

Background: Trafficking of AMPA receptors (AMPAR) to medium spiny neuron (MSN) synapses regulates synapse strength.
Results: The neurotransmitters dopamine and glutamate cooperate to induce MSN synaptic trafficking of the Ca2-permeable AMPAR, which stimulates further AMPAR trafficking.
Conclusion: Dopamine and glutamate cooperate in a feed-forward mechanism for MSN synaptic potentiation.
Significance: MSN synaptic strength regulation integrates convergent dopamine and glutamate signals.
Keywords: Dopamine; Glutamate; Glutamate Receptors; Neurons; Synaptic Plasticity; Dopamine, AMPA Receptor; Striatum; Nucleus Accumbens; GluA1; Medium Spiny Neurons; cGKII; cGMP
Abstract
Regulation of striatal medium spiny neuron synapses underlies forms of motivated behavior and pathological drug seeking. A primary mechanism for increasing synaptic strength is the trafficking of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) into the postsynapse, a process mediated by GluA1 AMPAR subunit phosphorylation. We have examined the role of converging glutamate and dopamine inputs in regulating biochemical cascades upstream of GluA1 phosphorylation. We focused on the role of Ca2+-permeable AMPARs (CPARs), which lack the GluA2 AMPAR subunit. Under conditions that prevented depolarization, stimulation of CPARs activated neuronal nitric oxide synthase and production of cGMP. CPAR-dependent cGMP production was sufficient to induce synaptic insertion of GluA1, detected by confocal microscopy, through a mechanism dependent on GluA1 Ser-845 phosphorylation. Dopamine D1 receptors, in contrast, stimulate GluA1 extra synaptic insertion. Simultaneous activation of dopamine D1 receptors and CPARs induced additive increases in GluA1 membrane insertion, but only CPAR stimulation augmented CPAR-dependent GluA1 synaptic insertion. This incorporation into the synapse proceeded through a sequential two-step mechanism; that is, cGMP-dependent protein kinase II facilitated membrane insertion and/or retention, and protein kinase C activity was necessary for synaptic insertion. These data suggest a feed-forward mechanism for synaptic priming whereby an initial stimulus acting independently of voltage-gated conductance increases striatal neuron excitability, facilitating greater neuronal excitation by a subsequent stimulus.
Introduction
Synapses onto striatal neurons are regulated by converging glutamate and dopamine inputs (1, 2). Plasticity at these synapses modulates the excitability of medium spiny projection neurons (MSNs),3 which are involved in the orchestration of motor programs, including the seeking of natural rewards and drugs of abuse (3–6). MSNs exhibit bistable membrane properties, suggesting a two-step process for action potential initiation; an excitatory stimulus causes a transition from a so-called “down” state at approximately −80 mV to an “up” state at roughly −50 mV, and a properly-timed subsequent stimulus triggers an action potential in the up state neuron (7). Therefore, receptors that function at hyperpolarized potentials are an important locus for regulating synaptic strength in MSNs. Importantly, voltage-gated conductance, including N-methyl-d-aspartate receptors (NMDARs), are inactive at hyperpolarized potentials. Initiation of up state transitions, therefore, requires voltage-independent conductance, and previous studies suggest that Ca2+-permeable AMPA receptors (CPARs) could be vitally important to this process because they conduct Ca2+ robustly in the MSN down state (8).
CPARs are AMPARs lacking the GluA2 subunit. They play a significant role in synaptic plasticity and neuronal dysfunction in disease states (9, 10). Long term potentiation (LTP) is a form of synaptic plasticity often considered a cellular correlate of learning and memory; it has been viewed most often as an NMDAR-dependent process (11–13). Some studies suggest that induction of LTP causes a transient increase in CPARs, which facilitates the subsequent incorporation of GluA2-containing AMPARs (14–16). Other studies suggest that although CPARs comprise a very small percentage of synaptic AMPARs (17), they are sufficient to induce LTP at spinal cord synapses and in interneurons of the amygdala, regulating nociception and fear conditioning, respectively (18–20). Nevertheless, the biochemical pathways by which CPARs regulate synaptic plasticity are unknown.
Ca2+ fluxes through NMDARs are known to trigger cascades that increase synaptic strength through membrane insertion of AMPARs (13, 21, 22). In hippocampal neurons, Ca2+-stimulated adenylyl cyclases induce CREB activation and stimulate membrane insertion of GluA1-containing AMPARs through protein kinase A (PKA)-dependent phosphorylation of GluA1 Ser-845 (23–25). In parallel, Ca2+ influx in the hippocampus regulates Ser-845 phosphorylation through the nitric oxide (NO)/cGMP pathway (26, 27). NMDAR regulation of neuronal nitric oxide synthase (nNOS) stimulates cGMP production and phosphorylation of Ser-845 by cGMP-dependent protein kinase II (cGKII) (26–30). Ser-845 phosphorylation has been shown to be necessary in stabilizing CPARs in the extra synaptic membrane (31), with additional synaptic activity necessary to facilitate synaptic insertion of GluA1-containing AMPARs (32, 33). Further studies have suggested that phosphorylation of GluA1 at Ser-818 by protein kinase C (PKC) is a key step in synaptic insertion of AMPARs during NMDAR-dependent LTP (34).
In striatal MSNs, cAMP production is stimulated primarily by dopamine receptor signal transduction rather than NMDAR activity, due to the prevalent expression of Ca2+-inhibited adenylyl cyclase isoforms (V and VI) in the striatum (35, 36). Activation of dopamine D1 receptors (D1Rs) has been shown to increase GluA1 Ser-845 phosphorylation and induce membrane insertion in striatal neurons through a PKA-dependent mechanism (37–40). Nitric oxide signaling plays a prevalent role in modulating MSN excitability (41), and cGMP plays an important role in this regulation (42, 43), but the mechanisms by which it regulates MSN synapses are unknown. In the present study we investigated the possibility of cooperation by these parallel pathways downstream of the D1 receptor and cGMP in promoting trafficking of GluA1-containing AMPARs. Additionally, we asked whether CPARs could serve as surrogates for NMDARs, specifically in highly hyperpolarized neurons such as MSNs, by facilitating Ca2+ fluxes that stimulate nitric oxide production and GluA1 synaptic insertion.
EXPERIMENTAL PROCEDURES
Primary Culture of Striatal Neurons
Striatal tissue was isolated from E19 fetal Sprague-Dawley rats from coronal slices bounded by the rostral extent of the olfactory tubercle and caudal extent of the Circle of Willis. The tissue was treated with trypsin (Invitrogen, 10×) for 15 min at 37 °C. Neurons were dissociated by trituration with fire-polished Pasteur pipettes and suspended in 5 ml of plating medium (Minimum Essential Medium (Invitrogen) containing 0.45% glucose, 5 ml penicillin/streptomycin (Invitrogen), sodium pyruvate (Invitrogen), 50 ml fetal bovine serum (Invitrogen), and 25 μm glutamate). Cells were counted with a hemocytometer and diluted for plating at the desired density. After incubation for 2 h at 37 °C in plating media, plating medium was removed and replaced with Neurobasal media (Invitrogen; containing glutamine (10×), penicillin/streptomycin (Invitrogen), and B18 supplement (as described previously (44)). Neurons were maintained by adding 50% volume of Neurobasal once per week.
cGMP/cAMP Measurement
Striatal neuronal cultures were prepared as described above and grown 1 × 106 cells/well on six-well dishes for 10 DIV. Cells were treated with combinations of NMDA (EMD Biosciences), AMPA (Sigma), SKF38393 (Sigma), SCH23390 (Sigma), NASPM (Sigma), BAPTA-AM (Sigma), quinpirole (EMD Biosciences), APV (Sigma), nNOS Inhibitor I (EMD Biosciences), CdCl2 (Sigma), and/or TTX (Sigma). cGMP or cAMP levels were assayed using GE Biosciences cGMP or cAMP Biotrak EIA kits (RPN 226 or RPN 225; acetylation protocols). Data were collected using a 96-well plate reader at 450 nm and analyzed using Student's t tests. Concentrations were determined from a standard curve created during each assay.
