dr. Natalie Ahn (University of Colorado) (45).
Polarography
Oxygen consumption of intact MCF7, MCF7-ERBB2, and BT474 cells was measured using a Clark oxygen electrode in a water-jacketed microcell, magnetically stirred, at 37 °C, and Oxygraph plus software (Hansatech Instruments Limited, Norfolk, UK). Respiratory buffer (0.3 m mannitol, 10 mm KCl, 5 mm MgCl2, and 10 mm K2PO4, pH 7.4) was equilibrated at 37 °C before adding freshly trypsinized cells (5–7 million cells). Endogenous oxygen consumption was recorded for 5 min, followed by addition of KCN at 1 min after (up to 700 μm) and KCN-insensitive respiration was normalized to the total cell number. The respiratory control ratio (RCR) was obtained by dividing state 3 respiration (CCCP) by state 4 respiration (oligomycin). Respiration was measured with or without addition of the following reagents directly into culture medium: Neuregulin-β1 (NRGβ1) (30 nm; 2 or 9 h), CCCP (5 μm; 30 min), oligomycin (5 μm, 15 min), Canertinib (1 μm, 2 or 9 h), and UO126 (25 μm, 2 or 9 h).
Single Cell Mitochondrial Membrane Potential Measurements and ROS Detection
MCF7 and MCF7-ERBB2 cells were harvested using Versene (PBS + 0.2% EDTA), seeded in 6-well plates, and treated with the indicated inhibitors or ligand for different time points (as indicated in the figures) followed by tetramethylrhodamine methyl ester (for mitochondrial membrane potential) (20 nm) and dihydroethidium (for ROS detection) (5 μm) for 30 min at 37 °C. Following two washes with PBS, images were acquired on a Zeiss Axiovert 200M. Fluorescence was analyzed for 200–300 individual cells for each dataset using Openlab software.
Real-time PCR
cDNA was synthesized from 500 ng of DNase-treated total RNA using Oligo(dT) primers (Invitrogen) and Moloney murine leukemia virus high performance reverse transcriptase (Epicenter Biotech.). Quantitative PCR were carried out with 10–20 ng of cDNA, and 200 nm gene-specific primers (UCP2 forward, CTGTGAAGTTTCTTGGGGCT, UCP2 reverse, TCAGAATGGTGCCCATCACA; 18 S rRNA forward, GTAACCCGTTGAACCCCATT, 18 S rRNA reverse, CCATCCAATCGGTAGTAGCG) in a 10-μl reaction containing SsoFast EvaGreen SuperMix (Bio-Rad). PCR amplification reactions were performed in triplicates on a CFX96 thermocycler (Bio-Rad) and quantified using the iCycler iQ software. Melting curves were included for every run to ensure that only one correct product was amplified. The relative quantities of UCP2 mRNA were determined for each sample based on the threshold cycle (CT) value normalized to the corresponding values for 18 S rRNA and data were plotted as normalized fold-expression. Error bars on the graphs represent the mean ± S.E. of each sample in triplicates.
Alamar Blue Proliferation Assay
Cells were seeded in RPMI 1640 with the indicated concentration of d-glucose and transfected with si-Scrambled RNA or si-UCP2 RNA. Cells were allowed to grow for 48 (MCF7) or 72 h (BT474) in the presence or absence of NRGβ1 (30 nm) at the indicated final glucose concentration. For conditions of changing glucose, media was exchanged from reservoirs containing different glucose concentrations but matching transfection reagents. Removed, conditioned media was retained and returned at later time points as needed to minimize non-glucose effects in media exchanges. Alamar blue assays were performed according to the manufacturer's protocol. Select data were compared by trypan blue exclusion assay and manual cell count.
Statistical Analysis
Unless otherwise stated, all quantitative data represent the average of triplicate datasets with indicated standard deviations (mean ± S.E. for mRNA data). Significance was calculated using an unpaired Student's t test for a null hypothesis of identical means compared with control. p Values below thresholds are indicated with asterisks (* for p < 0.05, ** for p < 0.01, and *** for p < 0.005). p Values above a threshold of 0.9, indicating a high probability of matching data sets, are marked with □.
Immunoblotting
Cells were lysed in SDS sample buffer and subjected to Western blot analysis using antibodies against UCP2 (sc-6525 (C-20) obtained from Santa Cruz Biotechnology). Other antibodies used were MAPK (9102), pMAPK (Thr-42/Tyr-44, 9101), GAPDH (5174), and pAKT (pS473, 9271) (Cell Signaling Technology), pY1139-ERBB2 (1991-1, Epitomics, Inc.), ERBB2 (MU134-UC, Biogenex), β-tubulin (ab52623), and VDAC1 (Porin) (ab154856) (Abcam Inc.), and phosphotyrosine (Tyr(P)) (05–321, EMD Millipore Corp.). For immunoprecipitation, phosphotyrosine antibody-conjugated beads (16-101) were obtained from EMD Millipore. Following Western blotting, images were acquired and quantified using the Odyssey® Fc dual-mode imaging system from LI-COR biosciences in chemiluminescence mode.
RESULTS
Neuregulin-activated Signaling Induces a Rapid Down-regulation of UCP2
For UCP2, but especially for ERBB2, non-pathogenic conditions are exemplified by low expression levels. We therefore minimized the use of recombinant overexpression in our analysis to better understand the basic mode of regulation before evaluating how it is derailed by the amplification of ERBB2 and surrounding chromosomal segments. With ∼10,000 ERBB2 and ERBB3 receptors but effectively undetectable EGFR (ERBB1) and ERBB4, MCF7 cells are a well established model system for ligand-responsive signaling through ERBB2/ERBB3 heterocomplexes after binding of neuregulin (NRGβ1) to ERBB3. MCF7 cells are considered “ERBB2 negative” by breast cancer standards. MCF7 cells frequently serve as a “normal” starting point for the study of mitochondrial activity in oncogenic progression and as a comparative model system for the enhanced oncogenic potential conferred by stably overexpressed ERBB2 (46). We observed that in MCF7 cells that were stimulated with NRGβ1 the rapid increase and subsequent attenuation of ERBB2 phosphorylation correlates with a loss of UCP2 protein (Fig. 1A). The ERBB2-specific therapeutic antibody Pertuzumab blocks the heterodimerization interface of ERBB2 and ligand-dependent activation. Pertuzumab completely blocked the ligand-dependent down-regulation of UCP2 (Fig. 1C) confirming that the relevant signaling indeed proceeds through ERBB2. The primary targets of NRGβ1-activated ERBB2/ERBB3 heterocomplexes are the PI3K/AKT and MAPK (ERK1/2) pathways. This will trigger cell growth, enhanced survival, migration, or suppression of apoptosis. The inhibition of MAPK (ERK1/2) with UO126 abrogates UCP2 down-regulation (Fig. 2A). Prior to ligand addition, non-serum starved MCF7 cells exhibit very low but detectable levels of MAPK(ERK1/2) activation (emphasized in Fig. 3). Inhibition of basal signaling by MAPK increases steady state levels of UCP2. Comparable data were obtained with the MAPK inhibitor PD98059, which has a different ERK isoform specificity (data not shown). Attempts to complement the pharmacological inhibition of MAPK (ERK1/2) by other means, such as the expression of dominant-negative mutants, resulted in very pronounced loss in cell viability over the time course needed to establish uniform protein expression. We therefore investigated the contribution of MAPK through the inverse approach. We introduced a constitutively active variant of MEK1 (ΔMEK1). This in turn results in constitutively phosphorylated and activated MAPK (ERK1/2) (45). Constitutive activation of MAPK (ERK1/2) was well tolerated and reduced UCP2 levels in a ERK1/2 activation level dependent manner (Fig. 2B). By contrast, the inhibition of AKT during ligand stimulation does not interfere with the down-regulation of UCP2 (Fig. 2C). The plant-derived glycoside genipin inhibits UCP2-mediated proton leakage in islet cells and UCP2 overexpressing cancer cells (47–49). It is expected to cross-link and block the central pore formed by UCP2. In doing so, it also blocks the impact of recombinantly expressed UCP2 on myocardial excitation-contraction coupling (26). However, genipin neither altered the steady state levels nor block the down-regulation of UCP2 after ligand addition (Fig. 2D). This suggests that the removal of UCP2 is not linked to its transport function. The removal of UCP2 does, however, require an intact membrane potential. Exposure to the pharmacological uncoupler CCCP blocked ligand-induced down-regulation of UCP2 (Fig. 2E).
