

Background: Cryptochromes (CRYs) are transcriptional repressors that are critical components of the circadian clock.
Results: We have identified a phosphorylation site in the CRY1 tail that is negatively regulated by the DNA repair enzyme DNA-dependent protein kinase.
Conclusion: Phosphorylation of CRY1 on Ser-588 increases its half-life and lengthens the circadian period.
Significance: The C-terminal tail of CRY1 modulates period length.
Keywords: Circadian Clock, Phosphorylation, Protein Kinases, Protein Stability, Transcription Repressor
Abstract
The Cryptochrome (CRY) proteins are critical components of the mammalian circadian clock and act to rhythmically repress the activity of the transcriptional activators CLOCK and BMAL1 at the heart of the clock mechanism. The CRY proteins are part of a large repressive complex, the components of which are not completely known. Using mass spectroscopy, we identified the catalytic subunit of DNA-dependent protein kinase as a CRY-interacting protein and found that loss or inhibition of this kinase results in circadian rhythms with abnormally long periods. We then identified serine 588 in the C-terminal tail of mouse CRY1 as a potential DNA-PK phosphorylation site but surprisingly found that the phosphomimetic mutation S588D also results in long period rhythms, similar to the loss of DNA-PK. Consistent with this, we found that phosphorylation of this site is increased in cells lacking DNA-PK, suggesting that DNA-PK negatively regulates the phosphorylation of this site most likely through indirect means. Furthermore, we found that phosphorylation of this site increases the stability of the CRY1 protein and prevents FBXL3-mediated degradation. The phosphorylation of this site is robustly rhythmic in mouse liver nuclei, peaking in the middle of the circadian day at a time when CRY1 levels are declining. Therefore, these data suggest a new role for the C-terminal tail of CRY1 in which phosphorylation rhythmically regulates CRY1 stability and contributes to the proper circadian period length.
Introduction
Circadian clocks are endogenous timekeeping mechanisms that drive daily oscillations in many aspects of behavior and physiology. In mammals, these clocks are distributed in cells throughout the body and control the rhythmic expression of thousands of genes (reviewed in Ref. 1). The core mechanism of the circadian clock is composed of a transcription/translation negative feedback loop in which a heterodimeric transcription factor composed of CLOCK and BMAL1 proteins activates expression of genes encoding components of a repressive complex, PERIOD (PER) and CRYPTOCHROME (CRY) (2). PER and CRY proteins form complexes with each other and with other proteins, translocate back into the nucleus, and repress the activity of CLOCK/BMAL1, thereby turning off their own synthesis. Eventual regulated degradation of the repressive components allows the CLOCK/BMAL1-driven transcription to begin again for the next cycle.
There are two CRY proteins in mammals, CRY1 and CRY2, and at least one of these proteins is required for a functioning clock; loss of both results in arrhythmicity (3, 4). Loss of CRY1 results in short circadian periods, whereas a loss of CRY2 results in long periods (3, 4), suggesting that these two proteins have opposing roles in setting the correct circadian period. The CRY proteins consist of well conserved N-terminal “photolyase homology regions” (PHR3; signifying their sequence similarity to the light-activated DNA repair enzymes, photolyases) and C-terminal tails that are much more highly divergent. Although the mechanism by which CRYs repress CLOCK/BMAL1 activity is not well understood, the PHR domain appears to be sufficient for this function as long as it includes a coiled coil domain near the junction between the PHR and the tail (5–7). The role of the C-terminal tails is more mysterious, although the CRY1 C terminus may have a role in nuclear localization and may facilitate BMAL1 binding (7, 8), and phosphorylation of the CRY2 C terminus destabilizes the protein (9, 10).
In Drosophila CRY, the C-terminal tail plays a regulatory role, and interactions of the tail with the PHR domain keep the protein in an inactive conformation in the dark, which can be reversed by loss of this interaction during light activation (11, 12). This model is supported by recent crystal structures, which show that the C-terminal helix docks in a groove of the PHR domain that is analogous to the DNA-binding groove in photolyase (13–15). Mammalian CRYs are not activated by light, and their C-terminal tails are not conserved with the Drosophila CRY C-terminal tail. It is unknown whether the tails play an analagous regulatory role. Recent crystal structures of mammalian CRY1 and CRY2 do not include the tail regions (13, 16).
Post-translational modifications of the CRY proteins play an important role in determining the period length of circadian rhythms. The stability of both CRY1 and CRY2 are regulated through ubiquitination by Skp1-Cul1-F-box protein (SCF) ubiquitin ligase complexes followed by proteasomal degradation. The SCF complex containing FBXL3 (SCFFBXL3) controls CRY degradation in the nucleus, whereas the FBXL21-containing complex (SCFFBXL21) mediates CRY ubiquitination in the cytoplasm and contributes to appropriate CRY degradation in the nucleus by antagonizing SCFFBXL3 activity on CRY (17–21). Phosphorylation of CRY1 by AMP-activated protein kinase (AMPK) promotes ubiquitination by SCFFBXL3 (22). Loss of function mutations in the Fbxl3 gene stabilize the CRY proteins and lengthen the circadian period (17, 19, 20), whereas mutation in Fbxl21 shortens circadian periods (18, 21). In addition, a mechanism that is specific for CRY2 includes the phosphorylation of CRY2 C-terminal tail, first at Ser-557 by the priming kinase DYRK1A and then at Ser-553 by GSK-3, which generates a degradation signal, resulting in proteasomal degradation of CRY2 through an undiscovered mechanism (9, 10). Knockdown of DYRK1A results in abnormal accumulation of CRY2 in the cytoplasm and a shortened circadian period (9).
Here we identify a phosphorylation site in the C-terminal tail of CRY1 on serine 588 that is regulated indirectly by the kinase DNA-PK. Phosphorylation of this residue causes the circadian period to lengthen and, unlike the previously identified phosphorylation sites, increases the stability of CRY1 by preventing FBXL3-dependent degradation. These data suggest that the CRY1 C-terminal tail is an important modulatory domain that contributes to period determination.
EXPERIMENTAL PROCEDURES
Animals
The animal experiments were conducted using protocols approved by the Animal Care and Use Committee of University of Texas Southwestern Medical Center. Eight-week-old male mice (C57BL/6J) were entrained to 12-h light/12-h dark cycles. After entrainment for at least 2 weeks, the animals were placed in constant dark conditions prior to tissue collection.
Cells and Cell Culture
U2OS-Bmal1-luciferase cells were a gift from Julie Baggs and John Hogenesch (23), and U2OS-Per2-luciferase cells were generated using the Per2 3.2-kb full promoter in pGL3 basic (24) followed by clonal selection. The cells were grown as previously described (25). The catalytic subunits of DNA-dependent protein kinase (DNA-PKcs) WT and KO MEF were grown in DMEM supplemented with 10% FBS, 100 units/ml penicillin, 100 mg/ml streptomycin and cultured at 37 °C in a humidified incubator with 5% CO2. Cells were synchronized with 100 nm dexamethasone, and real time bioluminescence was recorded as described (26). The rhythms were analyzed by performing base-line subtraction followed by sine curve fitting using the Lumicycle software (Actimetrics). NU7441 was purchased from Tocris Bioscience.