Immunocytochemistry
Striatal MSN 14 DIV were treated with 500 nm TTX, 50 μm CdCl2, and 10 μm APV for 30 min. Cells were then treated with the noted combination of AMPA, SKF38393, NASPM, ODQ (EMD Biosciences), BAPTA-AM, DEA/NO (A.G. Scientific, Inc.), 2,5-dideoxyadenosine (Sigma), chelerythrine (Sigma), (Rp)-8-bromo-PET-cyclic guanosine monophosphate salt (Rp-8-pCPT-cGMPS) (Sigma), or KT-5720 (Sigma). After drug treatment, coverslips were removed to a moisturized chamber in 37 °C and incubated with a polyclonal antibody for either the N terminus of GluA1 (EMD Biosciences, PC246) or a monoclonal antibody to the HA tag of exogenously expressed GluA1 (Sigma, S9658) for 10 min. Coverslips were washed, fixed, permeabilized, and stained for SV2 (Novus Biologicals), synaptophysin (EMD Biosciences), or PSD-95 (Santa Cruz Biotechnology) and D1R (Sigma). Confocal micrographs were taken of individual cells in a blinded fashion, and 30 by 60 pixel regions of juxta-soma dendrites were randomly selected and imported into the analysis program ImageJ, where the mean pixel intensities/micrograph region were measured for surface GluA1, D1Rs, PSD-95, GFP, and/or synaptophysin as described (27). For colocalization measurements, channels containing surface GluA1 and PSD-95 were merged using an ImageJ colocalization plug-in (rsbweb.nih.gov). The plug-in measured co-localized pixels in two different channels and represented the colocalized pixels individually in a third channel if pixel intensities were higher than the specified intensity threshold of the respective channels and if the ratio of intensities between channels exceeded 50%. Colocalized pixel intensities were then measured.
Western Blotting
Equal amounts of proteins were fractionated on 8% SDS-PAGE gels. Gels were blotted to PVDF membranes (Bio-Rad), and membranes were incubated for 1 h at room temperature in 3% dried milk in PBST to prevent nonspecific binding. Primary antibodies were added for 1 h at room temperature, including GluA1 (Chemicon), phosphor-845 GluA1 (Chemicon), or tubulin (Sigma). Membranes were washed three times with PBST and incubated with secondary antibodies (Millipore HRP-conjugated) for 1 h at room temperature. Developer (PerkinElmer Life Sciences Advanced Chemiluminescent Enhancer) was added for 5 min, and membranes were exposed to audioradiographic film.
Viral Infection
Cells were infected with Sindbis viruses expressing wild type GluA2, R607Q GluA2, wild type HA-tagged GluA1 as described previously (27, 45) or with a lentivirus expressing only the C terminus of GluA1, courtesy of Dr. Takuya Takahashi.
RESULTS
Regulation of Cyclic Nucleotide Production by Glutamate and Dopamine Receptors
To investigate signaling in striatal neurons as related to cyclic nucleotide-dependent GluA1 trafficking, we investigated the expression of pertinent signaling proteins in cultured striatal neurons. Previous studies suggest that MSNs in homogenous culture do not form dendritic spines (46). We analyzed confocal micrographs for co-localization of synaptic proteins involved in signaling upstream of GluA1 Ser-845 phosphorylation. MSN cultures were stained for dopamine D1Rs, nNOS, soluble guanylyl cyclase (sGC), adenylyl cyclase type V/VI (ACV/VI), and the neuronal marker MAP2 (Fig. 1A). Coexpression of these proteins suggested that this culture system could be utilized to investigate intracellular signaling cascades employing dopamine and cGMP in striatal neurons (47).
FIGURE 1.
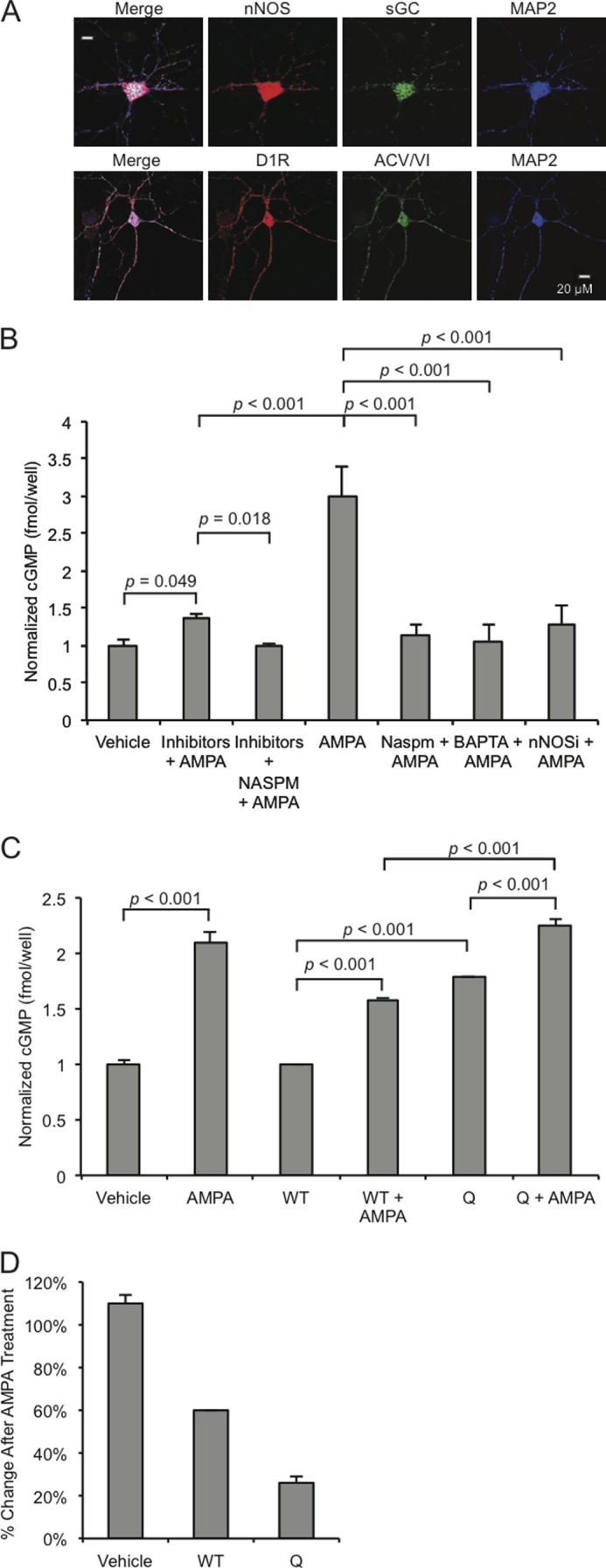
CPARs regulate cGMP production. A, accumbens MSNs (10 DIV) were stained for nNOS, adenylyl cyclase type VI (ACV/VI), soluble guanylyl cyclase (sGC), D1Rs, and MAP2. B, MSNs were either treated with vehicle or AMPA or with a mixture of inhibitors (APV, 10 μm; CdCl2, 50 μm; TTX, 500 nm) or BAPTA (500 μm) or nNOS Inhibitor (2 μm) or NASPM (200 μm) and then with AMPA (50 μm, 1 min) in the indicated combinations, and cGMP concentrations were measured by ELISA. C, MSNs (10 DIV) were infected overnight with Sindbis viruses expressing wild type GluA2 (WT) or GluA2-Q (Q) and treated with AMPA (50 μm, 1 min) or treated with vehicle as indicated, and cGMP concentrations were measured by ELISA. D, corresponding relative percent changes in the three infection conditions after AMPA treatment. Concentrations are fmol/well with n = 6 wells/test condition. Inhibitors or antagonists were added 30 min before treatment, and virus infections were performed 12 h before lysis. Data are represented as the mean ± S.E. normalized to vehicle treatment and analyzed using one-way ANOVA followed by Fisher's post hoc tests.
