

Background: Rhabdomyosarcoma (RMS) frequently harbor aberrant PI3K/mTOR pathway activation and are resistant to apoptosis.
Results: The mTOR inhibitor AZD8055 and the Bcl-2 inhibitor ABT-737 synergize to trigger apoptosis.
Conclusion: Cotreatment with AZD8055 and ABT-737 represents a new approach for apoptosis induction in RMS.
Significance: These findings have important implications for the development of novel treatment strategies for RMS.
Keywords: Apoptosis, Cancer, Cell Death, mTOR, Rhabdomyosarcoma
Abstract
The PI3K/mammalian Target of Rapamycin (mTOR) pathway is often aberrantly activated in rhabdomyosarcoma (RMS) and represents a promising therapeutic target. Recent evaluation of AZD8055, an ATP-competitive mTOR inhibitor, by the Preclinical Pediatric Testing Program showed in vivo antitumor activity against childhood solid tumors, including RMS. Therefore, in the present study, we searched for AZD8055-based combination therapies. Here, we identify a new synergistic lethality of AZD8055 together with ABT-737, a BH3 mimetic that antagonizes Bcl-2, Bcl-xL, and Bcl-w but not Mcl-1. AZD8055 and ABT-737 cooperate to induce apoptosis in alveolar and embryonal RMS cells in a highly synergistic fashion (combination index < 0.2). Synergistic induction of apoptosis by AZD8055 and ABT-737 is confirmed on the molecular level, as AZD8055 and ABT-737 cooperate to trigger loss of mitochondrial membrane potential, activation of caspases, and caspase-dependent apoptosis that is blocked by the pan-caspase inhibitor Z-VAD-fmk. Similar to AZD8055, the PI3K/mTOR inhibitor NVP-BEZ235, the PI3K inhibitor NVP-BKM120 and Akt inhibitor synergize with ABT-737 to trigger apoptosis, whereas no cooperativity is found for the mTOR complex 1 inhibitor RAD001. Interestingly, molecular studies reveal a correlation between the ability of different PI3K/mTOR inhibitors to potentiate ABT-737-induced apoptosis and to suppress Mcl-1 protein levels. Importantly, knockdown of Mcl-1 increases ABT-737-induced apoptosis similar to AZD8055/ABT-737 cotreatment. This indicates that AZD8055-mediated suppression of Mcl-1 protein plays an important role in the synergistic drug interaction. By identifying a novel synergistic interaction of AZD8055 and ABT-737, our findings have important implications for the development of molecular targeted therapies for RMS.
Introduction
Rhabdomyosarcoma (RMS)2 is the most frequent pediatric soft tissue sarcoma and can be classified into two major entities, i.e. alveolar RMS and embryonal RMS, based on histological and molecular features (1, 2). Despite aggressive therapies, including surgery, chemotherapy, and radiation, patients with high-risk or relapsed disease still have a poor prognosis (3). This highlights the need for novel, more efficient treatment approaches.
There have been many efforts to elucidate the molecular biology of sarcomas, and considerable progress has been made to understand signaling networks that regulate cancer progression and treatment resistance (4–6). For example, the PI3K/mTOR signaling pathway is often aberrantly activated in RMS, which has been linked to reduced survival (7). Hence, this pathway represents a promising target for therapeutic exploitation in RMS.
mTOR forms the catalytic subunit of two structurally and functionally distinct protein complexes, i.e. mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2), which are defined by unique components, namely RAPTOR (regulatory-associated protein of mTOR) for mTORC1 and RICTOR (rapamycin-insensitive companion of mTOR) for mTORC2 (8). mTOR complexes are regulated by various signals, including growth factors, nutrients and cellular stress (9). mTORC1 promotes translation, cell growth, and metabolism via the translational regulators eIF4E-binding protein 1 (4E-BP1) and S6 ribosomal protein (9, 10). mTORC2 phosphorylates and activates several AGC kinases, including Akt, and is also involved in the regulation of cell motility and invasion via actin cytoskeletal organization (8).
The first generation of allosteric mTORC1 inhibitors comprises rapamycin (sirolimus) and its analogues (rapalogues), including temsirolimus, everolimus (also known as RAD001), and ridaforolimus (11). In clinical trials, rapalogues turned out to have only limited success, which might be explained by loss of the S6K1-mediated negative feedback loop to IRS1 upon mTORC1 inhibition, leading to increased Akt phosphorylation (12) and/or by insufficient inhibition of downstream targets of mTOR such as 4E-BP1 (13). More specifically, a phase II trial of temsirolimus in children with solid tumors, including RMS, showed prolonged stable disease but failed to meet the primary objective efficacy threshold (14). By comparison, ATP-competitive pan-mTOR inhibitors effectively inhibit both mTOR complexes including suppression of 4E-BP1 phosphorylation, as they block mTOR kinase activity that is part of both mTORC1 and mTORC2 complexes (11). AZD8055, an ATP-competitive mTOR inhibitor (15), has recently been evaluated by the Preclinical Pediatric Testing Program. Although AZD8055 showed some in vivo antitumor activity against childhood solid tumors, including RMS, it did not cause objective tumor regression (16), suggesting that AZD8055-based combination therapies may be required to potentiate the antitumor activity of AZD8055.