FIGURE 1.
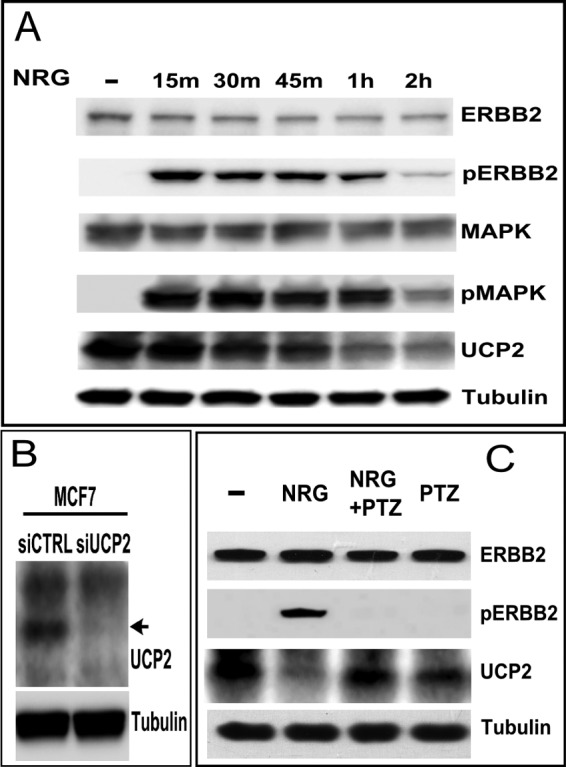
Ligand stimulation of ERBB2 results in the rapid and MAPK-dependent down-regulation of UCP2. A, stimulation of MCF7 cells with NRGβ1 (30 nm) for the times indicated above the lanes (NRG). Levels and activation states of ERBB2, MAPK, and UCP2 were evaluated by Western blotting. B, UCP2 levels in MCF7 are very low. To validate our detection system, we confirmed antibody specificity by siRNA knockdown of UCP2 in MCF7. C, ligand-induced down-regulation of UCP2 is blocked by the ERBB2-specific antibody, pertuzumab (PTZ), administered 1 h prior to a 2-h NRGβ1 stimulation.
FIGURE 2.

A, inhibition of MAPK with UO126 (3 h pretreatment with 25 μm) abrogates the NRG-induced down-regulation of UCP2. Protein levels were analyzed after 2 h of ligand stimulation. B, constitutive phosphorylation and activation of ERK1/2 by ΔMEK1 results in the down-regulation of UCP2. MCF7 cell lysates were harvested at the indicated time points post-transfection with ΔMEK1. C, the ligand-induced down-regulation of UCP2 does not involve AKT. The PI3K/AKT inhibitor LY294002 (50 μm) was added to MCF7 cells for 3 h with NRGβ1 addition after 1 h. D, the UCP2 inhibitor Genipin has no effect on the basal levels of UCP2 or its down-regulation. MCF7 cells were treated with NRGβ1 (30 nm) for 2 h in the presence or absence of Genipin (5 mm, added 1 h prior to ligand activation). E, an intact membrane potential is needed to facilitate the ligand-dependent down-regulation of UCP2. CCCP (5 μm) was used to depolarize mitochondria followed by treatment with NRGβ1 (30 nm) for the indicated time points.
FIGURE 3.

The down-regulation of UCP2 is transient and reflects competing regulatory events. A, UCP2 levels in MCF7 cells were evaluated at the indicated time points after stimulation with NRGβ1 (NRG, 30 nm) or alternatively, inhibition with UO126 (25 μm) or the irreversible ERBB2 inhibitor Canertinib (1 μm). B, NRGβ1 induced prolonged ERBB2/3 signaling causes a moderate increase in UCP2 protein levels in MCF7 cells. MCF7 cells were treated with NRGβ1 for 3 days. UCP2 protein levels were analyzed by Western blotting followed by densitometric analysis, which is represented as a bar graph of triplicates normalized to tubulin levels. C, inhibition of the basal level of ERBB2 signaling with the irreversible and ERBB specific kinase inhibitors Canertinib (CNT) reduces UCP2 levels, indicating a contribution to low levels of ERBB2 activation to the steady state level of UCP2. D, the basal level of ERBB2 signaling in non-serum-starved MCF7 cells is barely detectable but inhibited by Canertinib. L and H indicate low and high load levels, respectively. As a positive control of inhibitor activity, ligand-induced receptor activation was blocked. Immunoprecipitation was performed on MCF7 cell lysates using anti-phosphotyrosine antibody-conjugated beads followed by immunoblotting with anti-ERBB2 antibody.
The MAPK dependence in the short term regulation of UCP2 protein levels closely tracks the characteristic time profile of ERBB2 and MAPK signaling (Fig. 3). By approximately 4 h, the peak of ERBB2 signaling has subsided and UCP2 has returned to slightly above starting levels (Fig. 3A). However, in contrast to MAPK inhibition, the comparable rapid increase in UCP2 levels is followed by a steady decline to ∼20% below controls at 9 h. These data suggest that both ERBB2 and MAPK take part in the regulation of steady state levels of UCP2 protein, with comparable contributions to the short term reduction but additional signals from ERBB2 that are needed to sustain UCP2 levels long term. Consistent with the rebound that becomes apparent after 9 h of NRG treatment, sustained ligand stimulation for 48 h results in an increase in proliferation (42) as well as an increase in UCP2 protein levels (Fig. 3B). Without ligand stimulation, constitutive phosphorylation of ERBB2 in MCF7 cells is barely detectable (Fig. 3D). Nevertheless, the ERBB-specific and irreversible kinase inhibitor Canertinib (CI1033, CNT) reduces steady state levels of UCP2 (Fig. 3C), thereby indicating a contribution of basal ERBB2 signaling to UCP2 maintenance.
The Rapid Down-regulation of UCP2 Involves Translational Repression
To investigate whether ERBB2/ERBB3 stimulation by NRGβ1 accelerates UCP2 degradation, we inhibited protein synthesis with cycloheximide (CHX) (Fig. 4). Under those conditions, no difference in the rate of protein turnover was observed after ligand stimulation. The rate of turnover and overall depletion of UCP2 protein in the presence of cycloheximide mirrors that seen in the first 2 h of ligand stimulation. This points toward a halt in protein synthesis as the likely reason for the ligand-induced rapid decline of UCP2 levels. By contrast, mRNA levels are only reduced by 30% (Fig. 4B) at the 2-h time point when UCP2 protein is largely depleted by ligand stimulation (Figs. 1 and 2). However, the partial reduction in mRNA is overcompensated at a later point and this rebound does coincide with the rebound in overall protein levels, suggesting translational inhibition subsides at the same time that transcriptional activation begins to set in. The midpoint of ligand induced down-regulation occurs ∼45 min post-ligand addition. A quantitative comparison of protein levels at this point shows that despite the elevated staring levels of UCP2 after pretreatment with UO126, turnover still occurs at a similar and even slightly accelerated rate (Fig. 3C). This may explain why MAPK inhibition does not cause perpetual accumulation. Instead it may rapidly establish a higher steady level of UCP2 protein that is ultimately sustained by a matching, accelerated turnover.
FIGURE 4.