We employed the culture system to investigate the dynamics of cGMP production in MSNs. We demonstrated previously that in hippocampal neurons, NMDA treatment induces significant cGMP production through nNOS activation (26) and hypothesized that CPARs could act as surrogates for the NMDAR in stimulating cGMP production. Cultured MSNs (10 DIV) were treated with 50 μm AMPA for 1 min and lysed. Lysates were subjected to an ELISA assay measuring cGMP concentration. As shown in Fig. 1B, AMPA stimulation induced robust cGMP production (200.8 ± 40.3% increase in concentration (measured in fmol/well), n = 6, p < 0.001, Student's unpaired two-tailed t test). AMPA-induced increases were abolished when cultures were preincubated with either the cell-impermeable Ca2+-chelator BAPTA-AM, nNOS inhibitor I, or a specific polyamine inhibitor of CPARs, NASPM (Fig. 1B). To investigate whether the influx of Ca2+ that induced cGMP production required AMPA-induced depolarization, such as if the fluxes were through NMDARs or voltage-gated calcium channels or, alternatively, whether currents through Ca2+-permeable, GluA2-lacking AMPARs were sufficient, cultures were incubated with an inhibitor mixture containing APV (NMDAR antagonist) plus TTX (voltage-gated Na+ channel antagonist) plus CdCl2 (voltage gated Ca2+ channel inhibitor) (collectively “inhibitors”) to block non-CPAR-dependent Ca2+ currents. AMPA treatment increased cGMP production significantly in the presence of these inhibitors, and the increase was blocked if NASPM was added to the inhibitor mixture (Fig. 1B). This demonstrated that activation of CPARs is sufficient to trigger AMPA-induced cGMP production.
To investigate this question further, we infected cultures with Sindbis viruses expressing variants of the AMPAR subunit GluA2: 1) wild type GluA2 and 2) GluA2-Q, where Gln-607 has not undergone RNA editing to Arg, rendering the mutant permeable to Ca2+ (48–50). Base-line cGMP was significantly higher in GluA2-Q-expressing cultures than in GluA2 wild type-expressing cultures (79.3 ± 16.1% increase, n = 3, p < 0.001, Student's unpaired two-tailed t test), consistent with the capacity of Ca2+ currents arising from the spontaneous activity of CPARs to induce cGMP. Furthermore, treatment of GluA2-Q-expressing cultures with AMPA induced cGMP production that was significantly higher than with AMPA treatment of GluA2 wild type-expressing cultures (41.7 ± 9.0% increase, n = 3, p < 0.001, Student's unpaired two-tailed t test) (Fig. 1C), suggesting that Ca2+ permeability of AMPARs increases the capacity for cGMP production. Although the proportional increase obtained with GluA2-Q was not as great as with GluA2-WT (Fig. 1D), this is likely the result of the high base-line level of cGMP in the GluA2-Q-expressing cells.
To determine the role of AMPARs in cAMP production, we measured cAMP concentrations from the same MSN culture lysates where cGMP had been measured. We hypothesized that the presence of Ca2+-inhibited adenylyl cyclase type V in striatal neurons (35, 36) would attenuate cAMP production after AMPA treatment. Surprisingly, whereas treatment of cultures with AMPA induced a slight but not statistically significant increase in cAMP production (24.5 ± 3.5% increase n = 3), a significant increase was observed upon treatment with AMPA after preincubation with the Ca2+ flux inhibitors, either NASPM or the inhibitor mixture that blocks voltage gated Ca2+ conductance, APV, plus TTX plus CdCl2 (inhibitor) (14.3 ± 4.27 and 31.8 ± 2.15% increases, respectively, compared with NASPM or vehicle alone, n = 4, p < 0.05 and p < 0.01, Student's unpaired two-tailed t test) (Fig. 2A). This suggested that unlike cGMP production, cAMP production took place through a mechanism that did not require Ca2+ and may have been Ca2+-inhibited, also consistent with Na+ depolarization-induced stimulation of adenylate cyclase as reported in previous studies (51–53). Because this increase in cAMP was observed when CPARs were blocked by NASPM, the increase must have been induced by Ca2+-impermeable AMPA receptors, consistent with a role for Ca2+-inhibited adenylyl cyclase type V.
FIGURE 2.

cAMP production and CPAR activation. Lysates were prepared from accumbens MSNs (10 DIV), and intracellular cAMP was measured by ELISA. A, cAMP concentrations normalized to vehicle after treatment with AMPA (50 μm, 1 min) and/or NASPM (200 μm) and inhibitors (APV, 10 μm; CdCl2, 50 μm; TTX, 500 nm) as indicated. B, cAMP concentrations after treatment with NMDA (50 μm, 1 min) and/or SKF38393 (10 μm, 1 min) and SCH23390 (10 μm) as indicated. Concentrations are fmol/well with n = 6 wells/test condition. Inhibitors/antagonists were added 30 min before treatment. Data are represented as the mean ± S.E. normalized to vehicle treatment and analyzed using one-way ANOVA followed by Fisher's post hoc tests. N.S., not significant.
To determine further whether Ca2+ fluxes inhibited cAMP production in MSNs, we treated cultures with NMDA. Interestingly, treatment with 50 μm NMDA for 1 min significantly decreased the cAMP concentration in MSN lysates (33.8 ± 4.4% decrease, n = 3, p < 0.001, Student's unpaired two-tailed t test). As before, we attributed this result to the expression in striatal neurons of Ca2+-inhibited adenylyl cyclase type V in striatal neurons (35, 36). Stimulation of D1Rs with the agonist SKF38393 (10 μm for 1 min) increased cAMP concentration significantly, and this effect was attenuated by preincubation with the specific D1R-antagonist SCH23390 (10 μm) for 30 min or by co-administration with NMDA (Fig. 2B), suggesting that the inhibiting effect of NMDAR Ca2+ is dominant over D1R stimulation of adenylyl cyclase type V in MSN cultures.