The efficacy of most anticancer therapies largely depends on intact cell death pathways in cancer cells, for example apoptosis, which is one of the best characterized forms of programmed cell death (17). Engagement of the extrinsic (death receptor) or the intrinsic (mitochondrial) apoptosis pathways eventually leads to activation of caspases as effector molecules (17). Signal transduction to apoptosis is typically suppressed in human cancers, e.g. by aberrant activation of survival pathways such as the PI3K/mTOR cascade (18). In addition, antiapoptotic proteins such as Bcl-2, Bcl-xL, Bcl-w, and Mcl-1 are frequently expressed at high levels in human cancers, which contribute to evasion of apoptosis and treatment resistance (19). In RMS samples, overexpression of Bcl-2 and Mcl-1 was reported (20, 21). To neutralize antiapoptotic Bcl-2 proteins small-molecule inhibitors were developed that mimic the BH3-only proteins of the Bcl-2 family (19). One example is the BH3 mimetic ABT-737 that antagonizes the anti apoptotic proteins Bcl-2, Bcl-xL, and Bcl-w similarly to the BH3-only protein Bad and showed promising antitumor activity, especially in Bcl-2-dependent cancers (22).
Searching for novel strategies to enhance the efficacy of treatment options for RMS, in the present study, we investigated the question whether the antitumor activity of AZD8055 against RMS can be enhanced in combination therapies using either conventional chemotherapeutic drugs or molecular targeted agents such as the BH3 mimetic ABT-737.
EXPERIMENTAL PROCEDURES
Cell Culture and Chemicals
RMS cell lines were obtained from the American Type Culture Collection (Manassas, VA) and maintained in Invitrogen GlutaMAXTM-I medium (Invitrogen), supplemented with 10% FCS (Biochrom, Berlin, Germany), 1% penicillin/streptomycin (Invitrogen), and 1 mm sodium pyruvate (Invitrogen). ABT-737 was kindly provided by Abbott Laboratories (Abbott Park, IL) (22). AZD8055 was obtained from Selleck Chemicals (Houston, TX), N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone (Z-VAD-fmk) from Bachem (Heidelberg, Germany), and all other chemicals were from Sigma (Deisenhofen, Germany) unless indicated otherwise.
Determination of Cell Death, Apoptosis, and Loss of Mitochondrial Membrane Potential
Cell death was determined by analyzing plasma membrane permeability using propidium iodide (PI) staining. Briefly, cells were washed in PBS and incubated in PI solution (2 μg/ml PI in PBS) for 10 min. After washing with PBS, PI uptake was determined by flow cytometry (FACSCanto II, BD Biosciences). Apoptosis was determined by analysis of DNA fragmentation of PI-stained nuclei and flow cytometry as described previously (23). To determine mitochondrial transmembrane potential, cells were incubated with 1 μm tetramethylrhodamine methyl ester (Molecular Probes) at 37 °C and analyzed by flow cytometry.
Western Blot Analysis
Western blot analysis was performed as described previously (23) using the following antibodies: anti-4E-BP1, anti-Bid, anti-Bim, anti-caspase-3, anti-caspase-9, anti-poly(ADP-ribose) polymerase (PARP), anti-phospho-4E-BP1 (Thr-37/46), anti-phospho-Akt (Ser-473), anti-phospho-S6, anti-S6, anti-phospho-GSK3, and anti-GSK3 from Cell Signaling (Beverly, MA), anti-Akt, anti-Bcl-xL, anti-Bax, anti-Bak, and anti-Bcl-2 from BD Biosciences; anti-caspase-8 and anti-Noxa from Alexis (Grünberg, Germany), anti-Mcl-1 from Stressgene (Victoria, BC), anti-β-actin from Sigma, and anti-α-tubulin from Calbiochem (Darmstadt, Germany) were used as loading controls. Goat anti-mouse IgG, donkey anti-goat IgG, goat anti-rabbit IgG conjugated to horseradish peroxidase (Santa Cruz Biotechnology) and goat anti-mouse IgG1 or goat anti-mouse IgG2b (Southern Biotech, Birmingham, AL) conjugated to horseradish peroxidase were used as secondary antibodies, and enhanced chemiluminescence was used for detection (Amersham Bioscience). Alternatively, donkey anti-mouse IgG, donkey anti-rabbit IgG, or donkey anti-goat IgG labeled with IRDye infrared dyes were used for fluorescence detection (LI-COR Biotechnology, Bad Homburg, Germany).
Gene Silencing
Stable knockdown of Mcl-1 was performed by lentiviral shRNA vectors as described previously (24). Shortly, HEK293T cells were transfected with 12.5 μg of pCMV-dR8.91 (gag-pol), 1 μg of pMD2.G (env), and 7.5 μg of pGIPZ-shRNAmir vector using calcium phosphate transfection. All pGIPZ-shRNAmir vectors were purchased from Thermo Fisher Scientific (Dreieich, Germany) (non-silencing control, RHS4346; shMcl-1_1, RHS4430-99159062 and shMcl-1_2, RHS4430–98845167). Virus-containing supernatant was collected, filtered, and used for spin transduction at 30 °C in the presence of 8 μg/ml polybrene. Transduced cells were selected with 1 μg/ml puromycin (Sigma). For transient knockdown by siRNA, cells were reversely transfected with 15 nm SilencerSelect siRNA (Invitrogen) control siRNA (4390843) or targeting siRNA (s8583, s8585, and s8584 for Mcl-1 and s48409 and s48410 for Rictor) using Lipofectamine 2000 (Invitrogen) and OptiMEM (Invitrogen).