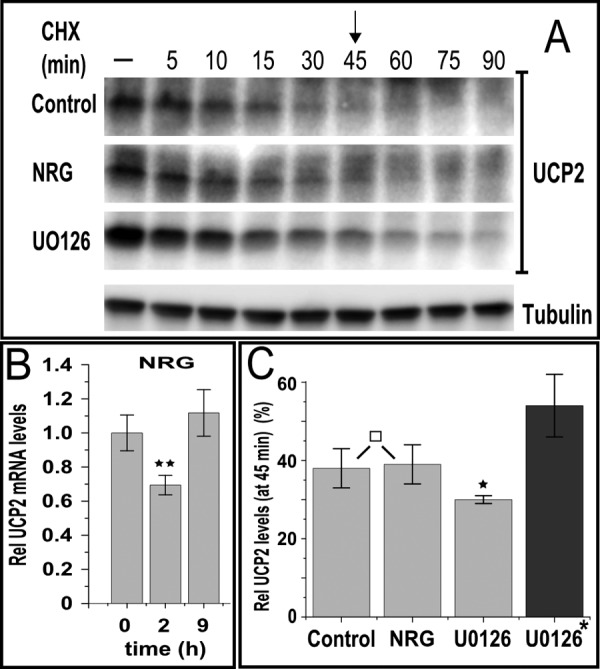
Ligand-mediated down-regulation of UCP2 occurs at the level of translation. A, cycloheximide treatment (CHX, 50 μm) results in the rapid decrease in UCP2 levels at a rate that is independent of ligand stimulation or UO126 pretreatment (added 1 h prior to CHX addition). B, compared with protein levels, ligand stimulation has little effect on UCP2 mRNA. Quantitative PCR derived data were normalized to 18 S rRNA and shown at 2 h (at which protein is almost completely depleted) or 9 h (protein levels are recovered). C, densitometric analysis of UCP2 levels at 45 min post-CHX addition. Data are normalized to tubulin levels and expressed as percent of starting levels prior to CHX addition. UO126 was present throughout the treatment as indicated. For UO126-treated samples marked with *, data are instead shown relative to the starting levels of non-pretreated samples. Error bars shown in B and C represent mean ± S.E. or S.D., respectively. p values <0.05, <0.01, <0.005, and >0.9 are marked *, **, ***, and □, respectively.
Sustained ERBB2 Signaling Increases UCP2 Protein through Transcriptional Regulation
Even in MCF7 cells, sustained ligand stimulation results in an increase in steady state protein levels of UCP2 (Fig. 3B). This raises the question of how ERBB2 gene amplification and constitutive signaling in an oncogenic setting impacts UCP2 levels. Stably ERBB2 overexpressing MCF7 cells are a long established model system for the oncogenic impact of receptor amplification. We therefore evaluated UCP2 regulation in a stable MCF7-ERBB2 line that expresses ∼2–3 × 105 receptors, resulting in high levels of constitutively phosphorylated ERBB2. MCF7-ERBB2 cells display a very pronounced increase in UCP2 protein (Fig. 5A) and mRNA levels (Control, Fig. 5C) compared with untreated MCF7 parental cells. ERBB2 overexpression renders MCF7-ERBB2 cells more sensitive to the inhibition of ERBB2 and MAPK, which is evident in overall cell viability at longer time points (>12 h). As was observed for parental cells, the Canertinib-induced decline in mRNA mirrors a decrease in UCP2 protein, which is very pronounced for MCF7-ERBB2. Inhibition with UO126 does result in a larger relative decrease in mRNA than was the case in MCF7, however, this decrease is smaller than that obtained with Canertinib. Moreover, the decrease in mRNA is in contrast to a relative increase in protein levels, which mirrors the response in MCF7.
FIGURE 5.

ERBB2 amplification drives UCP2 overexpression in stable ERBB2 overexpressing MCF7 cells. A, comparison of ERBB2 and UCP2 levels as well as associated constitutive signaling in MCF7 and MCF7-ERBB2 cells (data for ERBB2-amplified BT474 cells are compiled in Fig. 8). L and H represent low and high exposure levels needed to convey the vastly different levels of ERBB2 expression in both cell lines. B, the basic modes of regulation observed in MCF7 are also in place in MCF7-ERBB2 cells. The long term decrease in UCP2 levels in response to Canertinib (CNT) is more pronounced compared with MCF7. MAPK inhibition shows a relative increase of UCP2 that is comparable with that observed in MCF7 parental cells (Fig. 2). C, MCF7-ERBB2 have elevated levels of UCP2 mRNA, although not proportional to the elevated level of protein. For MCF7 cells, the inhibition of MAPK has only a modest impact on mRNA levels, which is not sustained, whereas Canertinib treatment results in likewise modest but a progressive decrease in mRNA levels. Inhibitor sensitivity is much more pronounced for MCF7-ERBB2 with Canertinib treatment achieving a close to 90% reduction in mRNA levels after 9 h of treatment despite the fact that UO126 shows a much more pronounced impact on cell viability at 9 h of treatment. All mRNA levels are normalized to 18 S rRNA and error bars represent S.E. p values <0.05, <0.01, <0.005, and >0.9 are marked *, **, ***, and □, respectively, and refer to comparisons with respective controls unless pairs are identified explicitly.
The Acute Down-regulation of UCP2 Correlates with a Rapid Change in Mitochondrial Physiology
Because UCP2 localizes to the inner mitochondrial membrane, we first evaluated the mitochondrial oxygen consumption by polarography as a function of ERBB2 signaling or inhibition (Fig. 6A). Maximal down-regulation of UCP2 by NRGβ1 occurs at 2 h and correlates with a 30% reduction in oxygen consumption. Although the physiological consequences of decreased cellular respiratory capacity depend upon every tissue, a 20–40% decrease in oxygen consumption is often seen in tissues from patients suffering from diseases related to mitochondrial defects as well as in elder individuals (50). Adjustment to changing environmental conditions, such as low oxygen tension or hypoxia can also involve adaptive reductions in mitochondrial metabolism in the 20–40% range, accompanied by decreased ROS leakage (51, 52). Hence, compared with known pathological and adaptive states, a 30% reduction represents a highly significant decrease. At the same time, this change is distinct from the above scenarios by the very rapid, reversible nature of the regulation. For the constitutively activated MCF7-ERBB2 cells, ligand stimulation does not present a viable means of controlling the ERBB2 activation status, but the inhibition of amplified ERBB2 with Canertinib provides a means to lower UCP2 mRNA levels and ultimately protein levels (Fig. 5B) in an ERBB2-dependent manner. Despite the different means by which UCP2 protein down-regulation is achieved in MCF7 and MCF7-ERBB2 cells, both scenarios result in a decrease in oxygen consumption (Fig. 6A). By contrast, the 1.8-fold elevation of UCP2 protein after MAPK inhibition is not associated with a consistent change in OXPHOS. For MCF7, oxygen consumption declines despite increased levels of total UCP2 protein. At the longest time point at which MCF7-ERBB2 cells can be exposed to continuous MAPK inhibition without a major impact on viability, elevated levels of UCP2 instead correlate with an increase in oxygen consumption. This and subsequent observations suggest that the changes in UCP2 levels, and possibly changes in its functional state, may translate into different net consequences to mitochondrial regulation at low physiological protein levels and scenarios of overexpression that can be found in pathological settings such as cancer cells (see compilation of data from various assays for ERBB2 gene-amplified BT474 (Fig. 8)).
FIGURE 6.

ERBB2 regulates mitochondrial activity in a UCP2-dependent manner. A, ligand activation of MCF7 reversibly reduces oxygen consumption as does the inhibition of ERBB signaling (and UCP2 down-regulation) by prolonged Canertinib treatment in MCF7-ERBB2 cells (compare Fig. 4, B and C). Relative endogenous oxygen consumption was measured in MCF7 and MCF7-ERBB2 cells after treatment with NRGβ1 (NRG, 30 nm), UO126 (25 μm), or Canertinib (CNT, 1 μm) for different time points as shown in the bar graph. For comparison, superimposed numbers in white boxes represent relative UCP2 protein levels obtained from immunoblots at those conditions (Figs. 2 and 4). B, the decrease in oxygen consumption upon ligand treatment of MCF7 cells is also reflected in a reduced membrane potential but not dependent on a down-regulation of UCP2. Data represent tetramethylrhodamine methyl ester (TMRM) fluorescence relative to respective controls with or without UO126 pretreatment (1 h prior to an additional 2-h ligand treatment as indicated). For graphic representation of fluorescence data, the residual tetramethylrhodamine methyl ester signal after CCCP treatment (approximately 50% of untreated samples) was subtracted as background. C, 2 h of ligand stimulation further reduces (already low) ROS levels in MCF7 cells. The indicated relative dihydroethidium fluorescence values are the average of ∼600 adherent live cells per data set, acquired by fluorescence microscopy and automated object identification. The observed decrease in ROS after NRG treatment is comparable in magnitude to the increase after inhibition with antimycin (AMyc, 10 μm) as a positive control (Ctrl). In contrast to NRG signaling with its associated UCP2 down-regulation, the isolated knockdown of UCP2 by siRNA results in an increase in ROS levels. D, the siRNA knockdown of UCP2 eliminates acute ligand responsiveness, measured as a decrease in membrane potential. Data are shown relative to their respective non-ligand stimulated controls. The assessment of the mitochondrial membrane potential and UCP2 protein levels was done 48 h post-transfection with siRNA or scramble control. Ligand stimulation (NRG, 10 nm) was carried out for 2 h. E, in contrast to the parallel decrease of membrane potential and oxygen consumption in ligand-stimulated and UCP2-depleted MCF7 parental cells, the down-regulation of UCP2 in MCF7-ERBB2 cells by Canertinib treatment or siRNA results in an increase of the membrane potential without increase in oxygen consumption, consistent with the removal of uncoupling capacity. Error bar represent S.D., except for C (S.E.). p values <0.05, <0.01, <0.005, and >0.9 are marked *, **, ***, and □, respectively.