CPARs and D1Rs Cooperate to Regulate GluA1 Trafficking
Recognizing that cGMP and cAMP can induce surface membrane trafficking of AMPARs through phosphorylation of GluA1 Ser-845 by cGKII and PKA, respectively (25, 27), we asked if AMPARs and, more specifically, CPARs, could cooperate with D1Rs through an additive mechanism, with each kinase stimulating surface membrane insertion of GluA1 through GluA1 serine 845 phosphorylation, respectively, by cGMP- and cAMP-dependent pathways. Stimulation had been shown previously to increase the rate of GluA1 surface trafficking (37). Cultured MSNs were pretreated with the voltage-gated Ca2+ conductance inhibitor mixture APV plus TTX plus CdCl2 to confine extracellular Ca2+ currents to CPARs as a source as described above. Neurons were then treated with SKF38393 and/or AMPA for 1 min and stained for surface GluA1 for D1Rs and for PSD-95. We determined total surface GluA1 by measuring immune fluorescence in randomly selected, equal size segments of juxta-soma dendrites in confocal micrographs of D1R-expressing MSNs and determined the synaptic component of GluA1 through measuring the fraction of this signal that colocalized with the synaptic marker PSD-95, as described under “Experimental Procedures.” Both AMPA and SKF38393 stimulation increased surface GluA1, and simultaneous addition of AMPA and SKF38393 increased GluA1 surface expression more greatly than stimulation of either receptor individually (46.9 ± 6.3% increase compared with D1R, 43.0 ± 6.3% increase compared with CPAR, n = 3, p < 0.05, Student's unpaired two-tailed t test) (Fig. 3, A and B), suggesting that CPARs and D1Rs stimulated GluA1 trafficking additively and in parallel through the cGMP and cAMP pathways, respectively. To investigate this further and to determine whether CPAR generation of cGMP rather than generation of cAMP was specifically involved after AMPA treatment, cultures were pretreated with the soluble guanylyl cyclase inhibitor 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), the adenylyl cyclase type V/VI inhibitor 2,5-dideoxyadenosine, or NASPM. The increase in surface GluA1 after AMPA treatment was blocked by pretreatment with NASPM and 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), whereas a slight but significant increase in surface GluA1 was observed after AMPA addition in 2,5-dideoxyadenosine-pretreated cultures, suggesting CPAR stimulation of cGMP production played a significant role in the induction of GluA1 surface expression by AMPA and that the cAMP pathway did not make a major contribution (34.3 ± 7.1% increase, n = 3, p < 0.037, Student's unpaired two-tailed t test) (Fig. 3C).
FIGURE 3.

CPARs selectively induce GluA1 trafficking through GluA1 Ser-845 phosphorylation. A, visualization of surface and synaptic GluA1 in cultured MSNs expressing D1 receptors (10 DIV) after treatment with AMPA (5 μm, 1 min) and/or SKF38393 (10 μm, 5 min) and 30 min pretreatment with APV (10 μm), CdCl2 (50 μm), TTX (500 nm). Neurons were stained for D1R, surface GluA1, and PSD-95 (synaptic marker). Scale bar = 20 μm. B, quantification of surface and synaptic GluA1 in cultured MSNs treated in A; n = 25 dendrite regions. C, MSNs (10 DIV) were treated with vehicle or AMPA (50 μm, 1 min) either alone or in cultures pretreated for 30 min with NASPM (200 μm), 2,3-dideoxyadenosine (DDA; 10 μm), or 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one or ODQ (5 μm), and surface GluA1 levels were determined and normalized to vehicle; n = 20 dendrite regions. D–F, MSN cultures were treated as in A and pretreated either with Rp-8-pCPT-cGMPS (D), chelerythrine (E), or KT-5720 (F). Data are represented as mean pixel intensity ± S.E. normalized to vehicle treatment and analyzed using one-way ANOVA followed by Fisher's post hoc tests; n = 20 dendrite regions for each condition.
Previous studies demonstrated that D1Rs stimulate trafficking of GluA1 to extrasynaptic sites (33). We next investigated whether CPAR stimulation acted differently and could facilitate synaptic insertion of GluA1, not just surface expression. AMPA treatment significantly increased synaptic GluA1 compared with vehicle or SKF38393 treatment (Fig. 3, A and B; n = 3, p < 0.02, Student's unpaired two-tailed t test). This indicated that AMPA was capable of inducing both surface expression at extrasynaptic sites and synaptic trafficking. Although treatment with SKF38393 alone elevated surface GluA1, it did not elevate synaptic GluA1, and simultaneous administration of AMPA and SKF38393 did not increase synaptic GluA1 compared with AMPA treatment alone (Fig. 3, A and B). This suggested that AMPA stimulation was capable of both extrasynaptic surface expression and synaptic expression, whereas D1R signaling was sufficient for induction of surface but not synaptic insertion.
Given that AMPA stimulation was sufficient to induce synaptic insertion of GluA1 but D1R stimulation was capable of only extrasynaptic surface expression, we investigated whether CPARs initiated an additional signal to trigger movement of GluA1 from extrasynaptic to synaptic sites. We hypothesized that CPAR Ca2+ could trigger PKC-dependent synaptic insertion of extrasynaptic GluA1 (34). Cultures were pretreated for 30 min with APV, TTX, and CdCl2 as described above and subsequently with either the cGKII inhibitor Rp-8-pCPT-cGMPS, the PKA inhibitor, KT-5720, or the PKC inhibitor chelerythrine. We then treated with AMPA or SKF38393 and measured GluA1 surface expression and synaptic localization. We observed that Rp-8-pCPT-cGMPS blocked surface and synaptic expression after AMPA treatment but had no effect on D1R-stimulated surface expression as induced by SKF38393, as expected (Fig. 3D). Rp-8-pCPT-cGMPS also prevented the additive cooperation by AMPA and D1Rs in induction of GluA1 surface expression. However, AMPA was still able to cooperate with SCF38393 to elevate synaptic GluA1 in the presence of Rp-8-pCPT-cGMPS, consistent with SCF38393 elevating surface GluA1 and AMPA trafficking this surface receptor to the synapse (Fig. 3D). Interestingly, PKC inhibitor chelerythrine had no effect on D1R- or CPAR-dependent surface membrane trafficking but prevented CPAR-induced synaptic insertion (Fig. 3E). These data suggest that CPARs induce surface and synaptic insertion of GluA1 through cGKII and PKC, respectively, which is in accordance with previous studies that demonstrate which Ser-845 phosphorylation is sufficient for plasma membrane retention/trafficking but not synaptic insertion (27, 33).
To determine whether AMPA induced trafficking of GluA1 relative to trafficking of GluA2, we measured surface GluA1:GluA2 ratios after stimulation with AMPA or a stimulator of cGMP production, the NO donor Diethylammonium (Z)-1-(N,N-diethylamino)diazen-1-ium-1,2-diolate (DEA/NO). Consistent with preferential trafficking of GluA1, treatment with AMPA or DEA increased the surface GluA1 to GluA2 ratio (Fig. 4, A and B), although a small increase in GluA2 was evident (Fig. 4A). To confirm that AMPA receptors induce GluA1 surface expression through Ser-845 phosphorylation, we biotin-labeled surface proteins in cultured MSNs after vehicle or AMPA treatment. We measured two ratios: 1) surface phospho-Ser-845 GluA1 to total GluA1 and 2) surface GluA1 to total GluA1. There were significant increases in surface phospho-Ser-845 to total GluA1 and surface GluA1 to total GluA1 after AMPA treatment compared with cultures treated with only vehicle or with AMPA + NASPM (Fig. 4, C–E). These data suggest that AMPA induces Ser-845 GluA1 phosphorylation and membrane insertion through a mechanism dependent in part on CPARs.