Statistical Analysis
Statistical significance was assessed by Student's t test (two-tailed distribution, two-sample, unequal variance). Drug interactions were analyzed by the combination index (CI) method based on that described by Chou (25) using CalcuSyn software (Biosoft, Cambridge, UK). CI < 0.9 indicates synergism, 0.9-1.1 indicates additivity, and >1.1 indicates antagonism.
RESULTS
AZD8055 Synergizes with ABT-737 to Induce Apoptosis in RMS Cells
To investigate whether the ATP-competitive mTOR inhibitor AZD8055 enhances apoptosis sensitivity of RMS cells, we initially tested AZD8055 alone and in combination with anticancer agents. To this end, we used conventional chemotherapeutic drugs that are currently included in clinical protocols for the treatment of RMS, i.e. vincristine, doxorubicin, and actinomycin D, or, alternatively, the BH3 mimetic ABT-737 that antagonizes antiapoptotic Bcl-2 proteins. To represent the two major histological subtypes of RMS, i.e. embryonal RMS and alveolar RMS, we selected two embryonal (RD and TE671) and two alveolar (RH30 and RMS13) RMS cell lines. Interestingly, we found that AZD8055 significantly increased ABT-737-induced cell death in all four RMS cell lines, whereas treatment with AZD8055 alone exerted minimal cytotoxicity (Fig. 1A). Calculation of CI revealed that the interaction of AZD8055 and ABT-737 is highly synergistic (Fig. 1B). In addition to determining cell death by PI uptake (Fig. 1A), we assessed DNA fragmentation as a typical biochemical feature of apoptosis (Fig. 1C). Similarly, the addition of AZD8055 significantly increased ABT-737-triggered DNA fragmentation compared with treatment with ABT-737 alone (Fig. 1C). In contrast to ABT-737, we found no cooperative effects of AZD8055 together with chemotherapeutics such as vincristine, doxorubicin, or actinomycin D (supplemental Fig. 1). This set of experiments shows that AZD8055 sensitizes RMS cells for ABT-737-induced apoptosis, whereas AZD8055 did not alter chemosensitivity to vincristine, doxorubicin, or actinomycin D.
FIGURE 1.

AZD8055 and ABT-737 cooperate to induce cell death in RMS cells. RMS cell lines were treated for 48 h with 1 μm AZD8055 and/or indicated concentrations of ABT-737. Cell death was determined by PI staining and flow cytometry (A), and corresponding CI values are shown for 1 μm AZD8055 and 1 μm ABT-737 (B). In C, apoptosis was determined by analysis of DNA fragmentation of PI-stained nuclei. Mean + S.D. of three independent experiments performed in triplicate are shown; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
AZD8055 and ABT-737 Cooperate to Trigger Caspase Activation, Mitochondrial Perturbations, and Caspase-dependent Apoptosis
For further mechanistic studies aiming at identifying the underlying molecular mechanisms of the synergistic interaction of AZD8055 and ABT737, we selected two RMS cell lines representing embryonal RMS (TE671) and alveolar RMS (RMS13). In a first step, we monitored activation of the caspase cascade by Western blotting. Combination treatment with AZD8055 and ABT-737 resulted in increased cleavage of caspases into active cleavage fragments compared with treatment with ABT-737 alone, i.e. cleavage of caspase-8 into p43/p41/p18 fragments, cleavage of caspase-3 into p17/p12 fragments and cleavage of caspase-9 into p37/p35 fragments, as well as enhanced cleavage of PARP (Fig. 2A). To check whether caspase activity is indeed required for the induction of cell death, we treated cells in the presence of the pan-caspase inhibitor Z-VAD-fmk. Of note, the addition of Z-VAD-fmk significantly reduced AZD8055/ABT-737-mediated DNA fragmentation and cell death (Fig. 2, B and C), demonstrating that caspase activity is required for apoptosis induction. Furthermore, we explored the involvement of the mitochondrial pathway of apoptosis by assessing the mitochondrial membrane potential. ABT-737 and AZD8055 cooperated to trigger loss of mitochondrial membrane potential in a time-dependent manner (Fig. 2D). Together, this set of experiments demonstrates that AZD8055 and ABT-737 act in concert to trigger caspase activation, mitochondrial perturbations, and caspase-dependent apoptosis.
FIGURE 2.

AZD8055 and ABT-737 cooperate to trigger caspase activation, mitochondrial perturbations and caspase-dependent apoptosis. TE671 and RMS13 cells were treated for indicated times (A and D) or for 48 h (B and C) with 1 μm AZD8055 and/or 1 μm ABT-737 in the presence or absence of 50 μm Z-VAD-fmk. In A, caspase activation and PARP cleavage were assessed by Western blotting; arrows indicate cleavage fragments, and an asterisk indicates unspecific bands. In B, cell death was determined by PI staining and flow cytometry. In C, apoptosis was determined by analysis of DNA fragmentation of PI-stained nuclei. In D, loss of mitochondrial membrane potential (MMP) was analyzed by flow cytometry using the dye tetramethylrhodamine methyl ester. Mean + S.D. of three independent experiments performed in triplicate are shown (B–D); **, p < 0.01; ***, p < 0.001.
