

Background: Glycine decarboxylase (P-protein) is essential for many vital processes, including nucleotide biosynthesis and photosynthesis.
Results: Disulfide formation drives conformational changes that inactivate the cyanobacterial P-protein, a model for plant and human glycine decarboxylase.
Conclusion: Glycine decarboxylase activity is regulated by cellular redox homeostasis.
Significance: This is the first molecular model for redox regulation of glycine decarboxylase.
Keywords: Crystal Structure, Decarboxylase, Disulfide, Pyridoxal Phosphate, Redox Regulation, P-protein, Glycine Decarboxylase
Abstract
Glycine decarboxylase, or P-protein, is a pyridoxal 5′-phosphate (PLP)-dependent enzyme in one-carbon metabolism of all organisms, in the glycine and serine catabolism of vertebrates, and in the photorespiratory pathway of oxygenic phototrophs. P-protein from the cyanobacterium Synechocystis sp. PCC 6803 is an α2 homodimer with high homology to eukaryotic P-proteins. The crystal structure of the apoenzyme shows the C terminus locked in a closed conformation by a disulfide bond between Cys972 in the C terminus and Cys353 located in the active site. The presence of the disulfide bridge isolates the active site from solvent and hinders the binding of PLP and glycine in the active site. Variants produced by substitution of Cys972 and Cys353 by Ser using site-directed mutagenesis have distinctly lower specific activities, supporting the crucial role of these highly conserved redox-sensitive amino acid residues for P-protein activity. Reduction of the 353–972 disulfide releases the C terminus and allows access to the active site. PLP and the substrate glycine bind in the active site of this reduced enzyme and appear to cause further conformational changes involving a flexible surface loop. The observation of the disulfide bond that acts to stabilize the closed form suggests a molecular mechanism for the redox-dependent activation of glycine decarboxylase observed earlier.
Introduction
P-protein (EC 1.4.4.2., glycine dehydrogenase (decarboxylating)) is one of four proteins of the glycine cleavage system (GCS).2 This enzyme system is essential for glycine degradation and one-carbon metabolism in bacteria, archaea, and all eukaryotes (e.g. see Refs. 1–6). Because the GCS-catalyzed reactions are reversible (7, 8), they can also perform de novo synthesis of glycine from one-carbon units, CO2, and NH3 (9, 10).
In addition to the pyridoxal 5′-phosphate (PLP)-dependent P-protein, GCS requires two more enzymes, namely the tetrahydrofolate (THF)-dependent T-protein (EC 2.1.2.10; aminomethyltransferase) and the NAD+-dependent L-protein (EC 1.8.1.4; dihydrolipoamide dehydrogenase). As the fourth component, a small lipoylated protein, the H-protein (initially named hydrogen carrier protein) interacts successively via its lipoyl arm with the P-, T-, and L-proteins (Fig. 1). The GCS reaction cycle comprises three reactions. First, in a transaldimination reaction (11), glycine reacts with the PLP on the P-protein to form an external aldimine that loses the carboxyl group as CO2 and donates the remaining aminomethylene moiety to the oxidized lipoamide arm of H-protein. Next, T-protein uses the S-aminomethyl-dihydrolipoyl H-protein and THF to produce CH2-THF, ammonia, and dihydrolipoyl H-protein. Finally, L-protein closes the GCS reaction cycle by the NAD+-dependent reoxidation of the H-protein's dihydrolipoyl arm to the dithiolane form (reviewed in Refs. 12 and 13). As a result, one molecule each of glycine, THF, and NAD+ is converted into CO2, NH3, CH2-THF, and NADH. The produced CH2-THF is used as a one-carbon donor in a variety of biosynthetic pathways, including the synthesis of serine and of S-adenosylmethionine as another universal one-carbon donor (reviewed in Ref. 14). The NADH is reoxidized to NAD+ by the mitochondrial electron chain with or without subsequent ATP production (15).
FIGURE 1.
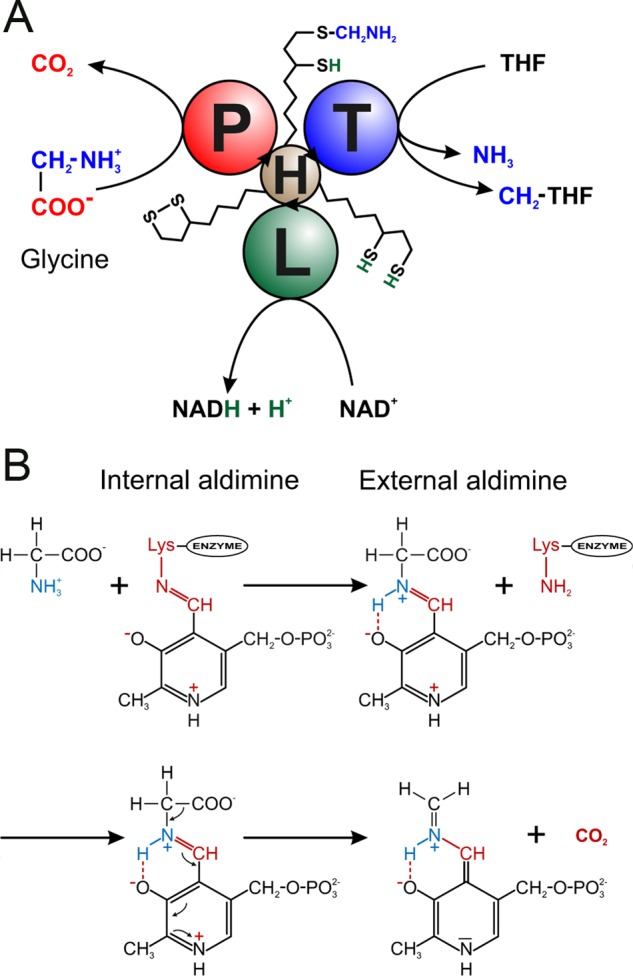
Schematic view of the reactions catalyzed by the GCS. A, the overall reaction catalyzed by the P-, T-, L-, and H-proteins of the GCS. First, the P-protein catalyzes the decarboxylation of glycine and the transfer of the aminomethylene moiety to the oxidized lipoamide arm of the H-protein. Next, the T-protein catalyzes the transfer of the methylene group to THF, thereby releasing NH3. Finally, the L-protein reoxidizes the reduced lipoamide group of the H-protein. B, outline of the involvement of the PLP cofactor in the decarboxylation reaction catalyzed by the P-protein. PLP first forms an internal aldimine with a lysine residue in the active site (Lys726 in the Synechocystis enzyme). Next the C4′ atom of PLP is attacked by glycine to form the external aldimine, thereby releasing the lysine. Finally CO2 is released from the external aldimine, leaving a quinoid intermediate ready to bind lipoamide.