FIGURE 4.

AMPA induces GluA1 trafficking and GluA1 Ser-845 phosphorylation. A and B, accumbens MSNs (10 DIV) were treated with an NO donor (DEA, 50 μm, 10 min) or AMPA (50 μm, 1 min), surface GluA1 and GluA2 were measured, and the surface GluA1:GluA2 ratio was determined. Scale bar = 10 μm; n = 20 dendrite regions. C, accumbens MSNs (10 DIV) were analyzed either directly or after treatment with AMPA (50 μm, 1 min) or NASPM (200 μm, 30 min pretreatment) as indicated. Surface proteins were labeled with biotin, isolated on streptavidin beads, analyzed by Western blot, and Ser(P)-845 GluA1, total GluA1, and tubulin were measured. D, the ratio of surface Ser(P)-845 GluA1:Total GluA1 was determined for the three conditions and normalized to vehicle; n = 4 samples/condition. E, the ratio of biotinylated, surface GluA1:total GluA1 was determined and normalized to vehicle; n = 4 samples/condition. Data are represented as the mean ± S.E. normalized to vehicle treatment and analyzed using one-way ANOVA followed by Fisher's post hoc tests.
To confirm by an additional method that AMPA induces GluA1 trafficking, we infected MSNs with a lentivirus expressing the C terminus of GluA1, a construct shown previously to prevent surface trafficking of GluA1 (54). We observed that expression of the GluA1 C terminus construct prevented both AMPA- and forskolin-induced surface trafficking of GluA1 (Fig. 5, A and B). We conclude that CPAR Ca2+ fluxes work via cGMP/cGKII in parallel with D1Rs to induce GluA1 extrasynaptic trafficking via a C terminus-dependent mechanism, consistent with a role for Ser-845 phosphorylation. CPARs also induce synaptic insertion through PKC stimulation. Although NMDARs play an important role in accumbens plasticity (55), these data identify additional NMDAR-independent mechanisms through which dopamine and glutamate can regulate GluA1 trafficking in MSNs and implicate CPARs as inducers of potentiation.
FIGURE 5.
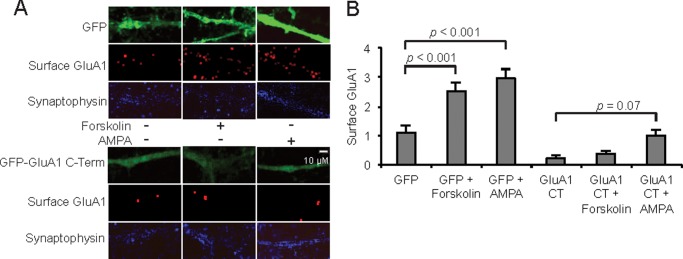
Confirmation of AMPA-induced GluA1 trafficking through genetic blockage of trafficking. Surface GluA1 was visualized (A) and quantified (B) in MSNs (10 DIV) infected with lentiviruses expressing GFP or the GluA1 C-terminal (CT) peptide tagged with GFP. Infections were carried out 4 days before treatment with AMPA or forskolin (25 μm, 10 min). Synaptophysin levels were unchanged among all conditions. Scale bar = 10 μm. AMPA treatment = 5 μm, 1 min in each case. Data are represented as the mean ± S.E. integrated density of surface GluA1; n = 20 dendrite regions, and data are represented as the mean ± S.E. normalized to vehicle treatment and analyzed using one-way ANOVA followed by Fisher's post hoc tests.
DISCUSSION
Excitability of striatal MSNs is regulated by dopamine transmission and glutamatergic regulation of the NO/cGMP pathway (43, 56). MSNs rest at a highly hyperpolarized down state where CPARs are responsible almost exclusively for Ca2+ conductance (8). For MSNs to generate action potentials, AMPAR activation first stimulates an up state transition, after which an action potential can be generated by additional stimulation (7, 57). This suggests that mechanisms by which synaptic strength can be increased independently of voltage, thereby increasing the probability of an up state transition, could be important for understanding synaptic potentiation in the striatum. In the present study we investigate production of cGMP and cAMP, second messengers important for increasing synaptic strength through trafficking of the AMPAR subunit GluA1 (25, 27). We observe that cyclic GMP and cyclic AMP are produced by parallel glutamate- and dopamine-dependent pathways. Our data show that voltage dependent Ca2+ fluxes, such as through the NMDA receptor and voltage gated Ca2+ channels can induce cGMP. Interestingly, stimulation of endogenous CPARs on their own was sufficient to induce cGMP production. In support, stimulation of neurons with exogenous expression of CPARs by AMPA elevated the level of cGMP more greatly than AMPA stimulation of neurons in which exogenous Ca2+-impermeable AMPA receptors have been expressed. We observed a numerically greater proportional increase in cGMP after AMPA treatment in GluA2-WT versus GluA2-Q infection conditions. We hypothesize that the proportional increase in cGMP after AMPA was less in GluA2-Q infection conditions because base-line cGMP was statistically higher; it is possible that we observed cell maximum capacity for cGMP production in the GluA2-Q + AMPA condition. Also, whereas NMDAR regulation of cAMP-dependent phosphorylation of GluA1 is important for hippocampal LTP (25), we found that NMDAR activation suppressed cAMP production. We then observed that CPARs, which signal through the NO/cGMP pathway, were sufficient to induce GluA1 synaptic insertion. We investigated the role of the GluA1 C terminus in the trafficking mechanism. AMPA receptor trafficking may take place by C-tail dependent and C-tail independent mechanisms (22). The insertion induced by the NO/cGMP pathway was blocked by a dominant negative peptide corresponding to the GluA1 C-terminal domain, consistent with a mechanism dependent on phosphorylation of Ser-845 in the GluA1 C-terminal domain. Additionally, whereas simultaneous activation of CPARs and D1Rs caused an additive increase in surface GluA1, there was not an additive increase in synaptic GluA1, and further experiments suggested that the CPAR Ca2+ signal was sufficient to induce a two-step process of GluA1 membrane insertion through cGKII and synaptic insertion through PKC. The demonstration of the ability of CPARs to mediate GluA1 trafficking via a cGMP-dependent pathway is a novel finding. The cGMP-dependent pathway provides a mechanism by which synaptic strength can be increased independently of voltage. Postsynaptic cell depolarization is not required, and thus it can operate when the NMDA receptor and voltage-gated calcium channels are inoperative.
Regulation of Cyclic Nucleotide Production
Although activation of AMPARs has been shown previously to induce cGMP production (58, 59), the mechanism for soluble guanylyl cyclase activation by AMPARs has been unclear. Here we demonstrate that CPAR Ca2+ is sufficient to induce cGMP production. This suggests that cGMP production can proceed independently of voltage-gated conductance, which is of particular importance in highly hyperpolarized cells such as MSNs in which NMDARs are inactive. We observed that CPAR-induced cGMP production is dependent on nNOS, providing the first evidence that CPARs regulate NO production. This provides a potential mechanistic link between CPARs and the pathologies often associated Ca2+ and non-NMDA-type glutamate receptors (45, 60, 61).