AZD8055 Inhibits Activation of the PI3K/mTOR Signaling Pathway
To explore the effects of AZD8055 on PI3K/mTOR signaling, we monitored the phosphorylation status of upstream (Akt) and downstream (S6 and 4E-BP1) pathway components by Western blot analysis. Treatment with AZD8055 alone or in combination with ABT-737 profoundly suppressed phosphorylation of Akt, S6, and 4E-BP1, whereas treatment with ABT-737 alone had no effect on phosphorylation levels of these proteins (Fig. 3). These findings demonstrate that AZD8055 is a potent inhibitor of upstream and downstream components of the PI3K/mTOR pathway.
FIGURE 3.
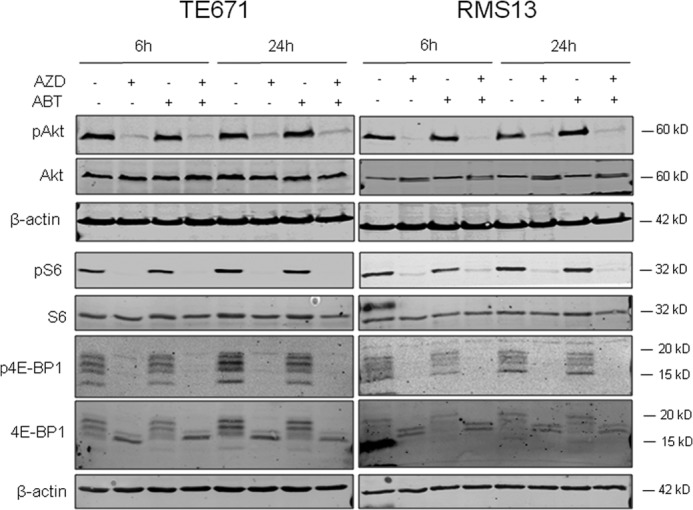
Effect of AZD8055 on PI3K/mTOR pathway activation. TE671 and RMS13 cells were treated for indicated times with 1 μm AZD8055 (AZD) and/or 1 μm ABT737 (ABT). Protein expression of phospho-Akt (Ser-473), Akt, phospho-S6 ribosomal protein, S6 ribosomal protein, phospho-4E-BP1, and 4E-BP1 was analyzed by Western blotting.
Sensitization to ABT-737-induced Apoptosis by mTORC1/2 or PI3K Inhibition
Because AZD8055 blocks both upstream (Akt) and downstream (S6 and 4E-BP1) elements of the PI3K/mTOR pathway, we next asked whether inhibition of PI3K, mTORC1, or mTORC2 is required for the synergistic interaction with ABT-737. To address this question, we used distinct inhibitors of the PI3K/mTOR cascade, including RAD001, an allosteric inhibitor of mTORC1, NVP-BKM120, an inhibitor of PI3K, and NVP-BEZ235, a dual PI3K/mTOR inhibitor that inhibits PI3K and both mTORC1/2 complexes (33). Interestingly, NVP-BEZ235 synergized with ABT-737 to induce DNA fragmentation to a similar extent as AZD8055/ABT-737 cotreatment, as demonstrated by CI values (Fig. 4, A and B). Consistently, NVP-BEZ235 profoundly suppressed phosphorylation of Akt, S6, and 4E-BP1 in a comparable manner with AZD8055 (Fig. 4C). By comparison, NVP-BKM120 was less effective than AZD8055 or NVP-BEZ235 to potentiate ABT-737-induced apoptosis, which was reflected by its decreased ability to reduce phosphorylation of 4E-BP1 and Akt (Fig. 4, A–C). RAD001 failed to act in concert with ABT-737 and even increased phosphorylation of Akt, whereas it exerted little inhibitory effects on 4E-BP1 phosphorylation (Fig. 4, A and C). Also, knockdown of Rictor did not cooperate with ABT-737 to trigger cell death, whereas the Akt inhibitor IV significantly enhanced ABT-737-induced apoptosis (supplemental Fig. 2). These findings suggest that inhibition of both mTORC complexes, PI3K, or Akt is required for sensitization to ABT-737-induced apoptosis.
FIGURE 4.

Effect of PI3K/mTOR inhibitors on ABT-737-induced apoptosis. TE671 and RMS13 cells were treated for 48 h (A and B) or 6 h (C) with indicated concentrations of ABT-737 and/or AZD8055, RAD001, NVP-BEZ235, or NVP-BKM120. In A, apoptosis was determined by analysis of DNA fragmentation of PI-stained nuclei. Mean + S.E. of three independent experiments performed in triplicate are shown. In B, corresponding CI values are shown for combinations affecting > 20% of the fraction (1 μm ABT-737 plus 1 μm AZD8055 or 1 μm NVP-BEZ235 or 1 μm NVP-BKM120). In C, protein expression of phospho-Akt (Ser-473), Akt, phospho-S6 ribosomal protein, S6 ribosomal protein, phospho-4E-BP1, and 4E-BP1 was analyzed by Western blotting.