FIGURE 8.

Characterization of UCP2 regulation and its consequences in BT474 cells, an ERBB2 gene-amplified breast cancer cell line (1–2 Mio receptors/cell) that is a model system for ERBB2 driven increases in metastasis and sensitivity to the therapeutic anti-ERBB2 antibody (Herceptin). A, comparison of ERBB2 and UCP2 levels as well as associated constitutive signaling in MCF7 and BT474 cells and transiently ERBB2-transfected MCF7 cells (MCF7-T). L and H represent low and high exposure levels needed to convey the vastly different levels of ERBB2 expression in both cell lines. Note that in contrast to stable MCF7-ERBB2 and BT474 cells UCP2 protein in MCF7-T is elevated but not at the levels seen in stable MCF7-ERBB2 or BT474 lines. This is a reflection of the high steady state levels of phosphorylated MAPK in transient expression settings that is not sustained in stable lines. B, ERBB2 overexpression in BT474 results in more than a 10-fold increase in UCP2 mRNA. C, the utilization of the large access of mRNA in BT474 is less efficient than in MCF7 (and MCF7-ERBB2) cells as indicated by a lower protein/mRNA ratio. D, inhibition of ERBB2 signaling results in a rapid decrease in mRNA levels that is not correlated with MAPK activity (compare with E). All mRNA levels are normalized to 18 S rRNA and error bars represent S.E. E, the long term decrease in UCP2 levels in response to Canertinib is more pronounced due to an over dependence on elevated mRNA levels, whereas MAPK inhibition shows a relative increase in UCP2 protein that is comparable with that observed in MCF7 and MCF7-ERBB2 cells. F, the growth of ERBB2 and UCP2 overexpressing BT474 cells is suppressed after UCP2 knockdown, even at physiological glucose concentrations, reflecting a stronger commitment of BT474 cells to glycolytic metabolism. All error bars represent S.D. p values <0.05, <0.01, and <0.005 are *, **, and ***, respectively.
A putative uncoupling contribution of UCP2 should be reflected in the mitochondrial membrane potential. To also gain insight into the causal involvement of UCP2 in the regulation of mitochondrial OXPHOS by ERBB2, we combined measurements of the mitochondrial membrane potential with an increase in UCP2 levels after short term MAPK inhibition (Fig. 6B) or the siRNA knockdown of endogenous UCP2 (Fig. 6D). At the point of maximum UCP2 down-regulation in ligand-stimulated MCF7 parental cells, the decrease in oxygen consumption coincides with a decrease in the membrane potential. Between datasets derived from different cell preparations (and fluorophore preparations) decreases in fluorescence intensities varied between 20 (e.g. Fig. 6B) and 40% (e.g. Fig. 6D) with CCCP-induced uncoupling being subtracted as a fluorescence baseline. However, within a given triplicate set of experiments, each containing large numbers of individual live cell measurements, fluorescence data were highly reproducible and differences were clearly statistically significant at p values of 0.01, and 0.005, respectively. This acute ligand response of the mitochondria in the presence of decreasing levels of UCP2 suggests that at low expression levels, UCP2 does not contribute an uncoupling activity that is of sufficient capacity to dictate the overall coupling state of the mitochondria. To determine whether the decline in UCP2 protein levels was needed to achieve the ligand-induced decrease in mitochondrial activity, we pretreated cells with UO126 (Fig. 6B). Inhibition of MAPK (ERK1/2) establishes elevated levels of UCP2 protein that are resistant to ligand stimulation, but are, nevertheless, actively turning over (see Fig. 4). Despite stable and elevated levels of UCP2, the ligand-induced decrease in mitochondrial membrane potential was unaltered. This indicates that the drop in membrane potential is not caused by the changes in UCP2 levels and is hence not in conflict with its ability to act qualitatively as an uncoupler at high expression levels. However, this raises the question of the causal involvement of UCP2 in the observed regulatory scheme. To address this, we compared the ligand response in the context of an siRNA knockdown of UCP2 (Fig. 6D). In contrast to the robust ligand-induced decrease of the membrane potential after ligand treatment of 40%, the knockdown of UCP2 effectively abrogates the ERBB2-mediated regulation of mitochondrial activity during the acute phase of signaling. In isolation, the comparison of the (non-normalized) mitochondrial potential after scramble siRNA and siUCP2 treatment does not show an elevated membrane potential but instead a slight decrease of insufficient statistical significance (data not shown). The reduced membrane potential after ligand treatment is expected to lower ROS levels. Although MCF7 cells show low basal ROS levels, those levels are further reduced by ligand stimulation (2 h) and increased on a comparable scale by antimycin as a positive control (Fig. 6C). In contrast to the mitochondrial membrane potential, the siRNA knockdown of UCP2 does in fact increase ROS at levels that are statistically significant, albeit less than the antimycin control. Hence, based on ROS levels, the removal of UCP2 in isolation by siRNA suggest a potential uncoupling contribution. However, its loss does not drive the net ligand response.
Previous studies had demonstrated that UCP2 at high expression levels does contribute a net uncoupling effect. We therefore evaluated the impact of UCP2 removal on oxygen consumption and membrane potential in MCF7-ERBB2 cells (Fig. 6E). In contrast to parental cells, the down-regulation of UCP2 by Canertinib treatment or siRNA knockdown results in an increase in membrane potential alongside a decreased oxygen consumption. This is consistent with a net uncoupling contribution of UCP2 at high expression levels.
The functional studies thus far indicate a regulatory contribution by low levels of UCP2 that are at least dominant over any direct or indirect uncoupling function of UCP2. Net uncoupling contributions emerge at high expression levels. To further characterize how changes in UCP2 levels impact the efficiency of mitochondrial oxygen consumption we determined the RCR by polarography on cell suspensions. The RCR represents the ratio of state III respiration (after pharmacological uncoupling with CCCP) to state IV respiration (inhibition of ATP synthesis with oligomycin). KCN insensitive oxygen consumption was subtracted as background (data not shown). At the peak of ligand-induced down-regulation of UCP2 in MCF7 cells, this yielded a highly reproducible decrease in RCR (−23 ± 1%). This is consistent with the above observation that any loss of UCP2 contributed uncoupling activity at low expression levels does not dominate the overall response in mitochondrial OXPHOS. However, in MCF7-ERBB2 and ERBB2 gene-amplified BT474 cells the decrease in UCP2 mRNA and protein levels after ERBB2 inhibition correlates with an increase in RCR of 22 and 12%, respectively, consistent with the loss of an uncoupling contribution that had been contributed by UCP2 at high expression levels.
Does the ability to regulate mitochondria as part of ERBB2 signaling have phenotypic consequences? To evaluate systemic consequences of the rapid regulation of UCP2 at low endogenous levels, we carried out ligand-induced cell proliferation studies at normal glucose concentrations (5 mm or 50% of standard RPMI), glucose-free, or high glucose stress conditions (25 mm). Proliferation was monitored by Alamar blue assays and results were confirmed by independent manual counts of viable cells to exclude artifacts derived from changes in mitochondrial activity at day 3 of stimulation (data not shown). The knockdown of UCP2 in MCF7 cells (Fig. 7A) induced a small but clearly discernible enhancement of growth in glucose-free media, similar to previous reports for UCP2−/− fibroblasts (24). By contrast, proliferation at high glucose was markedly suppressed. The impact of UCP2 knockdown on growth in high glucose conditions was comparable in the presence or absence of ligand. To evaluate the adaptability to simulated starvation and surplus cycles we altered glucose levels for 2 days between 0 (4 h), 25 (4 h), and 5 mm glucose (16 h). Under those conditions, we also observed a pronounced suppression of growth. However, distinct from conditions of constitutively high glucose, growth suppression was limited to conditions of ligand stimulation (Fig. 7B).