Plasmids and siRNA Transfections
Mammalian expression vectors encoding CRY and PER proteins were generated as described (27). All point mutations were introduced by QuikChange site-directed mutagenesis (Stratagene) and verified by sequencing. Transient transfections of HEK293 cells were carried out with X-tremeGENE 9 transfection reagent (Roche Applied Science) according to the manufacturer's instructions. siRNA oligonucleotides designed against DNA-PKcs (28) were transfected with RNAiMax (Invitrogen) according to the manufacturer's instructions.
Antibodies
We generated CRY1 antibodies (epitope: amino acids 496–606) in guinea pigs (Cocalico Biologicals). Other antibodies were obtained from the following commercial vendors: anti-DNA-PKcs 25-4,18-2 monoclonal antibody (NeoMarkers), RPA32 S33 (Bethyl Laboratories), anti-Myc and anti-tubulin (Cell Signaling), anti-FLAG (Sigma), and mPER1 and mPER2 (KeraFAST). For the CRY1 protein stability assay, the membranes were probed with goat anti-mouse IR-Dye 680RD labeled secondary antisera and were imaged using a LiCor Odyssey scanner (LiCor, Lincoln, NE).
Protein Extraction, Immunoblot, and Co-immunoprecipitation Experiments
Co-immunoprecipitation experiments were carried out with mouse liver nuclear extracts or with whole cell lysates. Proteins from mouse liver nuclei were prepared according to the NUN procedure (29). SDS-PAGE, immunoblotting, and immunoprecipitations were performed according to standard protocols as in Ref. 30. Signals were visualized by chemiluminescence using BM chemiluminescence blotting substrate from Roche Applied Science.
In Vitro Kinase Assay
In vitro kinase assays were performed as in Ref. 31. Phosphorylation reactions contained 25 mm Tris-HCl, pH 7.9, 25 mm MgCl2, 1.5 mm DTT, 50 mm KCl, 10% glycerol, 100 ng of sonicated herring DNA, 0.16 μm [γ-32P]ATP (6000 Ci/mmol), 8 nm DNA-PKcs, 20 nm Ku70/80, and GST-tagged mCRY1 carboxyl-terminal domain protein. The final volume was 10 μl. Reactions were incubated for 30 min at 30 °C and terminated by the addition of SDS-PAGE sample buffer.
Real Time Circadian Reporter Assay Using Cry1−/−/Cry2−/− Cells
Cry1−/−/Cry2−/− double knock-out MEF and the expression vector for Cry1 rescue were generous gifts from Dr. Hiroki R. Ueda and Dr. Andrew C. Liu, and real time circadian assays were performed according to their protocol (32).
RESULTS
CRY proteins are critical components of the large transcriptional repressive complex at the heart of the mammalian circadian clock mechanism, but the complete complement of proteins in this complex is not known. Therefore, to further characterize this complex, we immunoprecipitated CRY2-associated proteins from HEK293 cells expressing FLAG-tagged CRY2 and performed mass spectrometry to identify the interacting proteins. In addition to identifying peptides from proteins already known to interact with CRYs, such as PER1, FBXL3, and RACK1 (17, 19, 20, 33–36), we identified a number of peptides corresponding to proteins that had not been previously identified, including 19 peptides from DNA-PKcs (Table 1). DNA-PKcs belongs to a family of phosphatidylinositol 3-kinase-like protein kinases (PIKKs), other members of which include ATM, ATR, mTOR, hSMG-1, and TRRAP (37). In humans, DNA-PK plays an essential role in nonhomologous end joining-mediated double-stranded DNA break repair, and its kinase activity is activated by double-stranded breaks in DNA (38). However, it also plays roles in other processes, such as phosphorylation of transcription factors involved in the response to insulin signaling (39) and autoimmune regulation (40).
TABLE 1.
Peptides obtained from DNA-PKcs from mass spectroscopy analysis of CRY2-interacting proteins
The peptides are from gi 119607089 (protein kinase, DNA-activated, catalytic polypeptide, isoform CRA_d (Homo sapiens)).
| HGDLPDIQIK |
| LGLPGDEVDNK |
| QITQSALLAEAR |
| FMNAVFFLLPK |
| LNESTFDTQITK |
| DLLLNTMSQEEK |
| VVQMLGSLGGQINK |
| NLLIFENLIDLK |
| SLGPPQGEEDSVPR |
| ILELSGSSSEDSEK |
| NLLTVTSSDEMMK |
| YNFPVEVEVPMER |
| AQEPESGLSEETQVK |
| SDPGLLTNTMDVFVK |
| TVSLLDENNVSSYLSK |
| MEVQEQEEDISSLIR |
| TVGALQVLGTEAQSSLLK |
| ATQQQHDFTLTQTADGR |
| LTPLPEDNSMNVDQDGDPSDR |
To validate this interaction and to further investigate the role of DNA-PKcs in the mammalian circadian clock, we examined whether DNA-PKcs could interact with other repressive components of the circadian clock mechanism. HEK cells were transfected with tagged clock proteins and immunoprecipitated with an antibody to endogenous DNA-PKcs (Fig. 1A). In these experiments DNA-PKcs interacted most robustly with CRY1, although addition of ethidium bromide to the co-immunoprecipitation experiment to disrupt DNA-protein interactions (41) resulted in a marked increase in the interaction of DNA-PKcs and CRY2, PER1, and, to a lesser extent, PER2 (Fig. 1A). This increase in the presence of ethidium bromide suggests that DNA is not required for these interactions, and preventing interaction of DNA-PK with DNA may increase its ability to interact with the clock proteins.
FIGURE 1.

DNA-PKcs interacts with clock proteins and regulates circadian period length. A, co-immunoprecipitation (IP) shows interaction of DNA-PKcs with CRY and PER proteins with and without addition of ethidium bromide (EtBr). B, Western blot (WB) showing significantly reduced DNA-PKcs levels in U2OS cells following siRNA treatment. C, DNA-PKcs siRNA treatment of U2OS cells carrying the Per2-luc reporter results in rhythms with long periods. Cells were synchronized at time 0 with dexamethasone. The data shown are base line-subtracted luciferase measurements. D, quantitation of period lengths from four independent cultures of Per2-luc U2OS cells with control or DNA-PK siRNA treatment as in C. **, p < 0.01. E and F, inhibition of DNA-PK activity by DNA-PK inhibitor NU7441 also shows a dose-dependent period lengthening of the circadian clock in U2OS cells as measured in real time using the Bmal1-luc (E) and Per2-luc (F) reporters. G, inhibition of DNA-PKcs activity also affects endogenous circadian mRNA expression. U2OS cells were synchronized by a dexamethasone (Dex) shock, and RNAs were extracted at 4-h intervals starting 10 h after the shock. NU7441 (10 μm) was added 2 h after dexamethasone treatment. mRNA analysis was done by real time PCR using specific primers as in Ref. 69. The plotted values are the mean values ± S.D. from three independent experiments. DMSO, dimethyl sulfoxide.