AZD8055 and ABT-737 Cooperate to Down regulate Mcl-1
To further investigate the underlying mechanisms of the synergistic interaction of AZD8055 and ABT-737, we determined expression levels of pro- and antiapoptotic proteins. Interestingly, treatment with AZD8055 alone or in combination with ABT-737 decreased the expression of Mcl-1 protein (Fig. 5A). Also, protein expression of Noxa was reduced upon treatment with AZD8055 (Fig. 5A), indicating that in particular short-lived proteins are affected by AZD8055-mediated inhibition of 4E-BP1 phosphorylation and subsequently of protein translation. Also, AZD8055 cooperated with ABT-737 to trigger cleavage of Bid into truncated Bid, as evident by proteolytic turnover of full-length Bid and the appearance of the cleavage product truncated Bid (Fig. 5A).
FIGURE 5.

Down-regulation of Mcl-1 by mTORC1/2 or PI3K inhibition. A, TE671 and RMS13 cells were treated for indicated times with 1 μm AZD8055 and/or 1 μm ABT-737. Expression of pro- and antipoptotic proteins was analyzed by Western blotting. B, TE671 and RMS13 cells were treated for 6 h with 1 μm ABT-737 and/or 1 μm AZD8055 or 1 μm NVP-BEZ235 or 1 μm NVP-BKM120 or 1 μm RAD001. Expression of Mcl-1 protein was analyzed by Western blotting.
To determine whether the potency of the different PI3K/mTOR inhibitors to prime cells for ABT-737-induced apoptosis correlates with their ability to suppress Mcl-1, we monitored Mcl-1 levels upon exposure to these inhibitors. AZD8055, NVP-BEZ235, and BKM120, which all primed cells to ABT-737-mediated apoptosis (Fig. 4A), also reduced Mcl-1 protein expression (Fig. 5B). In contrast, RAD001 increased Mcl-1 protein levels (Fig. 5B), in line with its failure to potentiate apoptosis by ABT-737 (Fig. 4A). These findings suggest that down regulation of Mcl-1 by different PI3K/mTOR inhibitors correlates with their ability to enhance ABT-737-induced apoptosis. Although glycogen synthase kinase (GSK) 3 has been implicated in regulating Mcl-1 abundance (27), we did not observe differential effects of PI3K/mTOR inhibitors on GSK3 phosphorylation status (supplemental Fig. 3), indicating that GSK3 is not the key mediator of Mcl-1 down regulation by PI3K/mTOR inhibitors under these conditions.
Down regulation of Mcl-1 Sensitizes for ABT-737-induced Cell Death
To further investigate the role of Mcl-1 in the synergistic induction of apoptosis by AZD8055/ABT-737, we used an RNA interference-based approach to knock down Mcl-1. Efficient silencing of Mcl-1 protein with two different shRNA sequences was controlled by Western blotting (Fig. 6A). Importantly, Mcl-1 knockdown significantly increased ABT-737-induced cell death to a similar extent as treatment of control vector cells with the combination of ABT-737 and AZD8055 (Fig. 6B). Comparable results were obtained when cell death was assessed by analysis of DNA fragmentation (Fig. 6C). To confirm these results, we also monitored caspase cleavage in Mcl-1 knockdown cells. Interestingly, Mcl-1 knockdown cooperated with ABT-737 to trigger caspase-3, -8, and -9 cleavage to an extent comparable with that of cotreatment of control vector cells with ABT-737 and AZD8055 (Fig. 6D). In addition, we transiently silenced Mcl-1 by siRNA (Fig. 6E). Similarly, knockdown of Mcl-1 significantly enhanced ABT-737-triggered DNA fragmentation (Fig. 6F). These findings demonstrate that Mcl-1 is an important regulator of ABT-737-induced apoptosis in RMS cells and indicate that AZD8055-stimulated down regulation of Mcl-1 contributes to AZD8055-mediated sensitization to ABT-737.
FIGURE 6.

Down-regulation of Mcl-1 sensitizes for ABT-737-induced cell death. A–D, TE671 cells were stably transduced with two different shRNA sequences against Mcl-1 or control shRNA. In A, Mcl-1 expression was determined by Western blot analysis. In B and C, cells were treated for 48 h with 1 μm AZD8055 and/or 1 μm ABT-737, and cell death was determined by PI staining and flow cytometry (B) or apoptosis was determined by analysis of DNA fragmentation of PI-stained nuclei (C). Mean + S.D. of three independent experiments performed in triplicate are shown; **, p < 0.01; ***, p < 0.001. In D, cells were treated for 6 h with 1 μm AZD8055 and/or 1 μm ABT-737, and caspase activation was assessed by Western blotting; arrows indicate caspase cleavage fragments. E and F, TE671 cells were transiently transfected with three distinct siRNA against Mcl-1 (siMcl-1_1, siMcl-1_2, siMcl-1_3), or control siRNA (siControl). Mcl-1 expression was analyzed by Western blotting (E). Apoptosis was determined by analysis of DNA fragmentation of PI-stained nuclei and flow cytometry (F). Mean + S.D. of three independent experiments performed in triplicate are shown; *, p < 0.05; ***, p < 0.001.