FIGURE 7.
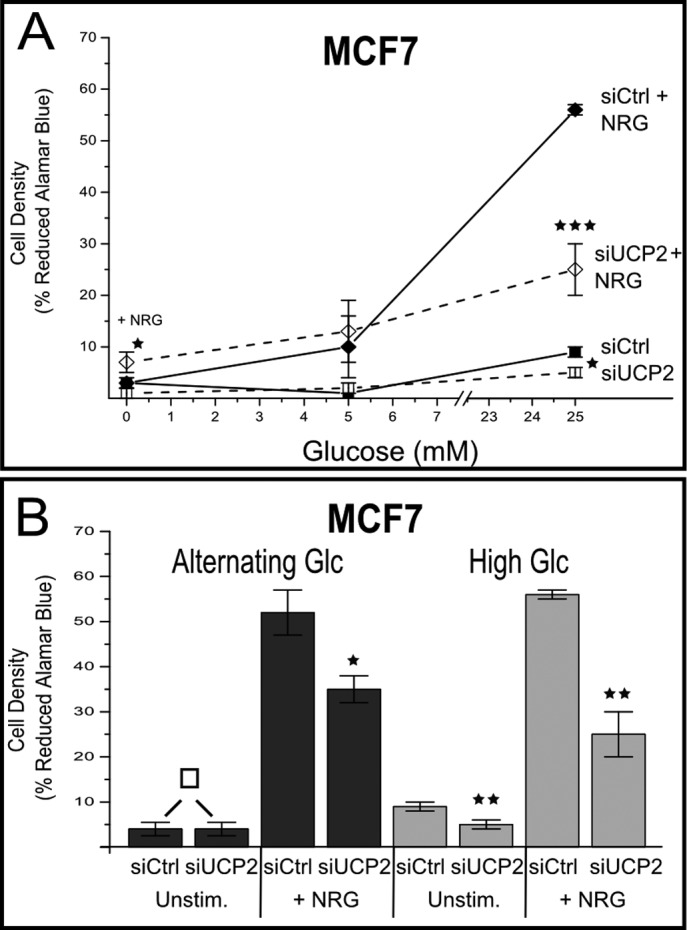
The contribution of UCP2 to ERBB2-mediated proliferation is dependent on glucose availability. A, MCF7 cells, transfected with si-UCP2 RNA or scramble control (siCtrl) were grown for 48 h at the indicated glucose concentrations in the presence or absence of NRGβ1 (NRG, 30 nm). Samples are identified in the graph. Proliferation was evaluated by Alamar blue assay (validated by manual cell count). The relative % of reduced dye is provided as the indicator of proliferation. B, at constitutively high glucose levels, the relative inhibition for si-UCP2 samples compared with scramble control is comparable independent of ligand stimulation. By contrast, at alternating glucose concentrations (5, 0, and 25 mm) the inhibition of growth was limited to conditions of ligand stimulation. All error bars represent S.D. p values <0.05, <0.01, <0.005, and >0.9 are *, **, ***, and □, respectively.
The isogenic nature of engineered MCF7-ERBB2 cells facilitates mechanistic dissection of the UCP2 regulation but does not reflect the full spectrum of changes that occurs in ERBB2-amplified cancers where the ERBB2 is gene amplified including surrounding chromosomal segments. BT474 cells, which are derived from ERBB2 amplified breast cancer, display well over one million ERBB2 receptors per cell (10 times more than the stable MCF7-ERBB2 cells used in our assays). The BT474 model system (compiled results in Fig. 8) effectively reproduce all aspects of ERBB2 UCP2 regulation observed for MCF7-ERBB2, including an effectively identical (1.8-fold) relative increase in UCP2 protein levels after inhibition with UO126. Notably, the reduction in UCP2 levels after inhibition of ERBB2 by Canertinib is much more pronounced on both the mRNA and protein levels, reducing protein levels effectively to those of MCF7 parental cells.
DISCUSSION
At the onset of our study, existing data on the properties of ERBB2 overexpressing cancer cells, as well as the impact of artificially stimulated or inhibited ERBB2 signaling in non-overexpressing tissues, had already pointed toward an involvement of ERBB2 signaling in mitochondrial function. However, especially with the broad changes in gene expression that accompany constitutive ERBB2 signaling upon gene amplification and the frequent progression of mitochondria in these cancer cells to a “dysfunctional state,” a direct functional linkage was not readily evident. Likewise, UCP2 overexpression had been observed in many of the same ERBB2 overexpressing cancers, and its own potency in cellular transformation demonstrated, but its integration with ERBB2 signaling was not known. Our data indicate that ERBB2 amplified cancer cells retain the basic components of a very dynamic and rapid mode of regulation that has distinctly different dynamics at very low levels of ERBB2 and UCP2. This regulation is based on the juxtaposition of transcriptional and translational control. At low levels of ERBB2 and UCP2 this regulation is linked to characteristic temporal aspects of growth factor signaling. Although the basic elements remain in place, the dynamic aspect of this regulation is lost upon overexpression. As a consequence, constitutive signaling by overexpressed ERBB2 results in a dominance of transcriptional regulation and highly elevated UCP2 levels. This in turn emphasizes functional contributions by UCP2 that result in net uncoupling.
For ligand controlled signaling at low levels of ERBB2 and UCP2, MAPK-dependent translational regulation coincides with the acute phase of growth factor signaling. Thus far, it was not clear that mitochondrial OXPHOS was indeed subject to direct growth factor regulation on such a short signaling time scale and that ERBB2 or UCP2 were part of such a regulatory scheme. A distinct contribution by UCP2 at low expression levels is also consistent with the previously reported paradox that overexpressed UCP2 strongly supports the aggressive growth of cancer cells, whereas the loss of normally low levels of UCP2 in the fibroblasts of UCP2−/− knock-out mice also contributes a growth enhancing effect. We therefore placed a strong emphasis on studying the regulation of mitochondrial function and UCP2 at low endogenous expression levels of both UCP2 and ERBB2.
Although fundamental to the study of UCP2 regulation, the analysis of UCP2 and ERBB2 at low levels and in the absence of recombinant overexpression is challenging, and the impact of constitutive ERBB2 signaling on the long term synthesis of UCP2 is more easily accessible. Both, sustained ligand activation of ERBB2/ERBB3 as well as constitutive ERBB2 signaling in MCF7-ERBB2 and BT474 cells, increase steady state levels of UCP2 mRNA and protein. For ERBB2 gene-amplified BT474 cells, this increase is especially pronounced and subject to strong inhibition by ERBB2-directed inhibitors. Given the demonstrated relevance of both ERBB2 and UCP2 in supporting an aggressive cancer phenotype, this regulatory link is of potential therapeutic importance. Mechanistically, we note that at conditions of UCP2 overexpression we can readily discern an uncoupling contribution. This is consistent with existing models for UCP2 function and is also reflected in an increase in RCR of 23%. Our study does not address the source of UCP2-dependent net uncoupling. It may be the consequence of direct UCP2-mediated proton leakage or the indirect consequence of transporting large quantities of charged metabolites, such as the export of pyruvate from mitochondria in support of anaerobic glycolysis.
In a parental MCF7 model system for the ligand response at low receptor (and UCP2) levels, we observe very rapid and all but complete down-regulation of UCP2 protein after ligand stimulation. Ligand responsiveness requires co-receptors, primarily ERBB3. However, sensitivity to ERBB kinase inhibitors and especially the ERBB2-selective therapeutic antibody pertuzumab demonstrates the specific requirement for ERBB2. Following the acute phase of signaling, steady state receptor phosphorylation is low compared with conditions of ERBB2 overexpression. Nevertheless, constitutive ligand stimulation over multiple days does result in a detectable but modest increase in UCP2 protein levels. Much of the normal, growth factor-specific outcome of stimulation is dependent on the characteristic time course of MAPK-mediated signaling during the initial, acute phase of signaling, a phenomenon that has long been known but is still poorly understood. The MAPK-dependent down-regulation of UCP2 results in a rapid depletion at the inherently fast rate of UCP2 turnover. It is noteworthy that in this instability, UCP2 differs sharply from the much longer half-lives of other UCP family members. Their longer half-lives effectively preclude them from taking part in a similar mode of regulation during the relatively narrow time window of acute signaling, that is from initial receptor stimulation to the onset of transcriptional regulation. This also explains the observation that UCP2 mRNA is consistently found across tissues at readily detectable and constant levels, whereas the resulting protein levels vary greatly and often pose a challenge for detection. For MCF7 cells, basal signaling by both ERBB2 and MAPK contribute to the maintenance of low steady state UCP2 levels. Although the inhibition of ERBB2 signaling in BT474 cells effectively reduces UCP2 levels to that of MCF7 parental cells, this inhibition is less pronounced in MCF7-ERBB2 cells or for the basal levels of UCP2 in parental cells. This would suggest that ERBB2-mediated regulation is capable of carrying out a rapid regulation of UCP2 levels that impacts mitochondrial function, but its contribution may be at least partially redundant with regard to the establishment of steady state levels.