To determine whether this interaction is biologically relevant, we tested the effect of inactivation or knockdown of DNA-PKcs on circadian rhythms. Knockdown of DNA-PKcs using siRNAs (Fig. 1B) in U2OS cells carrying the luciferase reporter gene under the control of the Per2 promoter (Per2-luc) and synchronized with dexamethasone significantly lengthened circadian periods (Fig. 1, C and D). Likewise, dose-dependent increases of the circadian period lengths of both Bmal1-luc (Fig. 1E) and Per2-luc (Fig. 1F) luciferase rhythms were observed when synchronized U2OS cells were treated with the specific DNA-PKcs inhibitor, NU7441 (42). Endogenous clock mRNAs from these cells also showed altered rhythms in the presence of the inhibitor (Fig. 1G).
DNA-PKcs is a nuclear serine/threonine protein kinase with specificity for serine/threonine-glutamine (S/T-Q) motifs and a preference for domains that contain clustered S/T-Q sites (37). Analysis of the clock protein sequences revealed several S/T-Q motifs, with a cluster of three of these sites in the C-terminal tail of CRY1 (Fig. 2A). Furthermore, one of these SQ sites (Ser-588) had previously been identified by phosphopeptide mapping in AD293 (HEK293 derivative) cells (22). Because of these observations and the fact that DNA-PK interacted more strongly with CRY1 than CRY2, we carried out in vitro kinase assays using the purified CRY1 C-terminal tail as the substrate and found that DNA-PK can phosphorylate the tail of CRY1 (Fig. 2B). Mutations in the three SQ sites revealed that Ser-551 is the preferred site but that mutation of all three SQ sequences was necessary to fully prevent phosphorylation (Fig. 2B), suggesting that all three sites can be used. However, when full-length CRY1 protein was used as substrate, no phosphorylation was observed, suggesting that the conformation of the intact protein makes the tail inaccessible under these conditions (Fig. 2C). We also could not detect significant phosphorylation of CRY2, PER1, PER2, CLOCK, or BMAL1 by DNA-PK in our assays (data not shown).
FIGURE 2.
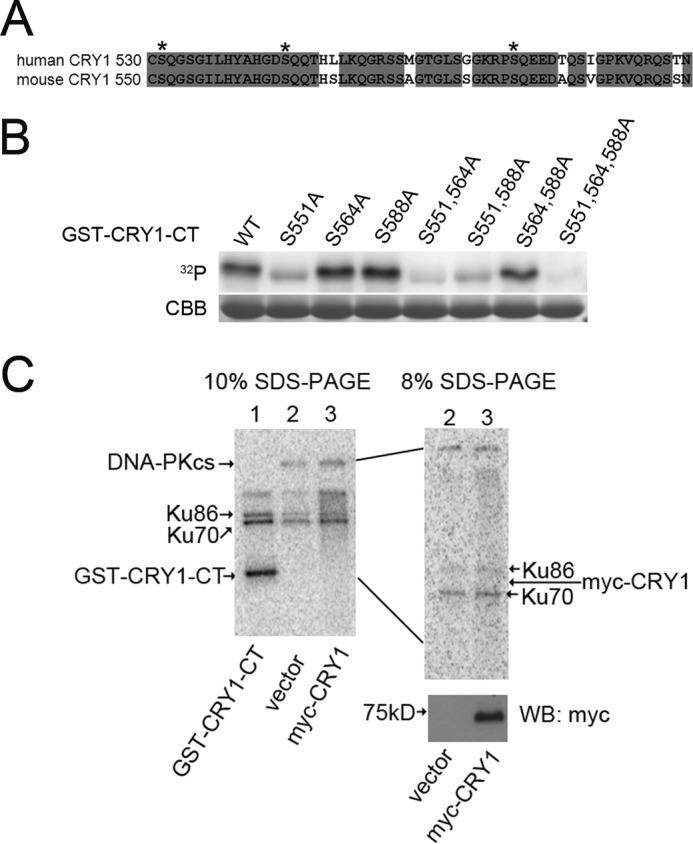
DNA-PK can phosphorylate isolated CRY1 C-terminal tails but not the intact CRY1 protein. A, sequence alignment of human (top row) and mouse (bottom row) CRY1 C-terminal tails. Asterisks mark the three serines in SQ motifs. Gray shading denotes conserved amino acids. B, in vitro DNA-PK kinase assays using isolated CRY1 C-terminal tails (WT and various mutants, as noted) as substrates. 32P shows phosphorylation. CBB is Coomassie Brilliant Blue staining of total GST-CRY1-CT showing equal loading in each lane. C, in vitro DNA-PK kinase assay on full-length CRY1 protein. The left panel shows autoradiography of a 10% polyacrylamide gel, which shows autophosphorylated DNA-PKcs (lanes 2 and 3), phosphorylated Ku86 and Ku70 (components of the DNA-PK complex that are also autophosphorylated), and phosphorylation of the C-terminal tail (lane 1). However, no phosphorylation of full-length CRY1 is observed. The right panel is an expanded view of the same samples as lanes 2 and 3 of the left panel, run on 8% gel to better separate the region where CRY1 would migrate (expected CRY1 position marked by an arrow). The bottom right panel is a Western blot (WB) of the samples in lanes 2 and 3, demonstrating that sample 3 does contain mCRY1 protein.
To determine whether phosphorylation of any of these three SQ sites in the CRY1 tail was important for its function, we generated mutant CRY1 proteins in which each of these three serines was replaced with an alanine (to prevent phosphorylation) or with an aspartate (to mimic the phosphorylated state). These six mutations (resulting in S551A, S551D, S564A, S564D, S588A, and S588D; alone and in combination) were introduced into the Cry1 sequence in an expression vector containing endogenous Cry1 regulatory sequences from the Cry1 promoter and intron 1, which result in appropriate rhythmic expression in a pattern like the endogenous Cry1 gene (32). Expression of WT CRY1 from this construct in a Cry1/Cry2 knock-out MEF cell line can rescue normal rhythmicity in these arrhythmic cells (Fig. 3) (6, 32). Expression of the CRY1 mutants also rescued rhythmicity, and in most cases these rhythms were indistinguishable from those achieved by WT CRY1 (Fig. 3). However, with the S588D mutant (or any combination that contained this mutation), the rescued periods were significantly longer than normal (Fig. 3). These results suggest that phosphorylation of Ser-588 in the tail of CRY1 can lengthen the period of the circadian clock.
FIGURE 3.

CRY1 mutations that mimic phosphorylation of Ser-588 cause lengthened circadian periods when used to rescue rhythms in Cry1−/−/Cry2−/− cells. A–E, traces show Per2-luc reporter rhythms (base line-subtracted) from cells transfected with WT or mutant CRY1 as noted in the legends. F shows quantitation of the period lengths from WT, S588A, and S588D rescues such as those shown in E. (n = 8, ***p < 0.001).