DISCUSSION
Novel treatment strategies are needed to improve survival rates of patients suffering from RMS. Rational approaches include inhibition of the PI3K/mTOR pathway, as it is often aberrantly activated in RMS (7). AZD8055 is an ATP-competitive mTOR inhibitor that has recently demonstrated antitumor activity in vivo against pediatric cancers, including RMS (16). However, AZD8055 did not induce objective tumor regression (16), suggesting that combinations of AZD8055, together with additional anticancer agents, may be required to potentiate its antitumor activity. In the present study, we therefore searched for rational AZD8055-based combination therapies.
Here, we identify a novel synergistic combination of AZD8055 together with the BH3 mimetic ABT-737 to induce apoptosis in RMS. This drug interaction is highly synergistic as demonstrated by CI values (CI < 0.2) and occurs in both major histological subtypes of RMS, i.e. embryonal RMS and alveolar RMS. By comparison, we found no additive or synergistic interaction of AZD8055 with chemotherapeutic drugs that are commonly used for the treatment of RMS such as actinomycin D, vincristine, or doxorubicin. A possible explanation for this phenomenon might be the AZD8055-mediated cell cycle arrest in G1 (data not shown), which may reduce the sensitivity to these chemotherapeutics.
Mechanistically, AZD8055 and ABT-737 cooperate to trigger caspase-dependent apoptosis, as shown by increased cleavage of caspase-3, -8, and -9 into active cleavage fragments, rescue of cell death by the pan-caspase inhibitor Z-VAD-fmk and enhanced loss of mitochondrial membrane potential in AZD8055/ABT-737 co-treated cells. In addition, we identify the AZD8055-mediated suppression of Mcl-1 as an important molecular event that contributes to the synergistic induction of apoptosis by AZD8055/ABT-737. This conclusion is underscored by data showing that genetic silencing of Mcl-1 cooperates with ABT-737 to trigger caspase activation and apoptosis similar to treatment with AZD8055. In addition, the ability of different PI3K/mTOR inhibitors to enhance ABT-737-induced apoptosis correlates with their ability to suppress phosphorylation of 4E-BP1 and to down-regulate Mcl-1 expression. Consistently, the 4E-BP1 pathway has recently been implicated to control Mcl-1 protein translation (26). Inhibition of protein synthesis by AZD8055 might also account for the observed AZD8055-mediated down-regulation of Noxa protein levels. Activation of GSK3β upon AZD8055-mediated inhibition of Akt activity does not likely contribute to down regulation of Mcl-1 protein levels because AZD8055 did not suppress GSK3β phosphorylation. The identification of Mcl-1 as an important mediator of the synergistic interaction of AZD8055/ABT-737 is of special interest for RMS, as Mcl-1 is often up-regulated in this malignancy (21). In addition, there is a rationale to combine ABT-737 with drugs that down-regulate Mcl-1 levels, as ABT-737 inhibits Bcl-2, Bcl-xL, and Bcl-w, but not Mcl-1 (22). Besides, down-regulation of Mcl-1 has previously been reported to enhance ABT-737-induced apoptosis (28–30).
Because a variety of PI3K/Akt/mTOR inhibitors have already been used in clinical trials for different tumor types (11), we went on to determine the CI values of various inhibitors together with ABT-737. As a result, we found strong potentiation of ABT-737-induced apoptosis, when both mTORC complexes (by AZD8055), PI3K-mTORC complexes (by NVP-BEZ235), or Akt (by Akt inhibitor) were inhibited, whereas inhibition of PI3K (by BKM120) proved to be less effective to synergize with ABT-737. The failure of the allosteric mTORC1 inhibitor RAD001 to enhance ABT-737-mediated apoptosis was associated with its inability to suppress 4E-BP1 phosphorylation and Mcl-1 expression in parallel with enhanced phosphorylation of Akt. These findings are consistent with previous reports showing that 1) mTORC1 inhibition results in increased Akt phosphorylation due to loss of the S6K1-mediated feedback inhibition of IRS1 (12) and that 2) allosteric mTORC1 inhibitors such as RAD001 incompletely inhibit phosphorylation of 4E-BP1 (13). By comparison, the allosteric mTORC1 inhibitor rapamycin has been reported to cooperate with ABT-263, a second-generation analogue of ABT-737, to induce apoptosis in lymphoma cells (31). In addition, cotreatment of rapamycin and ABT-737 was described to enhance the sensitivity of non-small cell lung carcinoma cells to radiation (32). Together, these data also imply a context-dependent role of mTORC1 in the regulation of ABT-737-mediated cell death.
The current work has important implications for the design of novel approaches for the treatment of RMS. Signal transduction modulators including inhibitors of PI3K/Akt/mTOR signaling and inhibitors of Bcl-2 proteins are currently tested in early clinical trials (33, 34). Because hyperactivation of the PI3K/Akt/mTOR pathway frequently occurs in RMS and bears a poor prognosis (7), this pathway represents a promising therapeutic target in RMS. By discovering a novel synergistic interaction of the second-generation mTOR inhibitor AZD8055 together with the BH3 mimetic ABT-737, our study lays the ground for further studies to exploit these classes of inhibitors for the treatment of RMS.