The ubiquitous occurrence of GCS already indicates its crucial importance for cellular metabolism. Indeed, the absence of or malfunction of GCS results in fatal metabolic dysfunctions, at least in eukaryotes. In humans, for example, such defects cause the inborn disease glycine encephalopathy (nonketotic hyperglycinemia), which leads to accumulation of glycine in the central nervous system. This terminal disease very often results from mutations in the P-protein-encoding gene (16) but can also be due to defective T-protein or H-protein genes (17). In plants, the artificial deletion of the two P-protein genes was lethal for seedlings of the model plant Arabidopsis thaliana during or shortly after germination (18). Plants generate very large daily amounts of glycine in the photorespiratory cycle, an essential complement to the photosynthetic Calvin-Benson cycle (reviewed in Ref. 19). This particular metabolic pathway requires a very high glycine-decarboxylating activity. Therefore, the GCS proteins amount to more than 30% of total matrix protein in green leaf mitochondria (20). Correspondingly, the artificial reduction of leaf GCS activity distinctly impairs the growth of plants in air (21), whereas an increased GCS activity resulted in an improved growth (22). Deletion of GCS in cyanobacteria, the predecessors of plant chloroplasts, is less catastrophic (23). Such mutants also accumulate glycine, which is somewhat toxic due to chelation of magnesium ions, but a bypass to the GCS reaction, the glycerate pathway, allows nearly normal growth (24, 25).
It has been suggested that the animal and plant GCS proteins form a labile enzyme complex loosely associated with the mitochondrial inner membrane (e.g. see Refs. 13 and 26). Although the formation of a moderately stable complex between two H-protein molecules per P-protein dimer was demonstrated (27) and the interaction between H- and T-protein was studied (28), the supramolecular structure of such a hypothetical complex comprising all GCS proteins is not known. On the other hand, three-dimensional structures of several individual GCS proteins were reported (29–31), including that of the P-protein from Thermus thermophilus HB8 (32). In contrast to eukaryotic P-proteins, which are all α2 homodimers of about 200 kDa (e.g. see Refs. 33–35), the P-protein from T. thermophilus consists of two smaller polypeptides, α and β, which assemble to an about 200-kDa heterotetrameric (αNβC)2 P-protein. Related P-proteins are present in other prokaryotes, such as the anaerobic Eubacterium acidaminophilum (36), but the majority of known P-proteins from aerobic bacteria, including Escherichia coli (37) and all cyanobacteria (38, 39), are homodimeric enzymes closely related to their eukaryotic homologs.
Following the biochemical characterization (38) of the homodimeric P-protein from the model cyanobacterium Synechocystis sp. PCC 6803 (hereafter referred to as Synechocystis) and preliminary structural characterization (40), we present here the first three-dimensional structure of a homodimeric P-protein. This knowledge is of particular interest because the Synechocystis P-protein is closely related to eukaryotic P-proteins. We also present structures of ternary complexes with the cofactor PLP and the substrate glycine. Based on the structural information, three cysteine residues that are potentially involved in redox-dependent regulation of P-protein and hence GCS activity were replaced by site-specific mutagenesis, and the P-protein variants were compared with the wild-type protein in biochemical assays.
EXPERIMENTAL PROCEDURES
Protein Expression, Directed Mutagenesis, and Purification
The P-protein gene (slr0293) from Synechocystis was cloned in the bacterial overexpression vector pBAD-slr0293, expressed, and purified as described elsewhere (38, 40). For the exchange of specific cysteines for serine by directed mutagenesis, we amplified the whole construct with Phusion polymerase (High-Fidelity DNA Polymerase, New England Biolabs) by PCR using phosphorylated primer pairs (C353S, forward (ATATCAGCACTGCCCAGGTTCTCTT) and reverse (TACTAGTGGCCTTGTCCCGACGGAT); C972S, forward (TAGTTAGCTCCTGTGAAGGTATGGA) and reverse (AATGGCGATCACCGTAGGTATTATT); C972S/C974S, forward (CTCCAGCGAAGGTATGGAGG) and reverse (CTAACTAAATGGCGATCACC). After circularization and multiplication in E. coli strain LMG194, the nucleotide sequence was confirmed for the regions between the restriction sites below. From the circularized plasmids carrying the mutation in Cys353, an XhoI-Bsp1407I fragment was excised and used to replace the corresponding sequence in the above mentioned wild-type slr0293 overexpression vector. Correspondingly, Bsp1407I-PvuII fragments carrying the individual or combined mutations in Cys972 and Cys974 were used to introduce these mutations into the wild-type overexpression vector or the C353S variant.
Activity Assays
The recombinant P- and H-proteins were affinity-purified as described earlier (38) and equilibrated in 20 mm Tris/HCl (pH 7.8) by using prepacked PD-10 columns (Sephadex G-25 M, GE Healthcare). The P-protein preparations were concentrated in Vivaspin 20 centrifugation units (Sartorius, Göttingen, Germany). After the addition of glycerol to a final concentration of 50% (v/v), protein concentrations were determined (41), and aliquots were stored at −80 °C. P-protein activity was measured by following the glycine-14C-bicarbonate exchange reaction (2) after a preincubation with 1 mm (“reduced” state) or zero (“oxidized” state) dithiothreitol (DTT) for 5 min at room temperature. Standard assays at 30 °C contained 100 mm sodium phosphate (pH 6.0), 0.1 mm PLP, 18 mm glycine, 1 mm or zero DTT, 18.5 μm H-protein, 0.6 μm P-protein dimer, and 3 mm NaH14CO3 (1.62 μCi) in a total volume of 300 μl for each time point. All enzyme assays were done in triplicates and repeated at least once with independent protein preparations. Immediately after starting the reactions by the addition of NaH14CO3 and every 10 min thereafter for a total time of 40 min, aliquots of 250 μl were mixed with 80 μl of 50% trichloroacetic acid. These samples were dried overnight to remove remaining NaH14CO3, dissolved in 500 μl of water, and analyzed in a liquid scintillation counter following a standard protocol. Rates were corrected by control reactions without glycine for all time points.
Crystallization of Synechocystis P-protein
The crystallization of the apo-P-protein has been described elsewhere (40). These crystals (named form 1) belong to the orthorhombic system, space group P212121, with unit cell dimensions a = 96.3 Å, b = 135.8 Å, c = 179.1 Å, α = β = γ = 90°, and one dimer in the asymmetric unit (Table 1). The subunits have been arbitrarily labeled by the chain identifications A and B in the coordinate files. A second crystal form was obtained using the hanging drop vapor diffusion method by mixing equal volumes of 15 mg ml−1 protein in 20 mm sodium phosphate buffer, pH 7.5, 50 mm NaCl, 5 mm tris(2-carboxyethyl)phosphine (TCEP), and 0.2 mm PLP with a reservoir solution of 100 mm Bicine, pH 9, 1.6 m (NH4)3PO4, 5 mm TCEP, and 0.2 mm PLP. Prism-shaped crystals (50 × 25 × 100 μm) were obtained 1–3 days after streak seeding at 20 °C. The form 2 crystals belong to space group P321, with unit cell dimensions a = b = 160.1 Å, c = 159.8 Å, α = β = 90°, γ = 120° and contain one dimer in the asymmetric unit.