To examine CRY1 Ser-588 phosphorylation in vivo, we studied the known epitopes (43) of several commercially available phospho-specific SQ antibodies, looking for similarity with the sequence surrounding the Ser-588–Gln-589 in CRY1. One of these, an antibody raised against RPA32 phosphorylated on Ser-33 (Bethyl Laboratories) had sequence with significant similarity to the sequence surrounding Ser-588 of CRY1 (Fig. 4A), and we found that it could recognize WT CRY1 but not the mutant S588A (Fig. 4B). Furthermore, treatment of the immunoprecipitate with protein phosphatase resulted in loss of immunoreactivity (Fig. 4C, second lane), whereas this was restored when a phosphatase inhibitor was added along with the phosphatase (Fig. 4C, third lane). Together, these results demonstrate that this antibody (which we will refer to as “P-CRY1(S588)”) is specific for the Ser-588-phosphorylated form of CRY1 and indicate that HEK293 cells contain an endogenous protein kinase capable of phosphorylating Ser-588 of CRY1.
FIGURE 4.

Phosphorylation of CRY1 Ser-588 is rhythmic in liver and is increased when DNA-PK is absent or depleted. A, alignment of CRY1 sequence surrounding the SQ site at 588–589 with epitope for antibody that recognizes Ser(P)-33 of RPA32. This information comes from consensus site analysis of other proteins recognized by this antibody (43). Red denotes required amino acids SQ at the phosphorylation site, blue denotes amino acids that are highly preferred at these positions, and green denotes amino acids that are also frequently found in protein sequences recognized by this antibody. B, P-CRY1(S588) antibody recognizes immunoprecipitated (IP) WT CRY1 but not S588A mutant CRY1. C, P-CRY1(S588) antibody recognizes immunoprecipitated WT CRY1, but not if lysate is treated with phosphatase. Signal is restored if phosphatase inhibitor sodium vanadate (Na3VO4) is added along with the phosphatase. D, CRY1 Ser-588 phosphorylation is rhythmic in mouse liver nuclei. Nuclear fractions were isolated from livers collected every 4 h from mice housed in constant darkness, and an aliquot (500 μg) of each was subjected to immunoprecipitation with P-CRY1(S588) antibody. These immunoprecipitates (top panel) as well as whole nuclear lysates (bottom panels) were then Western blotted (WB) with the antibodies shown. An asterisk denotes a nonspecific band. CT denotes circadian time, where CT12 is defined as time of activity onset. E, Ser-588 phosphorylation increases in the absence of DNA-PKcs. Endogenous CRY1 was immunoprecipitated from WT and DNA-PKcs KO MEF and then analyzed by Western blot using the P-CRY1(S588) antibody (top panel) and CRY1 antibody (bottom panel). Cells were treated with MG132 (10 μm) and okadaic acid (30 nm) for 5 h before harvesting. F, HEK293 cells were transfected with WT or S588A mutant Myc-CRY1 and with either control siRNAs or siRNAs to GAPDH or DNA-PKcs as labeled. Myc-CRY1 was immunoprecipitated and analyzed by Western blot as in E. Western blots showing the levels of DNA-PK, Myc-CRY1, and tubulin in the input cell lysates are also shown.
Using this antibody, we examined whether CRY1 is phosphorylated on this site in vivo and whether this varies in a circadian manner. We collected liver samples at different circadian times from mice maintained in constant darkness and isolated nuclear fractions from these livers. We found that the levels of the Ser-588-phosphorylated form of CRY1 exhibited a robust circadian rhythm with a peak at CT 8 (Fig. 4D, top panel). The protein levels of PER1, PER2, and CRY1 in the nucleus showed the expected robust daily variation, whereas the protein levels of DNA-PKcs were constant throughout the day (Fig. 4D, bottom panels).
Because rescue of rhythmicity by the phosphomimetic CRY1 S588D mutant had resulted in long period rhythms (Fig. 3), similar to those seen with loss or inhibition of DNA-PK (Fig. 1), we next used the P-CRY1(S588) antibody to further examine the role of endogenous DNA-PK in this phosphorylation in vivo. We immunoprecipitated CRY1 from DNA-PKcs WT and KO MEF and, surprisingly, found that the phosphorylation of Ser-588 of mCRY1 is increased in the DNA-PKcs KO MEF cells as compared with WT (Fig. 4E). Similar results were obtained when we depleted DNA-PKcs in HEK cells using siRNAs and then determined the Ser-588 phosphorylation of transfected CRY1 (Fig. 4F). The depletion of DNA-PKcs significantly increases the amount of Ser-588 phosphorylation (Fig. 4F). These data suggest that the Ser-588 phosphorylation of CRY1 is not due to direct kinase activity of DNA-PK, but instead DNA-PKcs somehow negatively regulates the phosphorylation of this site. To determine how the S588D causes a long circadian period, we next tested whether the S588D phosphomimetic mutation altered the ability of CRY1 to repress CLOCK/BMAL1-mediated activation of a Per1-luc reporter but found that CRY1 proteins carrying the S588D mutation had strong repressive activity, only subtly lower than WT CRY1 (Fig. 5A).
FIGURE 5.

The S588D phosphomimetic mutation causes increased CRY1 stability by antagonizing FBXL3 function. A, repression assays showing the level of CLOCK/BMAL1 activation of Per1-luc reporter in the presence of different doses of WT or mutant CRY1. HEK293 cells were transfected with Clock and Bmal1 expression plasmids and WT or mutant Cry1 expression plasmids as shown. The raw data were normalized such that the average level of activation of cells transfected with Clock and Bmal1, and the reporter control was equal to 100%. Each data point is averaged from the results of three replicates, with the error bars representing the S.E. B, triple phosphomimetic mutant (S551D,S564D,S588D) has a longer half-life than WT or the triple alanine mutant. Cycloheximide (CHX) was added to HEK293 cells expressing WT, S551A,S564A,S588A, or S551D,S564D,S588D mutant CRY1; cells were collected after 0, 2, 4, 6, or 8 h; and levels of CRY1 were detected by Western blot (WB). C, the single mutation S588D is sufficient to stabilize the mutant CRY1 compared with WT CRY1. Cycloheximide was added to HEK293 cells expressing WT or S588D mutant CRY1; cells were collected after 0, 2, 4, 6, or 8 h; and levels of CRY1 were detected by Western blot using the LiCor system for quantitation. D, the means of three replicates of experiment shown in C. The error bars represent S.E. E, WT CRY1 levels are reduced when FBXL3 is co-expressed in HEK293 cells, but this is not the case when the S588D mutation is present. Shown are Western blots for Myc-CRY1 (top panel) and FLAG-FBXL3 (bottom panel).
Because changes in CRY1 stability can result in changes in circadian period (17–21), we wondered whether phosphorylation of Ser-588 affected the stability of CRY1. Although phosphorylation is often linked to the promotion of degradation, when we assessed the protein half-life of CRY1 carrying the triple A and triple D mutations, we found that the CRY1 proteins with the triple phosphomimetic D mutations were more stable than the WT CRY1 or the triple A mutants (Fig. 5B). This increased stability was also seen in the single S588D mutant CRY1 (Fig. 5C), which was confirmed by quantitation of the half-lives (WT t½ = 2.48 h and S588D t½ = 5.06 h; Fig. 5D).