Acknowledgments
We thank R. Sauter for expert technical assistance and C. Hugenberg for expert secretarial assistance.
This work was supported in part by a grant from the Deutsche Krebshilfe (to S. F.).

This article contains supplemental Figs. 1–3.
- RMS
- rhabdomyosarcoma
- 4E-BP1
- eIF4E-binding protein 1
- CI
- combination index
- mTOR
- mammalian target of rapamycin
- mTORC1
- mTOR complex 1
- PARP
- poly(ADP-ribose) polymerase
- PI
- propidium iodide
- Z-VAD-fmk
- N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone.
REFERENCES
- 1. Malempati S., Hawkins D. S. (2012) Rhabdomyosarcoma: review of the Children's Oncology Group (COG) Soft-Tissue Sarcoma Committee experience and rationale for current COG studies. Pediatr. Blood Cancer 59, 5–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dagher R., Helman L. (1999) Rhabdomyosarcoma: an overview. Oncologist 4, 34–44 [PubMed] [Google Scholar]
- 3. Hayes-Jordan A., Andrassy R. (2009) Rhabdomyosarcoma in children. Curr. Opin. Pediatr. 21, 373–378 [DOI] [PubMed] [Google Scholar]
- 4. Fulda S. (2008) Targeting apoptosis resistance in rhabdomyosarcoma. Curr. Cancer Drug Targets 8, 536–544 [DOI] [PubMed] [Google Scholar]
- 5. Wan X., Helman L. J. (2007) The biology behind mTOR inhibition in sarcoma. Oncologist 12, 1007–1018 [DOI] [PubMed] [Google Scholar]
- 6. Fulda S. (2012) Cell death pathways as therapeutic targets in rhabdomyosarcoma. Sarcoma 2012, 326210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Petricoin E. F., 3rd, Espina V., Araujo R. P., Midura B., Yeung C., Wan X., Eichler G. S., Johann D. J., Jr., Qualman S., Tsokos M., Krishnan K., Helman L. J., Liotta L. A. (2007) Phosphoprotein pathway mapping: Akt/mammalian target of rapamycin activation is negatively associated with childhood rhabdomyosarcoma survival. Cancer Res. 67, 3431–3440 [DOI] [PubMed] [Google Scholar]
- 8. Zoncu R., Efeyan A., Sabatini D. M. (2011) mTOR: from growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 12, 21–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Laplante M., Sabatini D. M. (2012) mTOR signaling in growth control and disease. Cell 149, 274–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ma X. M., Blenis J. (2009) Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 10, 307–318 [DOI] [PubMed] [Google Scholar]
- 11. Benjamin D., Colombi M., Moroni C., Hall M. N. (2011) Rapamycin passes the torch: a new generation of mTOR inhibitors. Nat. Rev. Drug Discov. 10, 868–880 [DOI] [PubMed] [Google Scholar]
- 12. O'Reilly K. E., Rojo F., She Q. B., Solit D., Mills G. B., Smith D., Lane H., Hofmann F., Hicklin D. J., Ludwig D. L., Baselga J., Rosen N. (2006) mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 66, 1500–1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Choo A. Y., Yoon S. O., Kim S. G., Roux P. P., Blenis J. (2008) Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc. Natl. Acad. Sci. U.S.A. 105, 17414–17419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Geoerger B., Kieran M. W., Grupp S., Perek D., Clancy J., Krygowski M., Ananthakrishnan R., Boni J. P., Berkenblit A., Spunt S. L. (2012) Phase II trial of temsirolimus in children with high-grade glioma, neuroblastoma and rhabdomyosarcoma. Eur. J. Cancer 48, 253–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chresta C. M., Davies B. R., Hickson I., Harding T., Cosulich S., Critchlow S. E., Vincent J. P., Ellston R., Jones D., Sini P., James D., Howard Z., Dudley P., Hughes G., Smith L., Maguire S., Hummersone M., Malagu K., Menear K., Jenkins R., Jacobsen M., Smith G. C., Guichard S., Pass M. (2010) AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. 70, 288–298 [DOI] [PubMed] [Google Scholar]
- 16. Houghton P. J., Gorlick R., Kolb E. A., Lock R., Carol H., Morton C. L., Keir S. T., Reynolds C. P., Kang M. H., Phelps D., Maris J. M., Billups C., Smith M. A. (2012) Initial testing (stage 1) of the mTOR kinase inhibitor AZD8055 by the pediatric preclinical testing program. Pediatr. Blood Cancer 58, 191–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fulda S., Debatin K. M. (2006) Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 25, 4798–4811 [DOI] [PubMed] [Google Scholar]
- 18. Fulda S. (2009) Tumor resistance to apoptosis. Int. J. Cancer 124, 511–515 [DOI] [PubMed] [Google Scholar]
- 19. Fulda S., Galluzzi L., Kroemer G. (2010) Targeting mitochondria for cancer therapy. Nat. Rev. Drug Discov. 9, 447–464 [DOI] [PubMed] [Google Scholar]