TABLE 1.
Data collection and refinement statistics
| Apo-P-protein open flexible loop | P-protein-PLP-Gly | P-protein-PLP-Gly closed flexible loop | |
|---|---|---|---|
| Data collection | |||
| Wavelength (Å) | 0.9795 | 0.9334 | 0.9334 |
| Resolution (Å) | 54.1–2.0 (2.07-2.00)a | 99.0-1.9 (1.97-1.90) | 99.0-1.8 (1.86-1.80) |
| Space group | P212121 | P321 | P321 |
| Cell axes (Å) | 96.3, 135.8, 179.1 | 160.1, 160.1, 159.8 | 160.0, 160.0, 159.7 |
| No. of reflections | 1,119,901 | 1,010,518 | 999,624 |
| No. of unique reflections | 158,314 (15,696) | 185,454 (18,266) | 217,331 (20,709) |
| Completeness (%) | 99.8 (100) | 99.9 (99.1) | 99.3 (95.4) |
| Multiplicity | 7.1 (7.1) | 5.4 (4.4) | 4.6 (4.0) |
| Mosaicity (degrees) | 0.44 | 0.42 | 0.35 |
| Rmergeb | 0.083 (0.555) | 0.068 (0.371) | 0.094 (0.471) |
| Average I/σ(I) | 26.9 (3.5) | 18.4 (4.3) | 16.6 (3.3) |
| Wilson B-factor (Å2) | 32.9 | 17.9 | 16.2 |
| Refinement | |||
| Resolution (Å) | 54.1-2.0 (2.02-2.00) | 49.8-1.9 (1.92-1.90) | 49.8-1.8 (1.82-1.80) |
| R/Rfreec (%) | 16.1/19.2 (22.2/30.5) | 15.5/18.6 (19.7/22.4) | 16.3/19.1 (23.8/29.6) |
| Residues in structure | |||
| Subunit A | 20–757, 767–963, 966–978 | 15–339, 350–755, 767–977 | 15–755, 767–977 |
| Subunit B | 18–335, 348–757, 766–962, 968–975 | 14–337, 349–755, 767–977 | 15–337, 350–755, 767–977 |
| No. of non-hydrogen atoms | |||
| Protein | 14,582 | 14,674 | 14,694 |
| Ligands | 38 | 38 | |
| Solvent | 882 | 1524 | 1480 |
| B-factors (Å2) | |||
| Protein | 40.7 | 20.5 | 19.2 |
| Solvent | 41.9 | 30.0 | 28.1 |
| r.m.s. deviation bond lengths (Å) | 0.007 | 0.007 | 0.007 |
| r.m.s. deviation bond angles (degrees) | 1.06 | 1.08 | 1.10 |
| Ramachandran plot (%) | |||
| Favored | 97.77 | 97.41 | 97.25 |
| Allowed | 2.12 | 2.43 | 2.59 |
| Disallowed | 0.11 | 0.16 | 0.16 |
| PDB code | 4LGL | 4LHC | 4LHD |
a Numbers in parentheses indicate the outer resolution shell.
b Rmerge = Σhkl Σi|Ii(hkl) − 〈I(hkl)〉|/Σhkl Σi Ii(hkl), where 〈I(hkl)〉 is the average intensity of symmetry-equivalent reflections.
c R-factor = Σhkl‖Fo| − |Fc‖/Σhkl|Fo|, where Fo and Fc are the observed and calculated structure factor amplitudes, respectively.
Soaking of Ligands
Crystals of the binary complex with PLP were used to prepare ligand complexes by soaking crystals at 20 °C in solutions of the ligand. Glycine (1.5 μl, 3 m) was added to the drop containing the crystal (initial drop size 5 μl). After 60 min, the crystal was transferred for a few seconds to a cryosolution containing 0.3 m glycine, 17.5% ethylene glycol, 1.6 m (NH4)3PO4, 5 mm TCEP, 0.2 mm PLP; transferred to a nylon CryoLoop (Hampton Research); and then flash-cooled in liquid nitrogen and maintained at 100 K for data collection. Alternatively, lipoic acid (50 mg ml−1 in ethanol) was added to the drop prior to the addition of 1.5 μl of 3 m glycine. Soaking and flash-cooling were performed as described previously.
Data Collection and Structure Determination
X-ray diffraction data for the apoenzyme (without PLP) were collected to 2.0 Å resolution on a CCD detector (ADSC Q315r) at beam line ID14-4, European Synchrotron Radiation Facility (ESRF, Grenoble, France) at a wavelength of 0.98 Å. The crystals were soaked for a few seconds in the reservoir solution containing 20% (v/v) ethylene glycol as a cryoprotectant, transferred to a nylon CryoLoop (Hampton Research), and then flash-cooled in liquid nitrogen and maintained at 100 K for data collection. A data set to 2.0 Å resolution was collected from one single (form 1) crystal, and the data were processed with DENZO and SCALEPACK (42). The diffraction data statistics are shown in Table 1. The structure was solved by molecular replacement with PHASER (43) using one heterodimer (αNβC) of the P-protein from T. thermophilus (PDB code 1WYT (32)) as a search model. The first crude model was improved by automatic chain tracing in ARPwARP (44) using the sequence of the Synechocystis enzyme. The structure was further refined by manual rebuilding using O (45) alternated with refinement in REFMAC5 (46) and PHENIX (47). Solvent molecules were added using ARPwARP (48) and PHENIX and were manually inspected in O. Assignment of water molecules was made based on peak heights of residual electron density, hydrogen bonding patterns, and B-factors.
Diffraction data for the holoenzyme (binary complex containing PLP and ternary complexes containing PLP and glycine) were collected on a CCD detector (ADSC Q315r) at beam line ID14-1, ESRF, to a maximum resolution of 1.6 Å from one single crystal. The data were processed with DENZO and SCALEPACK (42) and the structures were solved by molecular replacement using PHASER (43) with the ligand-free structure as a search model. The models were improved by iterative manual rebuilding of missing loop regions and the C terminus in O (45) and refinement with REFMAC5 (46) and PHENIX (47). Solvent molecules were added using ARPwARP (48). Density for PLP was observed close to the side chain of Lys726 allowing the building of the cofactor into the model. Further refinement of the binary complex indicated that a mixture of PLP and phosphate ion from the crystallization buffer was bound in the active site. Several crystals were analyzed and were found to contain phosphate to various extents, ranging from 50 to 100%. For this reason, these data were not further pursued.