The physiological contributions of UCP2 remain controversial, and available data largely rely on scenarios of pre-existing or recombinant overexpression, or at least systems, such as macrophages and islet cells, with inherently elevated levels. By contrast, UCP2 (and ERBB2) expression at low levels is all but ubiquitous across tissues. Alternative models for UCP2 function emphasize its role as a metabolite sensor and transporter involved in regulating the relative utilization of glucose over other energy sources. Such activities could also contribute uncoupling activity, albeit indirectly based on the charge state of the metabolite involved. However, at the low UCP2 levels in most tissues, any direct or indirect uncoupling contribution may simply be too low to result in a noticeable increase in membrane potential or increased coupling efficiency by RCR. Yet, low UCP2 levels alone would not be sufficient to explain why we observe changes in mitochondrial parameters that are in the opposite direction to those that would be expected if UCP2 were to contribute detectable uncoupling contributions. Indeed our data do not indicate an inverse contribution of UCP2 at low levels. Instead, low endogenous levels of UCP2 are needed to ensure responsiveness to ligand, but the interference with UCP2 down-regulation does not abrogate the ligand-induced reduction in the mitochondrial membrane potential. Genipin neither interferes with the regulation of UCP2 protein levels (Fig. 2D) nor the decrease in oxygen consumption (data not shown). This would suggest that minimal levels of UCP2 are needed to facilitate a mode of regulation in a manner that is not associated with its transporter functions. This is in contrast to conditions of high UCP2 levels, where its transporter contributions become readily apparent. At present it is not clear what this “non-transport associated” function would be. Because it requires only small amounts of UCP2 and apparently does not require its transport ability, a regulated participation in a small number of critical protein assemblies would be consistent with such a function.
What could be the potential functions of the observed mode of UCP2 regulation? This may involve different answers for the long term adjustment of cellular metabolism versus the cells ability to adjust to short term fluctuations in glucose supply and external stimuli. The long term up-regulation of UCP2 or in reverse, the sustained down-regulation in macrophages have been linked to its uncoupling contributions and ability to enhance or decrease ROS levels, respectively. In the case of ROS increases in macrophages, it is noteworthy that this long term removal after LPS stimulation is MAPK dependent but utilizes sustained MAPK(p38) signaling, whereas explicitly excluding contributions from MAPK (ERK1/2) with its characteristic time profile of activation. Sustained increases in UCP2 in cancer cells are also consistent with a shift away from aerobic glycolysis for energy production. A large scale export of pyruvate, which is another function that has been assigned to UCP2, would constitute a de facto uncoupling contribution as well. Consistent with such a model, the knockdown of UCP2 suppresses BT474 growth across all external glucose concentrations, reflecting the commitment to glycolytic metabolism (Fig. 8). By contrast, MCF7 cells with their “intact” mitochondria and less glycolytic predisposition are especially impacted by UCP2 knockdown at elevated glucose levels. Interestingly, they display a reversal of UCP2 dependence around physiological levels of glucose (Fig. 7). Our observations for the long term regulation of UCP2 are therefore largely consistent with existing models for UCP2 function but establish for the first time that ERBB2 directly regulates UCP2 expression.
This leaves the question whether the very distinct regulation of UCP2 during the acute phase of growth stimulation serves a specific function. The associated regulation of mitochondrial properties is not merely correlated with UCP2 regulation, but instead is critically dependent on the presence of UCP2 for ligand responsiveness. The cornerstone of this regulation is the juxtaposition of transcriptional activation and translational suppression downstream of the same receptor. Combined with the exceptionally short half-life of UCP2 compared with other UCPs or mitochondrial transporters, this suggests a high emphasis on rapid response capability at the expense of creating a partially futile cycle. Fig. 9 places various regulatory events into the temporal context of the acute phase of ligand responsiveness. This time window between immediate consequences of receptor activation and time delayed and broad transcriptional responses also coincides with the characteristic temporal profile of proliferation-inducing MAPK (ERK1/2) signaling. ERBB2 rapidly relocalizes previously synthesized GLUT1, GLUT3, and GLUT4 to the cell membrane to stimulate glucose uptake. This response is very rapid and peaks at 1.5–2 h (53). By contrast, the transcriptional up-regulation of GLUT1, GLUT3, and the broader transcriptional response (including lactate dehydrogenase) is time delayed. This constitutes a cellular “adaptation gap.” During this time it may be beneficial to temporarily reduce mitochondrial activity. This would establish a buffering system between the rapid fluctuations of metabolites or growth factor signals and the slow and more permanent restructuring of mitochondria, which is only needed if the external stimulus (and metabolite supply) is sustained. This may also explain why the impact of UCP2 knockdown was only specific to ligand-stimulated growth under conditions of fluctuating glucose supply. A role of UCP2 in the ability to adjust to external changes in glucose supply and glycolytic dependence has recently been documented for human pluripotent stem cells. For human pluripotent stem cells, the reversal of their glycolytic dependence and ability to cope with changes in glucose supply is regulated through UCP2 in a manner that goes beyond a role of UCP2 as a suppressor of ROS stress (54). Hence the unanticipated observation that ERBB2 can directly regulate mitochondrial function via UCP2 may provide insight into the nature of ERBB2 overexpression mediated oncogenic transformation as well as the role of signaling by low levels of ERBB2 and UCP2.
FIGURE 9.
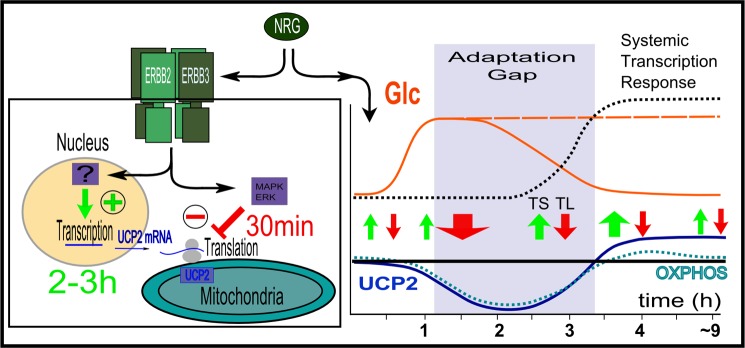
Left, UCP2 regulation as the balance of opposing transcriptional and translational regulation in non-overexpressing cells. Right, time course of changes in UCP2 protein levels and associated changes in mitochondrial OXPHOS. Transcriptional (TS) and translational (TL) regulation of UCP2 are shown relative to ERBB2-mediated and time-delayed systemic transcriptional responses as well as rapid changes in glucose uptake, described first for skeletal muscle (53). Depending on the glucose supply and subsequent up-regulation of GLUT transporter species, glucose supply may be sustained or not. The time window between the rapid relocation of previously synthesized GLUT transporters to the surface and ERBB-initiated transcriptional responses constitutes a potential “adaptation gap” during which mitochondrial activity is transiently reduced, thus favoring anaerobic utilization of glucose and minimizing spikes in ROS production.
This work was supported, in whole or in part, by National Institutes of Health Grant R01-CA98881 (to R. L.) from the NCI and the Braman Family Breast Cancer Institute (to R. L.).
- OXPHOS
- oxidative phosphorylation
- UCP
- uncoupling protein
- ERBB2
- erythroblastic leukemia viral oncogene homolog
- HER2
- human epidermal growth factor receptor 2
- CCCP
- carbonyl cyanide m-chlorophenyl hydrazone
- RCR
- respiratory control ratio
- NRGβ1
- neuregulin-β1
- CHX
- cycloheximide.