- 20. Armistead P. M., Salganick J., Roh J. S., Steinert D. M., Patel S., Munsell M., El-Naggar A. K., Benjamin R. S., Zhang W., Trent J. C. (2007) Expression of receptor tyrosine kinases and apoptotic molecules in rhabdomyosarcoma: correlation with overall survival in 105 patients. Cancer 110, 2293–2303 [DOI] [PubMed] [Google Scholar]
- 21. Pazzaglia L., Chiechi A., Conti A., Gamberi G., Magagnoli G., Novello C., Morandi L., Picci P., Mercuri M., Benassi M. S. (2009) Genetic and molecular alterations in rhabdomyosarcoma: mRNA overexpression of MCL1 and MAP2K4 genes. Histol. Histopathol. 24, 61–67 [DOI] [PubMed] [Google Scholar]
- 22. Oltersdorf T., Elmore S. W., Shoemaker A. R., Armstrong R. C., Augeri D. J., Belli B. A., Bruncko M., Deckwerth T. L., Dinges J., Hajduk P. J., Joseph M. K., Kitada S., Korsmeyer S. J., Kunzer A. R., Letai A., Li C., Mitten M. J., Nettesheim D. G., Ng S., Nimmer P. M., O'Connor J. M., Oleksijew A., Petros A. M., Reed J. C., Shen W., Tahir S. K., Thompson C. B., Tomaselli K. J., Wang B., Wendt M. D., Zhang H., Fesik S. W., Rosenberg S. H. (2005) An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435, 677–681 [DOI] [PubMed] [Google Scholar]
- 23. Fulda S., Sieverts H., Friesen C., Herr I., Debatin K. M. (1997) The CD95 (APO-1/Fas) system mediates drug-induced apoptosis in neuroblastoma cells. Cancer Res. 57, 3823–3829 [PubMed] [Google Scholar]
- 24. Gonzalez P., Mader I., Tchoghandjian A., Enzenmüller S., Cristofanon S., Basit F., Debatin K. M., Fulda S. (2012) Impairment of lysosomal integrity by B10, a glycosylated derivative of betulinic acid, leads to lysosomal cell death and converts autophagy into a detrimental process. Cell Death Differ. 19, 1337–1346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chou T. C. (1991) The median-effect principle and the combination index for quantitation of synergism and antagonism in Synergism and Antagonism in Chemotherapy (Chou T. C., ed.), pp. 61–102, Academic Press, San Diego [Google Scholar]
- 26. Coloff J. L., Macintyre A. N., Nichols A. G., Liu T., Gallo C. A., Plas D. R., Rathmell J. C. (2011) Akt-dependent glucose metabolism promotes Mcl-1 synthesis to maintain cell survival and resistance to Bcl-2 inhibition. Cancer Res. 71, 5204–5213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Maurer U., Charvet C., Wagman A. S., Dejardin E., Green D. R. (2006) Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol. Cell 21, 749–760 [DOI] [PubMed] [Google Scholar]
- 28. van Delft M. F., Wei A. H., Mason K. D., Vandenberg C. J., Chen L., Czabotar P. E., Willis S. N., Scott C. L., Day C. L., Cory S., Adams J. M., Roberts A. W., Huang D. C. (2006) The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 10, 389–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Konopleva M., Contractor R., Tsao T., Samudio I., Ruvolo P. P., Kitada S., Deng X., Zhai D., Shi Y. X., Sneed T., Verhaegen M., Soengas M., Ruvolo V. R., McQueen T., Schober W. D., Watt J. C., Jiffar T., Ling X., Marini F. C., Harris D., Dietrich M., Estrov Z., McCubrey J., May W. S., Reed J. C., Andreeff M. (2006) Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell 10, 375–388 [DOI] [PubMed] [Google Scholar]
- 30. Chen S., Dai Y., Harada H., Dent P., Grant S. (2007) Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation. Cancer Res. 67, 782–791 [DOI] [PubMed] [Google Scholar]
- 31. Ackler S., Xiao Y., Mitten M. J., Foster K., Oleksijew A., Refici M., Schlessinger S., Wang B., Chemburkar S. R., Bauch J., Tse C., Frost D. J., Fesik S. W., Rosenberg S. H., Elmore S. W., Shoemaker A. R. (2008) ABT-263 and rapamycin act cooperatively to kill lymphoma cells in vitro and in vivo. Mol. Cancer Ther. 7, 3265–3274 [DOI] [PubMed] [Google Scholar]
- 32. Kim K. W., Moretti L., Mitchell L. R., Jung D. K., Lu B. (2009) Combined Bcl-2/mammalian target of rapamycin inhibition leads to enhanced radiosensitization via induction of apoptosis and autophagy in non-small cell lung tumor xenograft model. Clin. Cancer Res. 15, 6096–6105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Engelman J. A. (2009) Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat. Rev. Cancer 9, 550–562 [DOI] [PubMed] [Google Scholar]
- 34. Thomas S., Quinn B. A., Das S. K., Dash R., Emdad L., Dasgupta S., Wang X. Y., Dent P., Reed J. C., Pellecchia M., Sarkar D., Fisher P. B. (2013) Targeting the Bcl-2 family for cancer therapy. Expert Opin. Ther. Targets 17, 61–75 [DOI] [PMC free article] [PubMed] [Google Scholar]