NMDAR-dependent activation of cAMP-dependent protein kinase (PKA) has been shown to regulate LTP induction in the hippocampus and amygdala (25, 62). In the striatum, however, where the predominant isoform is the Ca2+-inhibited type V, we demonstrate that NMDAR activation suppresses cAMP production over short time scales (1 min). Meanwhile, although AMPARs stimulated a modest and not significant increase in cAMP production, the increase was augmented and significant when CPARs and other Ca2+ conductances were blocked. Previous studies have demonstrated that adenylyl cyclases can be stimulated by depolarization (51–53), and our results suggest that Na+ depolarization and Ca2+ flux exert opposite effects on adenylyl cyclase type V in MSNs. We conclude that cAMP production is dopamine-dependent and NMDAR-suppressed in striatal neurons and that glutamate and dopamine pathways work in parallel to induce cGMP and cAMP production, respectively.
D1R and CPAR Regulation of GluA1 Trafficking
Phosphorylation of GluA1 Ser-845 promotes membrane trafficking and/or retention of AMPARs. Both PKA and cGKII phosphorylate Ser-845 (25, 27), and phosphorylation of Ser-818 by PKC has been shown to be necessary for synaptic insertion (34). We investigated whether trafficking of GluA1 could take place without the involvement of voltage-gated conductance. NMDARs, voltage-gated Ca2+ channels, and Na+ channels were blocked, and AMPARs and/or D1Rs were stimulated to induce cGMP and cAMP production. We observed that AMPARs stimulated GluA1 surface insertion through a mechanism dependent on CPARs, nNOS, and cGKII. Interestingly, synaptic insertion was dependent on PKC but not cGKII when the D1 receptor was stimulated, suggesting that cGKII or PKA phosphorylation of Ser-845 increases the extrasynaptic pool of AMPARs, and PKC phosphorylation of Ser-818 promotes synaptic incorporation. Further evidence for this hypothesis came from the observation that stimulation of D1Rs and CPARs together, although causing an additive increase in surface expression, did not lead to an additive increase in synaptic expression. We conclude that stimulation of Ser-845 phosphorylation by cGMP or cAMP increases extrasynaptic accumulation of GluA1-containing AMPARs, and these receptors are trafficked into synaptic areas through Ca2+-dependent activation of PKC, perhaps through subsequent phosphorylation of Ser-818 (34). In this mechanism, D1 receptors, which induce cAMP, and CPARs, which induce cGMP and activate PKC, can cooperate to regulate GluA1 synaptic trafficking. Also, although AMPA and an NO donor increased the ratio of surface GluA1 to GluA2, consistent with GluA1 surface trafficking by this pathway, the surface level of GluA2 also increased, although to a lesser extent than GluA1. This is consistent with the ability of GluA1/2 heteromers to traffic in response to the same signals as GluA1 homomers (63). Given our recent findings that CPAR activity is necessary for hyperactivity immediately after sucrose consumption (6), it is likely that the mechanisms described herein play a role in synaptic plasticity related to reward-driven behavioral modifications.
Conclusion
Our results suggest that cAMP- and cGMP-dependent pathways work in parallel in striatal MSNs to increase neuronal excitability through GluA1 phosphorylation and trafficking. Given that glutamate receptors did not promote cAMP production and dopamine D1 receptors did not promote cGMP production (data not shown), our results also suggest that dopamine and glutamate release regulate synaptic plasticity through distinct pathways in MSNs. Therefore, environmental stimuli that pair glutamate and dopamine release likely both promote GluA1 Ser-845 phosphorylation-dependent trafficking of AMPARs to the neuronal membrane, and it is likely additive compared with glutamate or dopamine alone. However, the observation that D1R signaling does not promote synaptic insertion of GluA1 suggests that paired glutamate and dopamine stimulation leads to an additive increase only in extrasynaptic AMPAR accumulation, with an additional Ca2+-dependent stimulus that activates PKC necessary for synaptic insertion. Physiologically, this mechanism could play an important role in lowering the threshold for highly hyperpolarized MSNs to undergo down-to-up state transitions and generate action potentials by means of a mechanism in which a voltage-independent increase in excitability facilitates greater neuronal excitation by a subsequent voltage-dependent stimulus. Additionally, given the role of dopamine in reward prediction error (64), this mechanism also potentially underlies synaptic plasticity related to natural rewards.
Acknowledgments
We thank Dr. Takuya Takahashi for the GluR1 C terminus vector and Latika Khatri, Julian Horwitz, and Laveria Lee for technical assistance. We also thank Dr. Gerald Rameau, Dr. Jainne Ferreira, Dr. Yafell Serulle, Dr. Joey Pick, and members of the Ziff laboratory for helpful discussions.
This work was supported, in whole or in part, by National Institutes of Health Grants R01NS06192 (CPAR studies) and R01MH67229 (cGKII studies) (to E. B. Z.)
- MSN
- medium spiny neuron
- NMDAR
- NMDA receptor
- CPAR
- Ca2+-permeable AMPA receptor
- LTP
- long term potentiation
- AMPA
- α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- nNOS
- neuronal NOS
- D1R
- dopamine D1 receptor
- cGKII
- cGMP-dependent protein kinase II
- DIV
- days in vitro
- ANOVA
- analysis of variance
- DEA
- diethylammonium (Z)-1-(N,N-diethylamino)diazen-1-ium-1,2-diolate
- NASPM
- 1-naphthylacetyl spermine
- TTX
- tetrodotoxin; (2R)-amino-5-phosphonovaleric acid.