Diffraction data for the ternary PLP-glycine complexes were collected at beam line ID14-1, ESRF, to resolutions of 1.8 and 1.9 Å. Processing, phasing, and model refinement were performed as described above. Electron density for PLP indicated that the cofactor bound exclusively at this site in the ternary complex. Toward the end of refinement, glycine was built into positive difference density close to PLP. The data collection and refinement statistics are presented in Table 1.
The structures were evaluated using PROCHECK (49) and MOLPROBITY (50). Analysis of dihedral angles indicated that Ser165 is in a disallowed region of the Ramachandran diagram. However, inspection of electron density showed this residue in well defined density in both subunits. Amino acid sequence alignments were made with ClustalW (51). Secondary structure assignments were made with DSSP (52), and structure-based sequence alignments were carried out using the least-square alignment function in the program O (45). Model figures were prepared with PyMOL (53).
Electrostatic Surface Analysis
Electrostatic potentials were calculated in PyMOL (53) using the vacuum electrostatics method. The analysis was performed on coordinates of one monomer each of (i) the apoenzyme with a closed C terminus and an extended mobile loop and (ii) the PLP complex with the C terminus in an open conformation and a closed mobile loop. Solvent molecules were removed from the coordinates before the analysis. The model of the apoprotein was extended with residues 964–965 in molecule A, which were built into trace electron density to provide a more realistic surface for the analysis. These residues are not present in the final model submitted to the PDB.
RESULTS
Recombinant and enzymatically active homodimeric P-protein from Synechocystis (38, 40) crystallized in an orthorhombic space group (P212121) (40) under mildly reducing conditions (10 mm 2-mercaptoethanol). The structure solved from these (form 1) crystals shows that the C terminus (residues 963–978) is folded over the active site opening and secured by a disulfide bond between Cys-972 in the C terminus and Cys-353 located in the active site cavity (Fig. 2). This effectively isolates the active site from solvent and excludes the binding of ligands (cofactor and substrates) in the active site. Form 1 crystals thus contain the apoenzyme in an inactive, locked enzyme form. By using a more powerful reducing agent, TCEP, a trigonal crystal form (space group P321) was obtained that diffracts X-rays to 1.6 Å resolution. In these (form 2) crystals, the disulfide is reduced, the C terminus is free and folded away from the active site entrance, and the enzyme is able to bind ligands, such as the cofactor PLP and the substrate glycine. This is the active, unlocked enzyme form.
FIGURE 2.
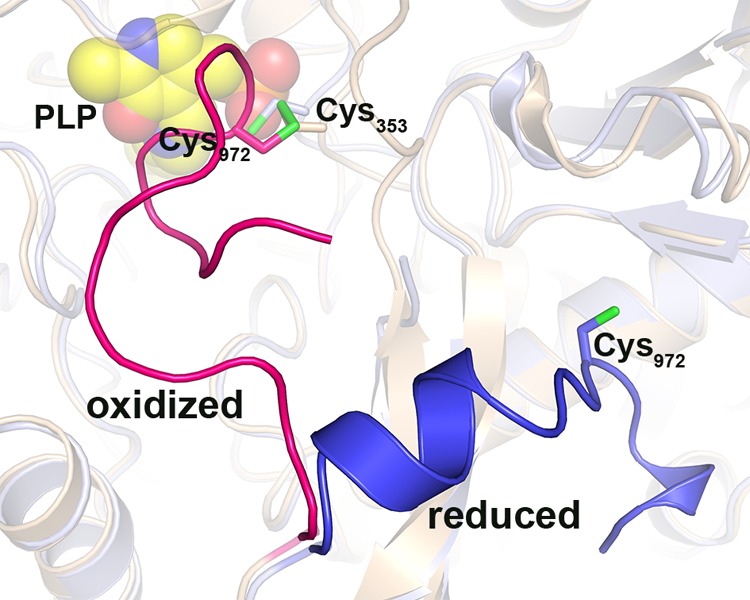
Conformational states of the C terminus in oxidized and reduced P-protein. Superposition of ribbon models of the oxidized, ligand-free P-protein (beige) and the reduced, PLP-bound P-protein (light blue) is shown. The C terminus (residues 962–978) of the oxidized and reduced P-protein is highlighted in red and blue, respectively. Sulfur atoms of cysteines 353 and 972 are colored green. PLP (bound in the reduced enzyme) is shown as spheres with carbon atoms colored yellow.
Aside from the C terminus, the loop comprising residues 337–349 (named the mobile loop according to Nakai et al. (32)) also adopts a number of different conformations: disordered, extended, and closed (see below). Apart from these two striking differences, continuous density was observed for most of the protein in both crystal forms. There was weak or no density for the first 19 N-terminal residues, for a surface loop (residues 756–766), and for the six C-terminal residues. Contents of the refined models are summarized in Table 1.
Quaternary Structure
The Synechocystis P-protein has a homodimeric quaternary structure (α2NC; Fig. 3), and each monomer harbors one active site. The N-terminal and C-terminal halves of the αNC monomer correspond to the αN and βC subunits, respectively, of the heterodimer of the tetrameric enzyme (32) (Figs. 4 and 5). The bulk of the αN and αC parts of the homodimeric enzyme fold into separate units that pack tightly. The N-terminal arms of each unit wrap around the opposite unit to make a compact monomer. Two monomers pack tightly to form the active dimer. The formation of the dimer is driven by the hydrophobic interaction between mainly helical elements across the local two-fold axis (Fig. 3). The dimer interface, calculated following Lee and Richards (54) with a probe radius of 1.4 Å, is 3894 Å2 and comprises 6.7% of the dimer total accessible surface area.
FIGURE 3.
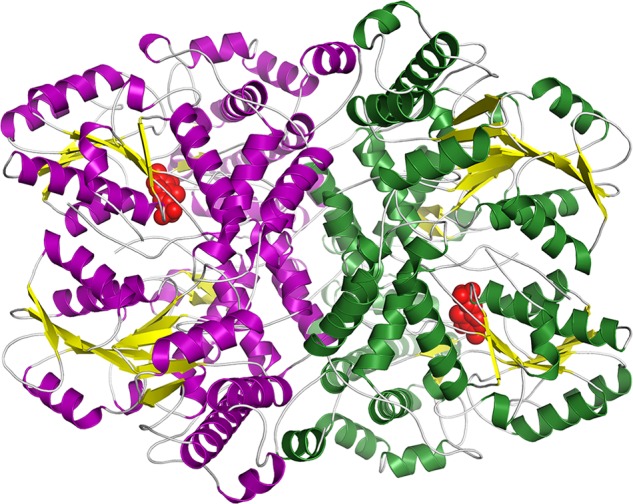
Overall structure of the Synechocystis P-protein homodimer in the reduced state with the cofactor PLP bound. Shown is a ribbon diagram with β-sheets in yellow and helices in purple in one subunit and green in the second subunit. PLP is shown as red spheres.
FIGURE 4.