Nuclear CRY1 half-life is regulated by the F box protein FBXL3, a component of the SCF E3 ubiquitin-ligase complex, which targets CRY1 for degradation (17–21). To determine whether phosphorylation of S588D affects FBXL3 action on CRY1, we co-expressed WT and mutant CRY1 with FBXL3. Although co-expression of FBXL3 with WT CRY1 resulted in significantly lower levels of CRY1 protein, we found that presence of the S588D mutation, either alone or in combination with the other mutations, results in higher levels of CRY1 accumulation, similar to levels found when in the absence of FBXL3 (Fig. 5E).
DISCUSSION
Post-translational regulatory mechanisms significantly contribute to the precision and stability of the circadian clock and provide means by which diverse signals can influence the phase of the rhythms (44, 45). Here we identify a phosphorylation site in the C-terminal tail of the CRY1 protein that contributes to determination of period length. Phosphorylation on Ser-588 stabilizes the CRY1 protein by antagonizing the destabilizing effects of FBXL3, resulting in a lengthened circadian period. The protein kinase DNA-PK interacts with CRY1 and other components of the clock mechanism and antagonizes phosphorylation of this site, most likely through an indirect mechanism.
Most of the phosphorylation events that have been discovered to occur on clock proteins, including those previously identified on CRY1 and CRY2, act to destabilize the proteins. These include Ser-71 and Ser-280 of CRY1, which are phosphorylated by AMPK, resulting in increased ubiquitination by FBXL3 and subsequent degradation (22), and Ser-557 and Ser-553 in CRY2, which are phosphorylated by DYRK1A and GSK3, respectively, and also trigger degradation (9). In contrast, the Ser-588 site we have identified prevents FBXL3 action and results in increased stability of the protein. Expression of the CRY1 S588D phosphomimetic mutant in Cry1−/−/Cry2−/− cells results in the generation of circadian rhythms with long periods, consistent with the long periods observed in cells or animals lacking normal FBXL3 function (17, 20). Phosphorylation of Ser-588 is rhythmic in mouse liver, peaking during the day at a time when CRY1 protein levels in the nucleus are declining. It is possible that transient daily phosphorylation of this site slows the rate of degradation to generate the proper waveform of CRY1 protein, which ultimately contributes to the waveform of circadian gene expression. Other examples of stabilizing phosphorylation events in the clock system include human PER2, which is also subject to stabilization by phosphorylation at Ser-659, the serine mutated in patients with familial advanced phase syndrome (46). Similarly, the Neurospora core clock protein FRQ is stabilized by phosphorylation by cyclic AMP-dependent protein kinase A (47).
Despite the strong phenotype observed in the S588D mutant CRY1, the lack of effect on circadian period in the S588A mutant suggests that there may be a compensatory regulatory mechanism when that site cannot be phosphorylated, as in the S588A mutant. There are many examples of compensatory phosphorylation events in the literature, including endothelial NOS, which is phosphorylated on multiple sites by Akt. Compensatory phosphorylation of other sites occurs when the preferred sites are mutated to alanine so that the phenotype (NO production) of some S to A mutants is the same as WT (48). Similar compensatory phosphorylation on secondary sites is also observed for the important neurogenesis regulator p27Kip1 and the transcription factor inhibitor IKBα, when the primary sites (Thr-187 and Ser-293, respectively) are mutated to alanine (49, 50). Finally, in the core circadian clock protein PERIOD, multiple phosphorylations regulate its stability and function and in both mammalian and fly systems, mutation of one kinase (CKIe or dbt) causes compensatory phosphorylation by other kinases (51, 52) Elucidation of the nature of the compensatory response for CRY1 S588A remains to be determined in future studies.
Several pieces of evidence demonstrate that DNA-PK plays a role in regulating the circadian period. This kinase is present in complexes with other core circadian clock proteins, and loss of DNA-PK activity, by knockdown or through the use of DNA-PK-specific inhibitors, results in abnormally long circadian periods. Surprisingly, our data suggest that at least some of this effect is due to suppression by DNA-PK of CRY1 Ser-588 phosphorylation. The phosphorylation of this residue is increased in DNA-PKcs knock-out MEF, and this is not likely to be a developmental compensatory effect because we see similar increases in phosphorylation in HEK293 cells in which we acutely knockdown DNA-PK levels with siRNAs. Furthermore, rescue of rhythms in Cry1−/−/Cry2−/− cells with the phosphomimetic S588D mutant also results in long periods, similar to the effect of knocking down DNA-PKcs.
The increase in phosphorylation when DNA-PK is reduced or absent indicates that DNA-PK antagonizes the phosphorylation of S588 by some mechanism. It is possible that DNA-PK phosphorylates and activates a protein phosphatase that then dephosphorylates CRY1. DNA-PK (either DNA-PKcs itself or the DNA-PK complex) has been shown to interact with several protein phosphatases, including PP6, PP2A, PP5, and PP1c (53–55), and PP5 and PP1c have previously been shown to interact with cryptochromes and other clock components (56–58). Alternatively, DNA-PK may phosphorylate and inhibit some other kinase that directly targets Ser-588 of CRY1. For example, DNA-PKcs has been shown to interact with the regulatory subunit of AMPK and regulate AMPK activation in response to glucose-free conditions (59). However, although CRY1 is a substrate for AMPK, this is unlikely to be the mechanism in this case, because DNA-PK activates AMPK (rather than inhibiting its activity) and because the Ser-588 site on CRY1 is part of an SQ motif, which is not predicted to be a phosphorylation site for AMPK.
Finally, DNA-PK is a member of the PIKK family of protein kinases, and it is possible that some other member of this family phosphorylates CRY1 on Ser-588, and loss of DNA-PK somehow enhances the ability of one of these kinases to act on CRY1. It has been proposed that one of the other PIKK family members can substitute for DNA-PK in DNA-PKcs null cells (60). Furthermore, most of the members of this family phosphorylate SQ motifs, and it has previously been reported that knockdown of one of these (SMG-1) can cause up-regulation of another (ATM), resulting in enhanced phosphorylation of the nonsense-mediated decay protein hUPF1 (61). Similarly, in our attempt to identify the kinase responsible for Ser-588 phosphorylation, we found that inhibition of ATM also caused an increase in Ser-588 phosphorylation (data not shown), suggesting that phosphorylation of this site may be regulated in a complex way, by more than one member of the PIKK family. However, neither ATM nor ATR could phosphorylate CRY1 in our in vitro kinase assays (data not shown).
The identification of DNA-PK as a regulator of circadian period is interesting because of a long-recognized intimate relationship between DNA repair and circadian rhythms, and it may be that circadian clocks evolved as a protective mechanism to protect DNA from light- and radiation-induced damage (62). Furthermore, loss of various clock components, including CLOCK, BMAL1, PER1, and PER2, have been linked to increased chronic sensitivity to DNA cross-linking reagents, increased tumor development, and deficiencies in DNA damage responses (63–65). In addition, the CLOCK protein is recruited to sites of DNA damage (66), and DNA damage can act as a resetting cue for the mammalian circadian clock (67). Therefore, a role for DNA-PK in the regulation of CRY1 stability and period control is another example of the relationships between these pathways.