REFERENCES
- 1. Klingenberg M., Winkler E. (1985) The reconstituted isolated uncoupling protein is a membrane potential driven H+ translocator. EMBO J. 4, 3087–3092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Winkler E., Klingenberg M. (1994) Effect of fatty acids on H+ transport activity of the reconstituted uncoupling protein. J. Biol. Chem. 269, 2508–2515 [PubMed] [Google Scholar]
- 3. Rial E., González-Barroso M., Fleury C., Iturrizaga S., Sanchis D., Jiménez-Jiménez J., Ricquier D., Goubern M., Bouillaud F. (1999) Retinoids activate proton transport by the uncoupling proteins UCP1 and UCP2. EMBO J. 18, 5827–5833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nedergaard J., Golozoubova V., Matthias A., Asadi A., Jacobsson A., Cannon B. (2001) UCP1. The only protein able to mediate adaptive non-shivering thermogenesis and metabolic inefficiency. Biochim. Biophys. Acta 1504, 82–106 [DOI] [PubMed] [Google Scholar]
- 5. Divakaruni A. S., Brand M. D. (2011) The regulation and physiology of mitochondrial proton leak. Physiology 26, 192–205 [DOI] [PubMed] [Google Scholar]
- 6. Azzu V., Affourtit C., Breen E. P., Parker N., Brand M. D. (2008) Dynamic regulation of uncoupling protein 2 content in INS-1E insulinoma cells. Biochim. Biophys. Acta 1777, 1378–1383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fleury C., Neverova M., Collins S., Raimbault S., Champigny O., Levi-Meyrueis C., Bouillaud F., Seldin M. F., Surwit R. S., Ricquier D., Warden C. H. (1997) Uncoupling protein-2. A novel gene linked to obesity and hyperinsulinemia. Nat. Genet. 15, 269–272 [DOI] [PubMed] [Google Scholar]
- 8. Pecqueur C., Alves-Guerra M. C., Gelly C., Levi-Meyrueis C., Couplan E., Collins S., Ricquier D., Bouillaud F., Miroux B. (2001) Uncoupling protein 2, in vivo distribution, induction upon oxidative stress, and evidence for translational regulation. J. Biol. Chem. 276, 8705–8712 [DOI] [PubMed] [Google Scholar]
- 9. Rousset S., Mozo J., Dujardin G., Emre Y., Masscheleyn S., Ricquier D., Cassard-Doulcier A. M. (2007) UCP2 is a mitochondrial transporter with an unusual very short half-life. FEBS Lett. 581, 479–482 [DOI] [PubMed] [Google Scholar]
- 10. Andrews Z. B., Horvath T. L. (2009) Uncoupling protein-2 regulates lifespan in mice. Am. J. Physiol. Endocrinol. Metab. 296, E621–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Basu Ball W., Kar S., Mukherjee M., Chande A. G., Mukhopadhyaya R., Das P. K. (2011) Uncoupling protein 2 negatively regulates mitochondrial reactive oxygen species generation and induces phosphatase-mediated anti-inflammatory response in experimental visceral Leishmaniasis. J. Immunol. 187, 1322–1332 [DOI] [PubMed] [Google Scholar]
- 12. Bugger H., Guzman C., Zechner C., Palmeri M., Russell K. S., Russell R. R., 3rd. (2011) Uncoupling protein down-regulation in doxorubicin-induced heart failure improves mitochondrial coupling but increases reactive oxygen species generation. Cancer Chemother. Pharmacol. 67, 1381–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Giardina T. M., Steer J. H., Lo S. Z., Joyce D. A. (2008) Uncoupling protein-2 accumulates rapidly in the inner mitochondrial membrane during mitochondrial reactive oxygen stress in macrophages. Biochim. Biophys. Acta 1777, 118–129 [DOI] [PubMed] [Google Scholar]
- 14. Andrews Z. B. (2010) Uncoupling protein-2 and the potential link between metabolism and longevity. Curr. Aging Sci. 3, 102–112 [DOI] [PubMed] [Google Scholar]
- 15. Teshima Y., Akao M., Jones S. P., Marbán E. (2003) Uncoupling protein-2 overexpression inhibits mitochondrial death pathway in cardiomyocytes. Circ. Res. 93, 192–200 [DOI] [PubMed] [Google Scholar]
- 16. Bodyak N., Rigor D. L., Chen Y. S., Han Y., Bisping E., Pu W. T., Kang P. M. (2007) Uncoupling protein 2 modulates cell viability in adult rat cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 293, H829–835 [DOI] [PubMed] [Google Scholar]
- 17. Dalgaard L. T. (2012) UCP2 mRNA expression is dependent on glucose metabolism in pancreatic islets. Biochem. Biophys. Res. Commun. 417, 495–500 [DOI] [PubMed] [Google Scholar]
- 18. Arsenijevic D., Onuma H., Pecqueur C., Raimbault S., Manning B. S., Miroux B., Couplan E., Alves-Guerra M. C., Goubern M., Surwit R., Bouillaud F., Richard D., Collins S., Ricquier D. (2000) Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat. Genet. 26, 435–439 [DOI] [PubMed] [Google Scholar]
- 19. Emre Y., Hurtaud C., Karaca M., Nubel T., Zavala F., Ricquier D. (2007) Role of uncoupling protein UCP2 in cell-mediated immunity. How macrophage-mediated insulitis is accelerated in a model of autoimmune diabetes. Proc. Natl. Acad. Sci. U.S.A. 104, 19085–19090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dalgaard L. T. (2011) Genetic variance in uncoupling protein 2 in relation to obesity, type 2 diabetes, and related metabolic traits. Focus on the functional −866G>A promoter variant (rs659366). J. Obes. 2011, 340241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dhamrait S. S., Stephens J. W., Cooper J. A., Acharya J., Mani A. R., Moore K., Miller G. J., Humphries S. E., Hurel S. J., Montgomery H. E. (2004) Cardiovascular risk in healthy men and markers of oxidative stress in diabetic men are associated with common variation in the gene for uncoupling protein 2. Eur. Heart J. 25, 468–475 [DOI] [PubMed] [Google Scholar]
- 22. Harper M. E., Antoniou A., Villalobos-Menuey E., Russo A., Trauger R., Vendemelio M., George A., Bartholomew R., Carlo D., Shaikh A., Kupperman J., Newell E. W., Bespalov I. A., Wallace S. S., Liu Y., Rogers J. R., Gibbs G. L., Leahy J. L., Camley R. E., Melamede R., Newell M. K. (2002) Characterization of a novel metabolic strategy used by drug-resistant tumor cells. FASEB J. 16, 1550–1557 [DOI] [PubMed] [Google Scholar]
- 23. Ayyasamy V., Owens K. M., Desouki M. M., Liang P., Bakin A., Thangaraj K., Buchsbaum D. J., LoBuglio A. F., Singh K. K. (2011) Cellular model of Warburg effect identifies tumor promoting function of UCP2 in breast cancer and its suppression by genipin. PloS One 6, e24792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pecqueur C., Bui T., Gelly C., Hauchard J., Barbot C., Bouillaud F., Ricquier D., Miroux B., Thompson C. B. (2008) Uncoupling protein-2 controls proliferation by promoting fatty acid oxidation and limiting glycolysis-derived pyruvate utilization. FASEB J. 22, 9–18 [DOI] [PubMed] [Google Scholar]
- 25. Trenker M., Malli R., Fertschai I., Levak-Frank S., Graier W. F. (2007) Uncoupling proteins 2 and 3 are fundamental for mitochondrial Ca2+ uniport. Nat. Cell Biol. 9, 445–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Turner J. D., Gaspers L. D., Wang G., Thomas A. P. (2010) Uncoupling protein-2 modulates myocardial excitation-contraction coupling. Circ. Res. 106, 730–738 [DOI] [PubMed] [Google Scholar]
- 27. Garlid K. D., Orosz D. E., Modrianský M., Vassanelli S., Jezek P. (1996) On the mechanism of fatty acid-induced proton transport by mitochondrial uncoupling protein. J. Biol. Chem. 271, 2615–2620 [DOI] [PubMed] [Google Scholar]