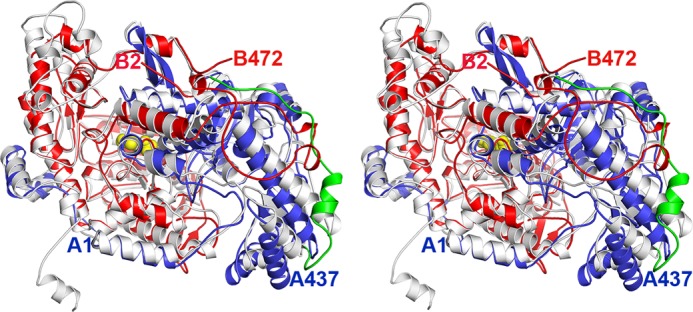
Stereo image showing a comparison of the homodimeric and heterotetrameric P-protein structures. The T. thermophilus heterodimer (32) in blue and red is superimposed on a monomer of the Synechocystis enzyme in white. The loop, residues 461–494, connecting the N-terminal and C-terminal parts of the Synechocystis monomer is colored green, and PLP is shown as yellow spheres. N-terminal and C-terminal residues of the α- and β-chains of the T. thermophilus enzyme are labeled in blue and red, respectively.
FIGURE 5.


Amino acid sequence alignment of P-proteins based on a structural alignment of the enzymes from Synechocystis sp. PCC 6803 and T. thermophilus (PDB code 1WYV). The sequences were obtained from the NCBI database. The sequence of Synechocystis P-protein was compared with homologous proteins from A. thaliana, Homo sapiens, and T. thermophilus. Residue numbering is according to the Synechocystis sequence. Because the N-terminal target peptides were omitted, the residue numbering of A. thaliana P-protein starts from position 40, and the H. sapiens numbering starts from position 57. To facilitate alignment, peptides αN and βC of T. thermophilus were fused to one polypeptide. Conserved residues are boxed, strictly conserved residues have a red background, residues well conserved within a group are indicated by red letters, and the remainder are in black letters. Symbols above blocks of sequences correspond to the secondary structure of the Synechocystis enzyme: α, α-helix; β, β-strand; η, 310-helix. Gray asterisks above the sequence indicate the presence of alternate conformations. The secondary structure assignment for the Synechocystis enzyme was made using DSSP (52). The figure was prepared using ESPript (70).
The Monomer
The homodimeric P-protein from Synechocystis shows about 35% sequence identity with the heterotetrameric enzyme from T. thermophilus (BLAST 2). Comparison of the homodimeric α2NC and the heterotetrameric (αNβC)2 structures reveals that the fold of the proteins is highly similar (Figs. 4 and 5), but there are some striking differences. Superposition of 373 equivalent Cα atoms of the homodimeric structure with the αN monomer from T. thermophilus gives a root mean square (r.m.s.) deviation of 1.55 Å, whereas superposition of 416 Cα atoms of the homodimeric structure with the βC monomer gives an r.m.s. deviation of 1.23 Å. The N-terminal αNC-N domain is conserved between the heterotetrameric and homodimeric enzymes and consists of a central β-sheet of seven strands flanked by helices on both sides. The N terminus is extended by 34 residues, half of which are visible in the final electron density maps. Residues 17–26 form an α-helix (α1) that packs against the first helix (η10, residues 471–476) in the C-terminal part (αNC-C) of the adjacent subunit of the homodimer. Residues 33–37 form an extra helix not present in the T. thermophilus enzyme, but apart from that, the N-terminal residues 38–100 adopt a conformation similar to that of the residues of the N-terminal arm of the αN subunit of the heterotetramer. The domain ends with a twisted β-hairpin (strands β9 and β10). Residues 337–349 of the loop between strand β10 and helix α13 are able to adopt a number of different conformations and appear to contribute to the closing of the active site during catalysis (see below). The remainder of the N-terminal part of the subunit, consisting of three helices (14–16) and an antiparallel β-sheet, is structurally homologous to the αN of the T. thermophilus enzyme. Helix 16, the last helix of αNC-N, connects to the C-terminal part of the αNC subunit and, as a consequence, is significantly shifted compared with the corresponding helix in the T. thermophilus enzyme. Residues 461–478 form a linker connecting the N- and C-terminal parts, and this linker loop is not structurally homologous to any part of the T. thermophilus structure.
The C-terminal part, from residue 479 onward, superimposes well with the T. thermophilus model. The central domain corresponds to the central domain of the N-terminal part of the βC subunit in the heterotetrameric P-protein. Exiting this domain, residues 756–767, which form a β-hairpin loop at the surface, are disordered in the Synechocystis enzyme structure. The structures deviate considerably at the C terminus. In the Synechocystis enzyme, helix α30 is extended by 7 residues at its C terminus, and the loop preceding helix α31 is extended by 5 residues. Residues 925–956 of the C terminus of the Synechocystis enzyme contain three short helices not present in the T. thermophilus enzyme. The following 22 residues, 957–978, constitute the mobile C terminus, which in the ligand-bound open structures is very similar to the C terminus of the T. thermophilus enzyme. The last 6 residues of the C terminus are not visible in the electron density maps. In the apoenzyme, helix α34 unfolds partly, covers the active site, and is secured by a disulfide bond between the C-terminal Cys972 and Cys353. Cys353 is located in the active site within van der Waals distance to the glycine substrate in the ternary complex (see below).
Transition from the Apo to the Holo Structure
The initial crystallization solution (40) contained 10 mm 2-mercaptoethanol, and the cofactor PLP was added to the enzyme solution prior to crystallization. However, there was no trace of bound PLP in the active site in the final electron density maps after refinement. Instead, a network of solvent molecules was observed lining the active site. A hydrogen bond was observed between Lys726, to which PLP normally binds and where transaldimination occurs (see below), and the hydroxyl group of Tyr131. Opposite to Lys726 in the active site, there was strong electron density close to the Sγ atom of Cys353, indicating that this cysteine was engaged in a disulfide bond with another cysteine residue (Fig. 2). This cysteine, identified as Cys972, is part of a short C-terminal peptide 966GDRHLVCSCEGME978 in one subunit of the dimer (subunit A) and 968RHLVCSCE975 in the second subunit (subunit B), for which there was well defined electron density. The C-terminal peptide covers the active site, effectively isolates it from solvent, and presumably hindered the access of the cofactor to the active site during the initial crystallization experiment.
In the apoenzyme, electron density for two other cysteines, Cys642 and Cys974, indicated that these residues were modified. The additional density was interpreted as mixed disulfides with 2-mercaptoethanol. Although this suggests that these cysteines are redox-active, we failed to find a suitable disulfide partner in these cases. The Sγ atoms of Cys642 and Cys974 are located more than 15 Å apart, making it unlikely that they would be part of the same disulfide. Cys642 is located close to the surface of the molecule, and there are no other cysteines within reasonable distance. Cys974 has its Sγ atom positioned only 6 Å from the Sγ atom of the active site residue Cys530, but there was no sign of oxidation or modification of Cys530 in any of the electron density maps. We note also that the position of the Sγ atom of Cys974 in the apoenzyme is located less than 2 Å from the position of the carboxyl group of the glycine substrate in the holoenzyme structure.