REFERENCES
- 1. Calabresi P., Picconi B., Tozzi A., Di Filippo M. (2007) Dopamine-mediated regulation of corticostriatal synaptic plasticity. Trends Neurosci. 30, 211–219 [DOI] [PubMed] [Google Scholar]
- 2. Wolf M. E., Sun X., Mangiavacchi S., Chao S. Z. (2004) Psychomotor stimulants and neuronal plasticity. Neuropharmacology 47, 61–79 [DOI] [PubMed] [Google Scholar]
- 3. Carlezon W. A., Jr., Thomas M. J. (2009) Biological substrates of reward and aversion. A nucleus accumbens activity hypothesis. Neuropharmacology 56, 122–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kalivas P. W., Volkow N. D. (2005) The neural basis of addiction. A pathology of motivation and choice. Am. J. Psychiatry 162, 1403–1413 [DOI] [PubMed] [Google Scholar]
- 5. Graybiel A. M. (2005) The basal ganglia. Learning new tricks and loving it. Curr. Opin. Neurobiol. 15, 638–644 [DOI] [PubMed] [Google Scholar]
- 6. Tukey D. S., Ferreira J. M., Antoine S. O., D'amour J. A., Ninan I., Cabeza de Vaca S., Incontro S., Wincott C., Horwitz J. K., Hartner D. T., Guarini C. B., Khatri L., Goffer Y., Xu D., Titcombe R. F., Khatri M., Marzan D. S., Mahajan S. S., Wang J., Froemke R. C., Carr K. D., Aoki C., Ziff E. B. (2013) Sucrose ingestion induces rapid AMPA receptor trafficking. J. Neurosci. 33, 6123–6132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Calabresi P., Mercuri N. B., Stefani A., Bernardi G. (1990) Synaptic and intrinsic control of membrane excitability of neostriatal neurons. I. An in vivo analysis. J. Neurophysiol. 63, 651–662 [DOI] [PubMed] [Google Scholar]
- 8. Carter A. G., Sabatini B. L. (2004) State-dependent calcium signaling in dendritic spines of striatal medium spiny neurons. Neuron 44, 483–493 [DOI] [PubMed] [Google Scholar]
- 9. Pellegrini-Giampietro D. E., Gorter J. A., Bennett M. V., Zukin R. S. (1997) The GluR2 (GluR-B) hypothesis. Ca2+-permeable AMPA receptors in neurological disorders. Trends Neurosci. 20, 464–470 [DOI] [PubMed] [Google Scholar]
- 10. Liu S. J., Zukin R. S. (2007) Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci. 30, 126–134 [DOI] [PubMed] [Google Scholar]
- 11. Morris R. G., Anderson E., Lynch G. S., Baudry M. (1986) Selective impairment of learning and blockade of long-term potentiation by an N-methyl-d-aspartate receptor antagonist, AP5. Nature 319, 774–776 [DOI] [PubMed] [Google Scholar]
- 12. Bliss T. V., Collingridge G. L. (1993) A synaptic model of memory. long-term potentiation in the hippocampus. Nature 361, 31–39 [DOI] [PubMed] [Google Scholar]
- 13. Nicoll R. A., Malenka R. C. (1999) Expression mechanisms underlying NMDA receptor-dependent long-term potentiation. Ann. N.Y. Acad. Sci. 868, 515–525 [DOI] [PubMed] [Google Scholar]
- 14. Plant K., Pelkey K. A., Bortolotto Z. A., Morita D., Terashima A., McBain C. J., Collingridge G. L., Isaac J. T. (2006) Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat. Neurosci. 9, 602–604 [DOI] [PubMed] [Google Scholar]
- 15. Liu S. J., Cull-Candy S. G. (2005) Subunit interaction with PICK and GRIP controls Ca2+ permeability of AMPARs at cerebellar synapses. Nat. Neurosci. 8, 768–775 [DOI] [PubMed] [Google Scholar]
- 16. Liu S. Q., Cull-Candy S. G. (2000) Synaptic activity at calcium-permeable AMPA receptors induces a switch in receptor subtype. Nature 405, 454–458 [DOI] [PubMed] [Google Scholar]
- 17. Lu W., Shi Y., Jackson A. C., Bjorgan K., During M. J., Sprengel R., Seeburg P. H., Nicoll R. A. (2009) Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach. Neuron 62, 254–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vikman K. S., Rycroft B. K., Christie M. J. (2008) Switch to Ca2+-permeable AMPA and reduced NR2B NMDA receptor-mediated neurotransmission at dorsal horn nociceptive synapses during inflammatory pain in the rat. J. Physiol. 586, 515–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hartmann B., Ahmadi S., Heppenstall P. A., Lewin G. R., Schott C., Borchardt T., Seeburg P. H., Zeilhofer H. U., Sprengel R., Kuner R. (2004) The AMPA receptor subunits GluR-A and GluR-B reciprocally modulate spinal synaptic plasticity and inflammatory pain. Neuron 44, 637–650 [DOI] [PubMed] [Google Scholar]
- 20. Mahanty N. K., Sah P. (1998) Calcium-permeable AMPA receptors mediate long-term potentiation in interneurons in the amygdala. Nature 394, 683–687 [DOI] [PubMed] [Google Scholar]
- 21. Barry M. F., Ziff E. B. (2002) Receptor trafficking and the plasticity of excitatory synapses. Curr. Opin. Neurobiol. 12, 279–286 [DOI] [PubMed] [Google Scholar]
- 22. Ziff E. B. (2007) TARPs and the AMPA receptor trafficking paradox. Neuron 53, 627–633 [DOI] [PubMed] [Google Scholar]
- 23. Nguyen P. V., Abel T., Kandel E. R. (1994) Requirement of a critical period of transcription for induction of a late phase of LTP. Science 265, 1104–1107 [DOI] [PubMed] [Google Scholar]
- 24. Poser S., Storm D. R. (2001) Role of Ca2+-stimulated adenylyl cyclases in LTP and memory formation. Int. J. Dev. Neurosci. 19, 387–394 [DOI] [PubMed] [Google Scholar]
- 25. Esteban J. A., Shi S. H., Wilson C., Nuriya M., Huganir R. L., Malinow R. (2003) PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat. Neurosci. 6, 136–143 [DOI] [PubMed] [Google Scholar]
- 26. Rameau G. A., Tukey D. S., Garcin-Hosfield E. D., Titcombe R. F., Misra C., Khatri L., Getzoff E. D., Ziff E. B. (2007) Biphasic coupling of neuronal nitric oxide synthase phosphorylation to the NMDA receptor regulates AMPA receptor trafficking and neuronal cell death. J. Neurosci. 27, 3445–3455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Serulle Y., Zhang S., Ninan I., Puzzo D., McCarthy M., Khatri L., Arancio O., Ziff E. B. (2007) A GluR1-cGKII interaction regulates AMPA receptor trafficking. Neuron 56, 670–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hayashi Y., Nishio M., Naito Y., Yokokura H., Nimura Y., Hidaka H., Watanabe Y. (1999) Regulation of neuronal nitric-oxide synthase by calmodulin kinases. J. Biol. Chem. 274, 20597–20602 [DOI] [PubMed] [Google Scholar]
- 29. Rameau G. A., Chiu L. Y., Ziff E. B. (2004) Bidirectional regulation of neuronal nitric-oxide synthase phosphorylation at serine 847 by the N-methyl-d-aspartate receptor. J. Biol. Chem. 279, 14307–14314 [DOI] [PubMed] [Google Scholar]
- 30. Serulle Y., Arancio O., Ziff E. B. (2008) A role for cGMP-dependent protein kinase II in AMPA receptor trafficking and synaptic plasticity. Channels 2, 230–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. He K., Song L., Cummings L. W., Goldman J., Huganir R. L., Lee H. K. (2009) Stabilization of Ca2+-permeable AMPA receptors at perisynaptic sites by GluR1-S845 phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 106, 20033–20038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Oh M. C., Derkach V. A., Guire E. S., Soderling T. R. (2006) Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J. Biol. Chem. 281, 752–758 [DOI] [PubMed] [Google Scholar]
- 33. Gao C., Sun X., Wolf M. E. (2006) Activation of D1 dopamine receptors increases surface expression of AMPA receptors and facilitates their synaptic incorporation in cultured hippocampal neurons. J. Neurochem. 98, 1664–1677 [DOI] [PubMed] [Google Scholar]
- 34. Boehm J., Kang M. G., Johnson R. C., Esteban J., Huganir R. L., Malinow R. (2006) Synaptic incorporation of AMPA receptors during LTP is controlled by a PKC phosphorylation site on GluR1. Neuron 51, 213–225 [DOI] [PubMed] [Google Scholar]