Finally, this work reveals new insight into the role of the CRY1 C-terminal tail. The function of this highly divergent portion of the CRY1 protein has been unclear, with some evidence that it is involved in nuclear localization or BMAL binding (7, 8, 68) and other evidence that it is dispensable for core clock function (6). However, even in the latter case, some role for the C terminus in period control was noted (6). Here we provide a mechanism for this period control through rhythmic phosphorylation of Ser-588, which causes increased stability of CRY1 and lengthening of the circadian period.
Acknowledgments
We thank Dr. Hiroki R. Ueda for the Cry1 rescue plasmid and Dr. Andrew C. Liu and Dr. Sanjoy K. Khan for the Cry1−/−/Cry2−/− double knock-out MEF and helpful advice.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 GM090247 (to C. B. G.) and R01 CA166777 (to B. P. C.).
- PHR
- photolyase homology region
- AMPK
- AMP-activated protein kinase
- DNA-PKcs
- catalytic subunit of DNA-dependent protein kinase
- MEF
- mouse embryonic fibroblast(s)
- PIKK
- phosphatidylinositol 3-kinase-related protein kinase.
REFERENCES
- 1. Mohawk J. A., Green C. B., Takahashi J. S. (2012) Central and peripheral circadian clocks in mammals. Annu. Rev. Neurosci. 35, 445–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lowrey P. L., Takahashi J. S. (2011) Genetics of circadian rhythms in Mammalian model organisms. Adv. Genet. 74, 175–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. van der Horst G. T., Muijtjens M., Kobayashi K., Takano R., Kanno S., Takao M., de Wit J., Verkerk A., Eker A. P., van Leenen D., Buijs R., Bootsma D., Hoeijmakers J. H., Yasui A. (1999) Mammalian Cry1 and Cry2 are essential for maintenance of circadian rhythms. Nature 398, 627–630 [DOI] [PubMed] [Google Scholar]
- 4. Vitaterna M. H., Selby C. P., Todo T., Niwa H., Thompson C., Fruechte E. M., Hitomi K., Thresher R. J., Ishikawa T., Miyazaki J., Takahashi J. S., Sancar A. (1999) Differential regulation of mammalian Period genes and circadian rhythmicity by cryptochromes 1 and 2. Proc. Natl. Acad. Sci. U.S.A. 96, 12114–12119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhu H., Conte F., Green C. B. (2003) Nuclear localization and transcriptional repression are confined to separable domains in the circadian protein CRYPTOCHROME. Curr. Biol. 13, 1653–1658 [DOI] [PubMed] [Google Scholar]
- 6. Khan S. K., Xu H., Ukai-Tadenuma M., Burton B., Wang Y., Ueda H. R., Liu A. C. (2012) Identification of a novel cryptochrome differentiating domain required for feedback repression in circadian clock function. J. Biol. Chem. 287, 25917–25926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chaves I., Yagita K., Barnhoorn S., Okamura H., van der Horst G. T., Tamanini F. (2006) Functional evolution of the photolyase/cryptochrome protein family. Importance of the C terminus of mammalian CRY1 for circadian core oscillator performance. Mol. Cell. Biol. 26, 1743–1753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Czarna A., Breitkreuz H., Mahrenholz C. C., Arens J., Strauss H. M., Wolf E. (2011) Quantitative analyses of cryptochrome-mBMAL1 interactions. Mechanistic insights into the transcriptional regulation of the mammalian circadian clock. J. Biol. Chem. 286, 22414–22425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kurabayashi N., Hirota T., Sakai M., Sanada K., Fukada Y. (2010) DYRK1A and glycogen synthase kinase 3β, a dual-kinase mechanism directing proteasomal degradation of CRY2 for circadian timekeeping. Mol. Cell. Biol. 30, 1757–1768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Harada Y., Sakai M., Kurabayashi N., Hirota T., Fukada Y. (2005) Ser-557-phosphorylated mCRY2 is degraded upon synergistic phosphorylation by glycogen synthase kinase-3β. J. Biol. Chem. 280, 31714–31721 [DOI] [PubMed] [Google Scholar]
- 11. Dissel S., Codd V., Fedic R., Garner K. J., Costa R., Kyriacou C. P., Rosato E. (2004) A constitutively active cryptochrome in Drosophila melanogaster. Nat. Neurosci. 7, 834–840 [DOI] [PubMed] [Google Scholar]
- 12. Rosato E., Codd V., Mazzotta G., Piccin A., Zordan M., Costa R., Kyriacou C. P. (2001) Light-dependent interaction between Drosophila CRY and the clock protein PER mediated by the carboxy terminus of CRY. Curr. Biol. 11, 909–917 [DOI] [PubMed] [Google Scholar]
- 13. Czarna A., Berndt A., Singh H. R., Grudziecki A., Ladurner A. G., Timinszky G., Kramer A., Wolf E. (2013) Structures of Drosophila cryptochrome and mouse cryptochrome1 provide insight into circadian function. Cell 153, 1394–1405 [DOI] [PubMed] [Google Scholar]
- 14. Levy C., Zoltowski B. D., Jones A. R., Vaidya A. T., Top D., Widom J., Young M. W., Scrutton N. S., Crane B. R., Leys D. (2013) Updated structure of Drosophila cryptochrome. Nature 495, E3–E4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zoltowski B. D., Vaidya A. T., Top D., Widom J., Young M. W., Crane B. R. (2011) Structure of full-length Drosophila cryptochrome. Nature 480, 396–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xing W., Busino L., Hinds T. R., Marionni S. T., Saifee N. H., Bush M. F., Pagano M., Zheng N. (2013) SCF(FBXL3) ubiquitin ligase targets cryptochromes at their cofactor pocket. Nature 496, 64–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Siepka S. M., Yoo S. H., Park J., Song W., Kumar V., Hu Y., Lee C., Takahashi J. S. (2007) Circadian mutant Overtime reveals F-box protein FBXL3 regulation of cryptochrome and period gene expression. Cell 129, 1011–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yoo S. H., Mohawk J. A., Siepka S. M., Shan Y., Huh S. K., Hong H. K., Kornblum I., Kumar V., Koike N., Xu M., Nussbaum J., Liu X., Chen Z., Chen Z. J., Green C. B., Takahashi J. S. (2013) Competing E3 ubiquitin ligases govern circadian periodicity by degradation of CRY in nucleus and cytoplasm. Cell 152, 1091–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Busino L., Bassermann F., Maiolica A., Lee C., Nolan P. M., Godinho S. I., Draetta G. F., Pagano M. (2007) SCFFbxl3 controls the oscillation of the circadian clock by directing the degradation of cryptochrome proteins. Science 316, 900–904 [DOI] [PubMed] [Google Scholar]
- 20. Godinho S. I., Maywood E. S., Shaw L., Tucci V., Barnard A. R., Busino L., Pagano M., Kendall R., Quwailid M. M., Romero M. R., O'Neill J., Chesham J. E., Brooker D., Lalanne Z., Hastings M. H., Nolan P. M. (2007) The after-hours mutant reveals a role for Fbxl3 in determining mammalian circadian period. Science 316, 897–900 [DOI] [PubMed] [Google Scholar]