To reduce the 353–972 disulfide and break the bond, we switched to the powerful reducing agent TCEP. PLP was included in the purification buffers to ensure that the cofactor was bound. The resulting protein crystallized in a different space group with significantly better diffraction. There was well defined density for PLP in the active site (Fig. 6), indicating that it was covalently linked to Lys726 via an internal aldimine bond (Schiff base linkage). Inspection of the C-terminal arm showed well defined density for residues 963–978, which were folded away and stabilized at the surface of the protein, thereby creating open access to the active site (Fig. 2). The Cys353–Cys972 disulfide was reduced, and there was well defined density for both free Sγ atoms. Breaking of the 353–972 disulfide bond allows the side chain of Cys353 to flip about 120º and the loop region carrying Cys353 to move ∼2–3 Å toward the active site. Apart from the 353–972 disulfide, the C terminus is stabilized in its closed conformation by a hydrogen bond between Asn318 and Arg975. Upon reduction of the 353–972 disulfide, the contact between residues 318 and 975 is also broken, allowing the C-terminal residues 963 and onward to fold away from the active site. In the open conformation, residues 963–969 adopt a helical conformation (Fig. 2), and the C terminus is anchored to the surface by several hydrogen bonds. As shown earlier (40) and below, the Synechocystis P-protein requires the presence of a reducing agent (usually DTT) in assays for full activity, and the structure presented here offers a structural explanation. There is no equivalent to Cys972 in the heterotetrameric enzyme, and the C terminus adopts an open conformation in the crystal structures of the apo and holo forms of the T. thermophilus enzyme.
FIGURE 6.
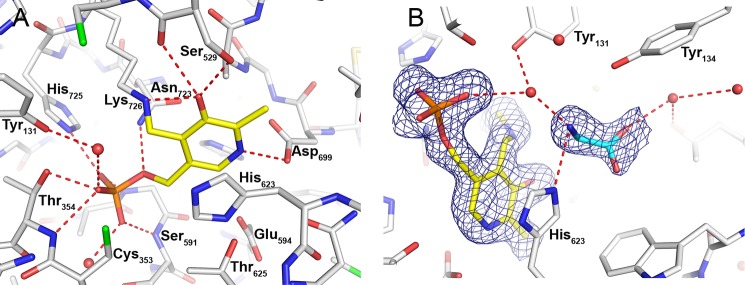
Binding of PLP and glycine in the active site of P-protein. A, residues in contact with PLP are shown as white sticks with hydrogen bonds indicated by red dashed lines. PLP (yellow) is covalently bound to Lys726. B, σA weighted difference density map (mFo − DFc) drawn at the level of 3 times the r.m.s. value of the map around PLP (yellow) and glycine (cyan) with hydrogen bonds indicated by red dashed lines. Water molecules are shown as red spheres.
A long loop, residues 337–349, is observed in two different conformations and also appears to participate in the transition from oxidized apoenzyme to reduced holoenzyme. In the apo structure, the loop adopts an extended conformation that stabilizes the C terminus in its closed conformation. This is observed in one of the two subunits (subunit A), whereas in the second subunit, the loop is disordered and not visible in the electron density maps. Upon reduction of the 353–972 disulfide and the folding of the C terminus away from the active site, the extended loop refolds and closes over the opening of the active site, shielding it partly. This conformation is observed in one of the subunits (subunit A) in the crystal structure of the ternary complex with PLP and glycine (Fig. 7). In the second subunit, residues 334LALQTR337 at one end and Thr349 at the other end of the loop are visible and assume a similar conformation. An identical conformation of the mobile loop is observed in the ligand-bound forms of the T. thermophilus enzyme (PDB codes 1WYV and 1WYU) and is thus likely to be a general structural feature of the holo forms of both heterotetrameric and homodimeric P-proteins. In its closed conformation in the Synechocystis enzyme, this loop interferes with the closed C terminus of the apo structure, and it is not likely that they could co-exist.
FIGURE 7.
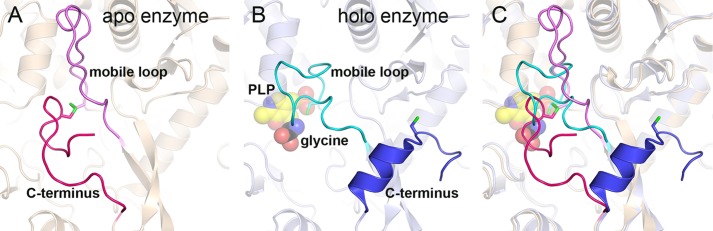
Conformation of the mobile loop and the C terminus in the homodimeric P-protein. A, ribbon model of the apo-P-protein (beige) with the C terminus (residues 962–978) and the mobile loop (residues 335–353) highlighted in red and pink, respectively. The disulfide bond between cysteines 353 and 972 is colored green. B, holo-P-protein (light blue ribbon) with PLP and glycine bound. The C terminus and the mobile loop are highlighted in blue and cyan, respectively, and PLP and glycine are shown as spheres. C, superposition of the models in A and B showing the overlap of the positions of the closed C terminus and the closed mobile loop.
The Active Site and PLP Binding
PLP makes an aldimine bond with Lys726 (Fig. 1B), whose Nζ atom is stabilized by a hydrogen bond to the backbone carbonyl oxygen atom of residue Ser529. PLP fits tightly in the active site and is stabilized by several hydrogen bonds (Fig. 6A). The oxygen atoms of the phosphate group are engaged in hydrogen bonding to the backbone nitrogen atoms of Thr354, Gly590, and Ser591; the side chains of Thr354, Asn723, and His725; and several solvent molecules. The pyridine ring of PLP is sandwiched on one side by His623 and on the other side by Ala701 and Asn723 and makes hydrogen bond interactions with Ser529 and Asp699. All of these residues are strictly conserved in the heterotetrameric enzyme despite their localization on separate polypeptides (αN and βC) and provide a good binding site for anion ligands, such as PLP.
The binding of PLP causes smaller rearrangements of the protein structure in the PLP binding site. The imidazole ring of His623 moves about 2 Å toward the pyridine ring for a better stacking interaction, although the angle of the imidazole ring is unaltered with respect to the PLP pyridine ring. Trp870, located on a loop region, moves ∼0.8 Å toward the bound PLP. Ala701 (positioned in another loop region) moves around 1.7 Å closer to the center of the pyridine ring. The loop region harboring Thr674 and His675 also adjusts, moving ∼1.2 Å toward PLP.