- 28. Rupprecht A., Sokolenko E. A., Beck V., Ninnemann O., Jaburek M., Trimbuch T., Klishin S. S., Jezek P., Skulachev V. P., Pohl E. E. (2010) Role of the transmembrane potential in the membrane proton leak. Biophys. J. 98, 1503–1511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hurtaud C., Gelly C., Chen Z., Lévi-Meyrueis C., Bouillaud F. (2007) Glutamine stimulates translation of uncoupling protein 2mRNA. Cell. Mol. Life Sci. 64, 1853–1860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Criscuolo F., Mozo J., Hurtaud C., Nübel T., Bouillaud F. (2006) UCP2, UCP3, avUCP, what do they do when proton transport is not stimulated? Possible relevance to pyruvate and glutamine metabolism. Biochim. Biophys. Acta 1757, 1284–1291 [DOI] [PubMed] [Google Scholar]
- 31. Bouillaud F. (2009) UCP2, not a physiologically relevant uncoupler but a glucose sparing switch impacting ROS production and glucose sensing. Biochim. Biophys. Acta 1787, 377–383 [DOI] [PubMed] [Google Scholar]
- 32. Pecqueur C., Alves-Guerra C., Ricquier D., Bouillaud F. (2009) UCP2, a metabolic sensor coupling glucose oxidation to mitochondrial metabolism? IUBMB Life 61, 762–767 [DOI] [PubMed] [Google Scholar]
- 33. Ding Y., Liu Z., Desai S., Zhao Y., Liu H., Pannell L. K., Yi H., Wright E. R., Owen L. B., Dean-Colomb W., Fodstad O., Lu J., LeDoux S. P., Wilson G. L., Tan M. (2012) Receptor tyrosine kinase ErbB2 translocates into mitochondria and regulates cellular metabolism. Nat. Commun. 3, 1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. García-Rivello H., Taranda J., Said M., Cabeza-Meckert P., Vila-Petroff M., Scaglione J., Ghio S., Chen J., Lai C., Laguens R. P., Lloyd K. C., Hertig C. M. (2005) Dilated cardiomyopathy in Erb-b4-deficient ventricular muscle. Am. J. Physiol. Heart Circ. Physiol. 289, H1153–1160 [DOI] [PubMed] [Google Scholar]
- 35. Crone S. A., Zhao Y. Y., Fan L., Gu Y., Minamisawa S., Liu Y., Peterson K. L., Chen J., Kahn R., Condorelli G., Ross J., Jr., Chien K. R., Lee K. F. (2002) ErbB2 is essential in the prevention of dilated cardiomyopathy. Nat. Med. 8, 459–465 [DOI] [PubMed] [Google Scholar]
- 36. Gordon L. I., Burke M. A., Singh A. T., Prachand S., Lieberman E. D., Sun L., Naik T. J., Prasad S. V., Ardehali H. (2009) Blockade of the erbB2 receptor induces cardiomyocyte death through mitochondrial and reactive oxygen species-dependent pathways. J. Biol. Chem. 284, 2080–2087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ewer M. S., Gibbs H. R., Swafford J., Benjamin R. S. (1999) Cardiotoxicity in patients receiving transtuzumab (Herceptin). Primary toxicity, synergistic or sequential stress, or surveillance artifact? Semin. Oncol. 26, 96–101 [PubMed] [Google Scholar]
- 38. Uray I. P., Connelly J. H., Thomázy V., Shipley G. L., Vaughn W. K., Frazier O. H., Taegtmeyer H., Davies P. J. (2002) Left ventricular unloading alters receptor tyrosine kinase expression in the failing human heart. J. Heart Lung Transplant. 21, 771–782 [DOI] [PubMed] [Google Scholar]
- 39. Rohrbach S., Niemann B., Silber R. E., Holtz J. (2005) Neuregulin receptors erbB2 and erbB4 in failing human myocardium. Depressed expression and attenuated activation. Basic Res. Cardiol. 100, 240–249 [DOI] [PubMed] [Google Scholar]
- 40. Lemmens K., Doggen K., De Keulenaer G. W. (2011) Activation of the neuregulin/ErbB system during physiological ventricular remodeling in pregnancy. Am. J. Physiol. Heart Circ. Physiol. 300, H931–942 [DOI] [PubMed] [Google Scholar]
- 41. Schafer Z. T., Grassian A. R., Song L., Jiang Z., Gerhart-Hines Z., Irie H. Y., Gao S., Puigserver P., Brugge J. S. (2009) Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 461, 109–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Warren C. M., Kani K., Landgraf R. (2006) The N-terminal domains of Neuregulin 1 confer signal attenuation. J. Biol. Chem. 281, 27306–27316 [DOI] [PubMed] [Google Scholar]
- 43. Landgraf R., Eisenberg D. (2000) Heregulin reverses the oligomerization of HER3. Biochemistry 39, 8503–8511 [DOI] [PubMed] [Google Scholar]
- 44. Landgraf R., Pegram M., Slamon D. J., Eisenberg D. (1998) Cytotoxicity and specificity of directed toxins composed of diphtheria toxin and the EGF-like domain of heregulin β1. Biochemistry 37, 3220–3228 [DOI] [PubMed] [Google Scholar]
- 45. Kortenjann M., Shaw P. E. (1995) Raf-1 kinase and ERK2 uncoupled from mitogenic signals in rat fibroblasts. Oncogene 11, 2105–2112 [PubMed] [Google Scholar]
- 46. Pegram M. D., Finn R. S., Arzoo K., Beryt M., Pietras R. J., Slamon D. J. (1997) The effect of HER-2/neu overexpression on chemotherapeutic drug sensitivity in human breast and ovarian cancer. Oncogene 15, 537–547 [DOI] [PubMed] [Google Scholar]
- 47. Zhang C. Y., Parton L. E., Ye C. P., Krauss S., Shen R., Lin C. T., Porco J. A., Jr., Lowell B. B. (2006) Genipin inhibits UCP2-mediated proton leak and acutely reverses obesity- and high glucose-induced beta cell dysfunction in isolated pancreatic islets. Cell Metab. 3, 417–427 [DOI] [PubMed] [Google Scholar]
- 48. Zhou H., Zhao J., Zhang X. (2009) Inhibition of uncoupling protein 2 by genipin reduces insulin-stimulated glucose uptake in 3T3-L1 adipocytes. Arch. Biochem. Biophys. 486, 88–93 [DOI] [PubMed] [Google Scholar]
- 49. Mailloux R. J., Adjeitey C. N., Harper M. E. (2010) Genipin-induced inhibition of uncoupling protein-2 sensitizes drug-resistant cancer cells to cytotoxic agents. PloS One 5, e13289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wallace D. C. (2005) A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer. A dawn for evolutionary medicine. Annu. Rev. Genet. 39, 359–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yang J., Staples O., Thomas L. W., Briston T., Robson M., Poon E., Simões M. L., El-Emir E., Buffa F. M., Ahmed A., Annear N. P., Shukla D., Pedley B. R., Maxwell P. H., Harris A. L., Ashcroft M. (2012) Human CHCHD4 mitochondrial proteins regulate cellular oxygen consumption rate and metabolism and provide a critical role in hypoxia signaling and tumor progression. J. Clin. Invest. 122, 600–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tello D., Balsa E., Acosta-Iborra B., Fuertes-Yebra E., Elorza A., Ordóñez Á., Corral-Escariz M., Soro I., López-Bernardo E., Perales-Clemente E., Martínez-Ruiz A., Enríquez J. A., Aragonés J., Cadenas S., Landázuri M. O. (2011) Induction of the mitochondrial NDUFA4L2 protein by HIF-1alpha decreases oxygen consumption by inhibiting Complex I activity. Cell Metab. 14, 768–779 [DOI] [PubMed] [Google Scholar]
- 53. Suárez E., Bach D., Cadefau J., Palacin M., Zorzano A., Gumá A. (2001) A novel role of neuregulin in skeletal muscle. Neuregulin stimulates glucose uptake, glucose transporter translocation, and transporter expression in muscle cells. J. Biol. Chem. 276, 18257–18264 [DOI] [PubMed] [Google Scholar]
- 54. Zhang J., Khvorostov I., Hong J. S., Oktay Y., Vergnes L., Nuebel E., Wahjudi P. N., Setoguchi K., Wang G., Do A., Jung H. J., McCaffery J. M., Kurland I. J., Reue K., Lee W. N., Koehler C. M., Teitell M. A. (2011) UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J. 30, 4860–4873 [DOI] [PMC free article] [PubMed] [Google Scholar]