- 21. Hirano A., Yumimoto K., Tsunematsu R., Matsumoto M., Oyama M., Kozuka-Hata H., Nakagawa T., Lanjakornsiripan D., Nakayama K. I., Fukada Y. (2013) FBXL21 regulates oscillation of the circadian clock through ubiquitination and stabilization of cryptochromes. Cell 152, 1106–1118 [DOI] [PubMed] [Google Scholar]
- 22. Lamia K. A., Sachdeva U. M., DiTacchio L., Williams E. C., Alvarez J. G., Egan D. F., Vasquez D. S., Juguilon H., Panda S., Shaw R. J., Thompson C. B., Evans R. M. (2009) AMPK regulates the circadian clock by cryptochrome phosphorylation and degradation. Science 326, 437–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Baggs J. E., Price T. S., DiTacchio L., Panda S., Fitzgerald G. A., Hogenesch J. B. (2009) Network features of the mammalian circadian clock. PLoS Biol. 7, e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yoo S. H., Ko C. H., Lowrey P. L., Buhr E. D., Song E. J., Chang S., Yoo O. J., Yamazaki S., Lee C., Takahashi J. S. (2005) A noncanonical E-box enhancer drives mouse Period2 circadian oscillations in vivo. Proc. Natl. Acad. Sci. U.S.A. 102, 2608–2613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen Z., Yoo S. H., Park Y. S., Kim K. H., Wei S., Buhr E., Ye Z. Y., Pan H. L., Takahashi J. S. (2012) Identification of diverse modulators of central and peripheral circadian clocks by high-throughput chemical screening. Proc. Natl. Acad. Sci. U.S.A. 109, 101–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yamazaki S., Takahashi J. S. (2005) Real-time luminescence reporting of circadian gene expression in mammals. Methods Enzymol. 393, 288–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McCarthy E. V., Baggs J. E., Geskes J. M., Hogenesch J. B., Green C. B. (2009) Generation of a novel allelic series of cryptochrome mutants via mutagenesis reveals residues involved in protein-protein interaction and CRY2-specific repression. Mol. Cell. Biol. 29, 5465–5476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Peng Y., Zhang Q., Nagasawa H., Okayasu R., Liber H. L., Bedford J. S. (2002) Silencing expression of the catalytic subunit of DNA-dependent protein kinase by small interfering RNA sensitizes human cells for radiation-induced chromosome damage, cell killing, and mutation. Cancer Res. 62, 6400–6404 [PubMed] [Google Scholar]
- 29. Lavery D. J., Schibler U. (1993) Circadian transcription of the cholesterol 7a hydroxylase gene may involve the liver-enriched bZIP protein DBP. Genes Dev. 7, 1871–1884 [DOI] [PubMed] [Google Scholar]
- 30. Asher G., Gatfield D., Stratmann M., Reinke H., Dibner C., Kreppel F., Mostoslavsky R., Alt F. W., Schibler U. (2008) SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell 134, 317–328 [DOI] [PubMed] [Google Scholar]
- 31. Davis A. J., Lee K. J., Chen D. J. (2013) The N-terminal region of the DNA-dependent protein kinase catalytic subunit is required for its DNA double-stranded break-mediated activation. J. Biol. Chem. 288, 7037–7046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ukai-Tadenuma M., Yamada R. G., Xu H., Ripperger J. A., Liu A. C., Ueda H. R. (2011) Delay in feedback repression by cryptochrome 1 is required for circadian clock function. Cell 144, 268–281 [DOI] [PubMed] [Google Scholar]
- 33. Sun Z. S., Albrecht U., Zhuchenko O., Bailey J., Eichele G., Lee C. C. (1997) RIGUI, a putative mammalian ortholog of the Drosophila period gene. Cell 90, 1003–1011 [DOI] [PubMed] [Google Scholar]
- 34. Robles M. S., Boyault C., Knutti D., Padmanabhan K., Weitz C. J. (2010) Identification of RACK1 and protein kinase Cα as integral components of the mammalian circadian clock. Science 327, 463–466 [DOI] [PubMed] [Google Scholar]
- 35. Tei H., Okamura H., Shigeyoshi Y., Fukuhara C., Ozawa R., Hirose M., Sakaki Y. (1997) Circadian oscillation of a mammalian homologue of the Drosophila period gene. Nature 389, 512–516 [DOI] [PubMed] [Google Scholar]
- 36. Kume K., Zylka M. J., Sriram S., Shearman L. P., Weaver D. R., Jin X., Maywood E. S., Hastings M. H., Reppert S. M. (1999) mCRY1 and mCRY2 are essential components of the negative limb of the circadian clock feedback loop. Cell 98, 193–205 [DOI] [PubMed] [Google Scholar]
- 37. Abraham R. T. (2004) PI 3-kinase related kinases. “Big” players in stress-induced signaling pathways. DNA Repair 3, 883–887 [DOI] [PubMed] [Google Scholar]
- 38. Weterings E., Chen D. J. (2008) The endless tale of non-homologous end-joining. Cell Res. 18, 114–124 [DOI] [PubMed] [Google Scholar]
- 39. Wong R. H., Chang I., Hudak C. S., Hyun S., Kwan H. Y., Sul H. S. (2009) A role of DNA-PK for the metabolic gene regulation in response to insulin. Cell 136, 1056–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liiv I., Rebane A., Org T., Saare M., Maslovskaja J., Kisand K., Juronen E., Valmu L., Bottomley M. J., Kalkkinen N., Peterson P. (2008) DNA-PK contributes to the phosphorylation of AIRE. Importance in transcriptional activity. Biochim. Biophys. Acta 1783, 74–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lai J. S., Herr W. (1992) Ethidium bromide provides a simple tool for identifying genuine DNA-independent protein associations. Proc. Natl. Acad. Sci. U.S.A. 89, 6958–6962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Leahy J. J., Golding B. T., Griffin R. J., Hardcastle I. R., Richardson C., Rigoreau L., Smith G. C. (2004) Identification of a highly potent and selective DNA-dependent protein kinase (DNA-PK) inhibitor (NU7441) by screening of chromenone libraries. Bioorg. Med. Chem. Lett. 14, 6083–6087 [DOI] [PubMed] [Google Scholar]
- 43. Matsuoka S., Ballif B. A., Smogorzewska A., McDonald E. R., 3rd, Hurov K. E., Luo J., Bakalarski C. E., Zhao Z., Solimini N., Lerenthal Y., Shiloh Y., Gygi S. P., Elledge S. J. (2007) ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316, 1160–1166 [DOI] [PubMed] [Google Scholar]
- 44. Harms E., Kivimäe S., Young M. W., Saez L. (2004) Posttranscriptional and posttranslational regulation of clock genes. J. Biol. Rhythms 19, 361–373 [DOI] [PubMed] [Google Scholar]