Binding of Glycine
Soaking of the substrate glycine into crystals of the PLP complex produced a ternary complex with glycine and PLP bound in the active site. Overall, the structure of the ternary complex is very similar to the structure of the co-crystallized PLP complex. Superposition of the two models gives an r.m.s. deviation of 0.139 for 1900 Cα atoms. Glycine binds in a pocket close to PLP; both ligands are bound with high occupancy in well defined electron density (Fig. 6B). The glycine-binding site is located on the side of the pyridine ring that faces the outside and is otherwise occupied by solvent (water or ethylene glycol). The amino group of glycine makes a hydrogen bond to the imidazole ring of His623. It also makes solvent-mediated interactions with the phosphate group and the aldimine C4′ carbon of PLP and with the hydroxyl group of Tyr131. There is no indication of the formation of an external aldimine with PLP; the amino nitrogen of glycine is located 3.7 Å away from the C4′ carbon of the pyridine ring, and there is well defined density indicating that the internal aldimine bond to Lys726 is maintained. The carboxyl group of glycine is stabilized by contact with the hydroxyl group of Tyr134 and by a solvent-mediated contact with Thr867. Apart from the lack of a covalent bond to PLP, the orientation of the substrate is appropriate for decarboxylation with the scissile bond of glycine oriented approximately perpendicular to the π-orbital system of the pyridine ring of PLP (corresponding to conformation III in Ref. 55).
Directed Mutation of Cysteine Residues Involved in Disulfide Formation
To test for the potential roles of the cysteines forming the disulfide bond, we made the C353S, C972S, C353S/C972S, C353S/C972S/C974S, and C972S/C974S variants and assayed them in the presence and absence of DTT using the wild-type enzyme for comparison (Fig. 8). In the presence of DTT, wild-type P-protein catalyzed the glycine-bicarbonate exchange reaction with a specific activity of 1.04 ± 0.12 μmol s−1 g−1. The C353S variant showed an ∼20-fold decrease in activity, whereas the C972S variant displayed a distinctly smaller, about 2-fold, decrease in activity. The activity of the double and triple mutants C353S/C972S and C353S/C972S/C974S was 20-fold reduced and did not significantly differ from the C353S P-protein variant. Similarly, the additional C974S exchange in the double-mutated P-protein C972S/C974S did not further decrease the specific activity relative to C972S. In the absence of DTT, the activity of the wild-type Synechocystis P-protein was more than 30-fold diminished, but the introduced single and combined mutations resulted in only 2–3-fold further decreases in specific activity. Under these conditions, the specific activity of the C972S/C974S variant was only slightly lower relative to the wild-type control, which suggests that the additional C974S exchange counteracts the effect of the C972S exchange. A similar although less strong effect was observed with C353S/C972S/C974S relative to C353S/C972S under non-reducing conditions. Taken together, the combined site-directed mutagenesis and structural information strongly support our hypothesis that formation of the Cys353–Cys972 bond could be linked to the redox status of the cell and hence redox-dependent activity regulation of the P-protein.
FIGURE 8.

Specific activities of P-protein variants at different redox conditions relative to the wild-type protein. The columns show mean activities of recombinant P-protein ± S.D. (error bars) (n = 4) preincubated with 1 mm (black) or zero (gray) dithiothreitol in the glycine-bicarbonate exchange reaction. The numbers indicate percentage activities relative to the corresponding wild-type control. The number in parentheses shows the percentage relative to fully activated wild-type P-protein.
Electrostatic Surface Analysis
The conformational transition from the apo- to the holoenzyme leads to a dramatic change in the charge of the molecular surface in the vicinity of the active site. The surface charge of the active site region of the apo-P-protein is mainly negative (Fig. 9A). Upon breaking of the Cys353–Cys972 disulfide, the C terminus moves from its closed state to an extended open state, which permits the mobile loop to move to its closed conformation. This change in conformation also alters the surface charge in this area, which becomes remarkably more positive (Fig. 9B). The closed loop conformation permits the basic residues of the mobile loop, Arg339, Arg344, Arg345, and Lys347, to be fully exposed to the surrounding environment. Together with Arg313 and Lys314, located on an adjacent short helix, they form a positively charged surface close to the active site entrance. In contrast, in its extended form (Fig. 9A), when the mobile loop is folded away from the active site opening, these residues form hydrogen bonds with residues at the surface far from the active site. Arg344 hydrogen-bonds to the carbonyl oxygens of residues Ser186 and Tyr181 (and to a water molecule), Arg345 interacts with the carbonyl oxygen of Glu340 and the carboxylate of Glu341, and Arg313 forms a salt link to Glu340. Lys347 hydrogen-bonds to the side chain of Asn189. When folded away from the active site, as in the PLP- and glycine-bound structures, the C terminus forms a convex protrusion, which effectively elongates the surface that a potential binding partner, such as the H-protein or lipoic acid, could use for interaction. Arg968 and His969, located at the C terminus and fully exposed in the PLP structure, add to the already existing positively charged surface provided by residues in the mobile loop. On the other hand, when the C terminus closes over the active site, Arg968 and His969 are directed inward, toward the active site. In its closed state, the C terminus bears a clear negative charge, formed by Glu975 and Asp967 and exposed backbone carbonyl oxygen atoms, which are likely to enhance the negatively charged surface in the vicinity.
FIGURE 9.
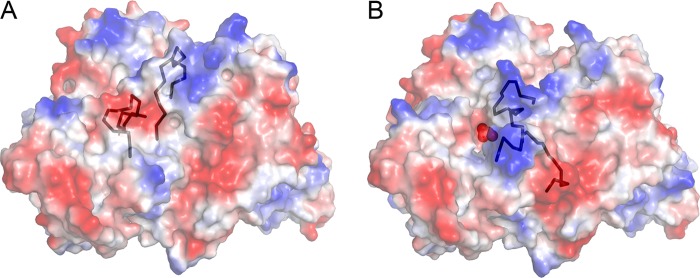
Electrostatic surface analysis of apoenzyme (A) and holoenzyme (B). Electrostatic surfaces are colored blue in the positive region and red in the negative region. The C terminus and the mobile loop are shown in a black ribbon representation. PLP is indicated as red spheres in B.
Several residues in the mobile loop are conserved in the heterotetrameric enzyme (32), but the difference in electrostatic potential between the apo and holo forms is rather small in this case (data not shown). There is no equivalent of the acidic surface in the T. thermophilus enzyme nor of the acidic residues at the C terminus (Glu975 and Asp967). This difference is in line with the observation that this particular conformation of the C terminus is not conserved between the homodimeric and heterotetrameric enzymes.