- 35. Mons N., Cooper D. M. (1994) Selective expression of one Ca2+-inhibitable adenylyl cyclase in dopaminergically innervated rat brain regions. Brain Res. Mol. Brain. Res. 22, 236–244 [DOI] [PubMed] [Google Scholar]
- 36. Kheirbek M. A., Beeler J. A., Ishikawa Y., Zhuang X. (2008) A cAMP pathway underlying reward prediction in associative learning. J. Neurosci. 28, 11401–11408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mangiavacchi S., Wolf M. E. (2004) D1 dopamine receptor stimulation increases the rate of AMPA receptor insertion onto the surface of cultured nucleus accumbens neurons through a pathway dependent on protein kinase A. J. Neurochem. 88, 1261–1271 [DOI] [PubMed] [Google Scholar]
- 38. Chao S. Z., Lu W., Lee H. K., Huganir R. L., Wolf M. E. (2002) D1 dopamine receptor stimulation increases GluR1 phosphorylation in postnatal nucleus accumbens cultures. J. Neurochem. 81, 984–992 [DOI] [PubMed] [Google Scholar]
- 39. Snyder G. L., Allen P. B., Fienberg A. A., Valle C. G., Huganir R. L., Nairn A. C., Greengard P. (2000) Regulation of phosphorylation of the GluR1 AMPA receptor in the neostriatum by dopamine and psychostimulants in vivo. J. Neurosci. 20, 4480–4488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Carr K. D., Chau L. S., Cabeza de Vaca S., Gustafson K., Stouffer M., Tukey D. S., Restituito S., Ziff E. B. (2010) AMPA receptor subunit GluR1 downstream of D-1 dopamine receptor stimulation in nucleus accumbens shell mediates increased drug reward magnitude in food-restricted rats. Neuroscience 165, 1074–1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sammut S., Dec A., Mitchell D., Linardakis J., Ortiguela M., West A. R. (2006) Phasic dopaminergic transmission increases NO efflux in the rat dorsal striatum via a neuronal NOS and a dopamine D(1/5) receptor-dependent mechanism. Neuropsychopharmacology 31, 493–505 [DOI] [PubMed] [Google Scholar]
- 42. Sammut S., Threlfell S., West A. R. (2010) Nitric oxide-soluble guanylyl cyclase signaling regulates corticostriatal transmission and short-term synaptic plasticity of striatal projection neurons recorded in vivo. Neuropharmacology 58, 624–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. West A. R., Grace A. A. (2004) The nitric oxide-guanylyl cyclase signaling pathway modulates membrane activity states and electrophysiological properties of striatal medium spiny neurons recorded in vivo. J. Neurosci. 24, 1924–1935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Osten P., Khatri L., Perez J. L., Köhr G., Giese G., Daly C., Schulz T. W., Wensky A., Lee L. M., Ziff E. B. (2000) Mutagenesis reveals a role for ABP/GRIP binding to GluR2 in synaptic surface accumulation of the AMPA receptor. Neuron 27, 313–325 [DOI] [PubMed] [Google Scholar]
- 45. Mahajan S. S., Ziff E. B. (2007) Novel toxicity of the unedited GluR2 AMPA receptor subunit dependent on surface trafficking and increased Ca2+ permeability. Mol. Cell Neurosci. 35, 470–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Segal M., Greenberger V., Korkotian E. (2003) Formation of dendritic spines in cultured striatal neurons depends on excitatory afferent activity. Eur. J. Neurosci. 17, 2573–2585 [DOI] [PubMed] [Google Scholar]
- 47. Yung K. K., Bolam J. P., Smith A. D., Hersch S. M., Ciliax B. J., Levey A. I. (1995) Immunocytochemical localization of D1 and D2 dopamine receptors in the basal ganglia of the rat. Light and electron microscopy. Neuroscience 65, 709–730 [DOI] [PubMed] [Google Scholar]
- 48. Burnashev N., Monyer H., Seeburg P. H., Sakmann B. (1992) Divalent ion permeability of AMPA receptor channels is dominated by the edited form of a single subunit. Neuron 8, 189–198 [DOI] [PubMed] [Google Scholar]
- 49. Greger I. H., Khatri L., Ziff E. B. (2002) RNA editing at arg607 controls AMPA receptor exit from the endoplasmic reticulum. Neuron 34, 759–772 [DOI] [PubMed] [Google Scholar]
- 50. Greger I. H., Khatri L., Kong X., Ziff E. B. (2003) AMPA receptor tetramerization is mediated by Q/R editing. Neuron 40, 763–774 [DOI] [PubMed] [Google Scholar]
- 51. Cooper D. M., Schell M. J., Thorn P., Irvine R. F. (1998) Regulation of adenylyl cyclase by membrane potential. J. Biol. Chem. 273, 27703–27707 [DOI] [PubMed] [Google Scholar]
- 52. Reddy R., Smith D., Wayman G., Wu Z., Villacres E. C., Storm D. R. (1995) Voltage-sensitive adenylyl cyclase activity in cultured neurons. A calcium-independent phenomenon. J. Biol. Chem. 270, 14340–14346 [DOI] [PubMed] [Google Scholar]
- 53. Billups D., Billups B., Challiss R. A., Nahorski S. R. (2006) Modulation of Gq-protein-coupled inositol trisphosphate and Ca2+ signaling by the membrane potential. J. Neurosci. 26, 9983–9995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mitsushima D., Ishihara K., Sano A., Kessels H. W., Takahashi T. (2011) Contextual learning requires synaptic AMPA receptor delivery in the hippocampus. Proc. Natl. Acad. Sci. U.S.A. 108, 12503–12508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Schotanus S. M., Chergui K. (2008) Long-term potentiation in the nucleus accumbens requires both NR2A- and NR2B-containing N-methyl-d-aspartate receptors. Eur. J. Neurosci. 27, 1957–1964 [DOI] [PubMed] [Google Scholar]
- 56. Hernández-López S., Bargas J., Surmeier D. J., Reyes A., Galarraga E. (1997) D1 receptor activation enhances evoked discharge in neostriatal medium spiny neurons by modulating an L-type Ca2+ conductance. J. Neurosci. 17, 3334–3342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Calabresi P., Mercuri N. B., Bernardi G. (1990) Synaptic and intrinsic control of membrane excitability of neostriatal neurons. II. An in vitro analysis. J. Neurophysiol. 63, 663–675 [DOI] [PubMed] [Google Scholar]
- 58. Garthwaite J., Southam E., Boulton C. L., Nielsen E. B., Schmidt K., Mayer B. (1995) Potent and selective inhibition of nitric oxide-sensitive guanylyl cyclase by 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one. Mol. Pharmacol. 48, 184–188 [PubMed] [Google Scholar]
- 59. Marcoli M., Maura G., Cervetto C., Giacomini C., Oliveri D., Candiani S., Pestarino M. (2006) Nitric oxide-evoked cGMP production in Purkinje cells in rat cerebellum. An immunocytochemical and pharmacological study. Neurochem. Int. 49, 683–690 [DOI] [PubMed] [Google Scholar]
- 60. Kwak S., Kawahara Y. (2005) Deficient RNA editing of GluR2 and neuronal death in amyotropic lateral sclerosis. J. Mol. Med. 83, 110–120 [DOI] [PubMed] [Google Scholar]
- 61. Corona J. C., Tapia R. (2007) Ca2+-permeable AMPA receptors and intracellular Ca2+ determine motoneuron vulnerability in rat spinal cord in vivo. Neuropharmacology 52, 1219–1228 [DOI] [PubMed] [Google Scholar]
- 62. Huang Y. Y., Kandel E. R. (1998) Postsynaptic induction and PKA-dependent expression of LTP in the lateral amygdala. Neuron 21, 169–178 [DOI] [PubMed] [Google Scholar]
- 63. Shi S., Hayashi Y., Esteban J. A., Malinow R. (2001) Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell 105, 331–343 [DOI] [PubMed] [Google Scholar]
- 64. Mirenowicz J., Schultz W. (1996) Preferential activation of midbrain dopamine neurons by appetitive rather than aversive stimuli. Nature 379, 449–451 [DOI] [PubMed] [Google Scholar]