- 45. Mehra A., Baker C. L., Loros J. J., Dunlap J. C. (2009) Post-translational modifications in circadian rhythms. Trends Biochem. Sci. 34, 483–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vanselow K., Vanselow J. T., Westermark P. O., Reischl S., Maier B., Korte T., Herrmann A., Herzel H., Schlosser A., Kramer A. (2006) Differential effects of PER2 phosphorylation. Molecular basis for the human familial advanced sleep phase syndrome (FASPS). Genes Dev. 20, 2660–2672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Huang G., Chen S., Li S., Cha J., Long C., Li L., He Q., Liu Y. (2007) Protein kinase A and casein kinases mediate sequential phosphorylation events in the circadian negative feedback loop. Genes Dev. 21, 3283–3295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bauer P. M., Fulton D., Boo Y. C., Sorescu G. P., Kemp B. E., Jo H., Sessa W. C. (2003) Compensatory phosphorylation and protein-protein interactions revealed by loss of function and gain of function mutants of multiple serine phosphorylation sites in endothelial nitric-oxide synthase. J. Biol. Chem. 278, 14841–14849 [DOI] [PubMed] [Google Scholar]
- 49. Zheng Y. L., Li B. S., Rudrabhatla P., Shukla V., Amin N. D., Maric D., Kesavapany S., Kanungo J., Pareek T. K., Takahashi S., Grant P., Kulkarni A. B., Pant H. C. (2010) Phosphorylation of p27Kip1 at Thr187 by cyclin-dependent kinase 5 modulates neural stem cell differentiation. Mol. Biol. Cell 21, 3601–3614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schwarz E. M., Van Antwerp D., Verma I. M. (1996) Constitutive phosphorylation of IκBα by casein kinase II occurs preferentially at serine 293. Requirement for degradation of free IκBα. Mol. Cell. Biol. 16, 3554–3559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Preuss F., Fan J. Y., Kalive M., Bao S., Schuenemann E., Bjes E. S., Price J. L. (2004) Drosophila doubletime mutations which either shorten or lengthen the period of circadian rhythms decrease the protein kinase activity of casein kinase I. Mol. Cell. Biol. 24, 886–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fan J. Y., Preuss F., Muskus M. J., Bjes E. S., Price J. L. (2009) Drosophila and vertebrate casein kinase Iδ exhibits evolutionary conservation of circadian function. Genetics 181, 139–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Douglas P., Zhong J., Ye R., Moorhead G. B., Xu X., Lees-Miller S. P. (2010) Protein phosphatase 6 interacts with the DNA-dependent protein kinase catalytic subunit and dephosphorylates γ-H2AX. Mol. Cell. Biol. 30, 1368–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mi J., Dziegielewski J., Bolesta E., Brautigan D. L., Larner J. M. (2009) Activation of DNA-PK by ionizing radiation is mediated by protein phosphatase 6. PLoS One 4, e4395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wechsler T., Chen B. P., Harper R., Morotomi-Yano K., Huang B. C., Meek K., Cleaver J. E., Chen D. J., Wabl M. (2004) DNA-PKcs function regulated specifically by protein phosphatase 5. Proc. Natl. Acad. Sci. U.S.A. 101, 1247–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Partch C. L., Shields K. F., Thompson C. L., Selby C. P., Sancar A. (2006) Posttranslational regulation of the mammalian circadian clock by cryptochrome and protein phosphatase 5. Proc. Natl. Acad. Sci. U.S.A. 103, 10467–10472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhao S., Sancar A. (1997) Human blue-light photoreceptor hCRY2 specifically interacts with protein serine/threonine phosphatase 5 and modulates its activity. Photochem. Photobiol. 66, 727–731 [DOI] [PubMed] [Google Scholar]
- 58. Lee H. M., Chen R., Kim H., Etchegaray J. P., Weaver D. R., Lee C. (2011) The period of the circadian oscillator is primarily determined by the balance between casein kinase 1 and protein phosphatase 1. Proc. Natl. Acad. Sci. U.S.A. 108, 16451–16456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Amatya P. N., Kim H. B., Park S. J., Youn C. K., Hyun J. W., Chang I. Y., Lee J. H., You H. J. (2012) A role of DNA-dependent protein kinase for the activation of AMP-activated protein kinase in response to glucose deprivation. Biochim. Biophys. Acta 1823, 2099–2108 [DOI] [PubMed] [Google Scholar]
- 60. Ruis B. L., Fattah K. R., Hendrickson E. A. (2008) The catalytic subunit of DNA-dependent protein kinase regulates proliferation, telomere length, and genomic stability in human somatic cells. Mol. Cell. Biol. 28, 6182–6195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Brumbaugh K. M., Otterness D. M., Geisen C., Oliveira V., Brognard J., Li X., Lejeune F., Tibbetts R. S., Maquat L. E., Abraham R. T. (2004) The mRNA surveillance protein hSMG-1 functions in genotoxic stress response pathways in mammalian cells. Mol. Cell 14, 585–598 [DOI] [PubMed] [Google Scholar]
- 62. Gehring W., Rosbash M. (2003) The coevolution of blue-light photoreception and circadian rhythms. J. Mol. Evol. 57, S286–S289 [DOI] [PubMed] [Google Scholar]
- 63. Gorbacheva V. Y., Kondratov R. V., Zhang R., Cherukuri S., Gudkov A. V., Takahashi J. S., Antoch M. P. (2005) Circadian sensitivity to the chemotherapeutic agent cyclophosphamide depends on the functional status of the CLOCK/BMAL1 transactivation complex. Proc. Natl. Acad. Sci. U.S.A. 102, 3407–3412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Fu L., Pelicano H., Liu J., Huang P., Lee C. (2002) The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell 111, 41–50 [DOI] [PubMed] [Google Scholar]
- 65. Gery S., Komatsu N., Baldjyan L., Yu A., Koo D., Koeffler H. P. (2006) The circadian gene per1 plays an important role in cell growth and DNA damage control in human cancer cells. Mol. Cell 22, 375–382 [DOI] [PubMed] [Google Scholar]
- 66. Cotta-Ramusino C., McDonald E. R., 3rd, Hurov K., Sowa M. E., Harper J. W., Elledge S. J. (2011) A DNA damage response screen identifies RHINO, a 9-1-1 and TopBP1 interacting protein required for ATR signaling. Science 332, 1313–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Oklejewicz M., Destici E., Tamanini F., Hut R. A., Janssens R., van der Horst G. T. (2008) Phase resetting of the mammalian circadian clock by DNA damage. Curr. Biol. 18, 286–291 [DOI] [PubMed] [Google Scholar]
- 68. Tamanini F., Chaves I., Bajek M. I., van der Horst G. T. (2007) Structure function analysis of mammalian cryptochromes. Cold Spring Harb. Symp. Quant. Biol. 72, 133–139 [DOI] [PubMed] [Google Scholar]
- 69. Zhang E. E., Liu A. C., Hirota T., Miraglia L. J., Welch G., Pongsawakul P. Y., Liu X., Atwood A., Huss J. W., 3rd, Janes J., Su A. I., Hogenesch J. B., Kay S. A. (2009) A genome-wide RNAi screen for modifiers of the circadian clock in human cells. Cell 139, 199–210 [DOI] [PMC free article] [PubMed] [Google Scholar]