DISCUSSION
We present here the structure of the glycine decarboxylase or P-protein from the cyanobacterium Synechocystis, which can be obtained as enzymatically active recombinant protein (38). This is the first structural information provided for a homodimeric glycine decarboxylase. The protein displays over 70% similarity and about 60% identity with plant and mammalian P-proteins and therefore can serve as an excellent model for these eukaryotic enzymes. Our models show strict identity of key residues with the counterparts in the eukaryotic enzymes, including all known active site residues and most cysteine residues (Fig. 5). When purified in the presence of 2-mercaptoethanol (40), crystallization of the Synechocystis P-protein in the presence of 0.2 mm PLP resulted in orthorhombic crystals (space group P212121). The resulting structure was of the unliganded form, devoid of the cofactor. Binding of the cofactor may have been prevented by the C-terminal lid closing over the active site as a part of the regulation of catalytic activity. As a less likely explanation, PLP may also have been hindered from binding by crystal packing restraints at the conditions used during the crystallization experiment. The disulfide between cysteine residues 353 and 972 firmly anchors the C terminus over the active site and was also observed in crystals obtained in the complete absence of reducing agents. However, the quality of the structure was inferior under these oxidizing conditions, and the structure was not analyzed further.
In the presence of the strong reducing agent TCEP, the C terminus is folded away from the active site, allowing the cofactor PLP and the substrate glycine to bind. This switch from a closed to an open conformation of the C terminus corresponds to the change from low to high enzymatic P-protein activity at oxidized versus reduced test conditions (see Fig. 8).
Glycine binds close to PLP in a pocket facing the opening toward solvent (Fig. 6B). To react, the amino group of glycine must form a Schiff base with PLP, upon which the carboxyl group of glycine is lost as CO2, and the remaining methylamine moiety is transferred to the lipoyl group of the H-protein (Fig. 1). There was no trace of a covalent bond between glycine and PLP in the crystal structures presented here, but several bicarbonate molecules were observed to bind in the crystals of the ternary complex with glycine. This suggests there may be a slow turnover of the substrate, resulting in bicarbonate formation at the pH of the crystallization. This is in line with the earlier demonstration of CO2 evolution in the absence of H-protein or lipoic acid by recombinant Synechocystis P-protein (38). The external aldimine may be too transient to be observed. It is also possible that productive binding may be hindered by crystal lattice forces preventing the mobile loop from closing effectively and that bicarbonate binding is an artifact of crystallization. However, no bicarbonate was found in crystals of the apoenzyme, suggesting that PLP and glycine are required for bicarbonate binding in the crystal.
Earlier reports (56) and our studies (40) (this work) have pointed to a strong dependence of reducing agents, mainly DTT, for full activity of the P-protein during in vitro assays. Our observations of the closing of the active site by the movement of the C terminus and the stabilization of the closed conformation in a disulfide bond suggest that the Synechocystis glycine decarboxylase is a target for regulatory redox-active proteins. The structures of inactive and active forms of the P-protein presented here show how this regulation could happen on the molecular level. It should be noted that all structural requirements for the redox-dependent conformational change are conserved in homodimeric P-proteins from eukaryotes, suggesting that the Synechocystis P-protein represents a model also for the regulation of eukaryotic glycine decarboxylases. Dithiol/disulfide exchange catalyzed by thioredoxins has been extensively studied in the light-dependent regulation of several processes in plants and is important also for other organisms. In plants, the processes of photosynthesis and respiration are separated in chloroplasts and mitochondria, respectively. Thioredoxin-mediated light-dependent regulation of each of the control points of the Calvin-Benson cycle enables the functional separation of diurnal and nocturnal processes in the chloroplast and was among the first discovered examples of this type of regulation (57). Although these enzymes are regulated by a common ferredoxin-thioredoxin system, structural studies have shown that the molecular mechanisms for activation of the target enzymes are different in each case, adapted to the reaction catalyzed by the enzyme (58), and there is no consensus regulatory sequence. In most cases, however, structural information of the different regulatory states of the target enzymes is lacking. More recently, numerous thioredoxin-linked proteins have been identified in plant mitochondria, among these all four GCS proteins, including the P-protein (59–62), but the molecular details of activation are yet unknown. There are also indications that communication between chloroplasts and mitochondria may be mediated by thioredoxin-linked processes (59, 63). Unlike plants, cyanobacteria cannot physically separate oxygenic photosynthesis and glycine decarboxylation in different organelles. Comparable with land plants, however, the simultaneous activation by light of photosynthetic electron transport, oxygen evolution, and CO2 fixation in cyanobacteria also employs thioredoxins. Proteomic investigations have been published describing potential thioredoxin targets in cyanobacteria, including Synechocystis (64, 65). These studies identified numerous thioredoxin-regulated photosynthetic proteins (66) and listed the GCS L-protein as a glutaredoxin target (65).
Cysteines 353 and 972 are well conserved in the P-protein from plants, animals, and many aerobic bacteria but absent in the enzymes from archaea and certain anaerobic bacteria (such as E. acidaminophilum (36)). As a consequence, a covalent bond between the corresponding residues 353 and 972 cannot form in the P-protein from these microorganisms. This is consistent with information from the crystal structures of T. thermophilus P-protein that shows the C terminus is in the open (unlocked) conformation in all complexes, including the ligand-free apoenzyme (32). It is striking that of 15 cysteines in Synechocystis P-protein, only one, Cys530, is conserved in the T. thermophilus enzyme (Fig. 5). The apparent absence of a redox switch in this particular case could be related to the extreme environment in which this thermophile lives (67), which is unfavorable to the transient formation of free cysteine residues (68). In the ternary complex of the Synechocystis P-protein, Cys353 is within van der Waals distance to (the carboxylate of) the glycine substrate. This may explain the low enzymatic activity of the C353S variant and the other examined P-protein variants containing this particular mutation. The influence of the C972S substitution on activity is more difficult to rationalize, but it is likely that the mutation influences the mobility of the C terminus and that this may in turn influence substrate binding and catalysis. Although this needs to be studied in more detail, the structural data and experiments with artificial enzyme variants strongly support the conclusion that the P-protein is redox-regulated.
The overlap of the C-terminal arm and the mobile loop in their closed conformations indicates that they cannot be closed at the same time and that, consequently, closure of the mobile loop can only happen when the C terminus is in its extended conformation (Fig. 7). Loop closure, too, is thus indirectly dependent on the redox conditions in the cell. There may be additional signal(s) that trigger loop closure, but these are currently unknown.
The dislocation of the C terminus from the active site and the closing of the mobile loop over the active site entrance have a dramatic effect on the surface charge in the area around the active site. The apoprotein features a mainly negatively charged surface, which becomes positive upon formation of the holoenzyme (Fig. 9). Compared with the C terminus, which closes completely over the active site, the mobile loop does not occupy as large a volume in its closed state and would allow the H-protein to simultaneously deliver its lipoyl arm to the active site (see Fig. 7 in Ref. 32). Surface potential analysis of the lipoylated H-proteins from Pisum sativum and T. thermophilus (31) shows two main surface areas on each side of the lipoylated residue, Lys63, with clear negative surface charge (see Fig. 7 in Ref. 31). The structure of the Synechocystis H-protein is not known, but the high homology between H-proteins of diverse origins (sequence identity 38–55% (31)) indicates that a similar negatively charged area may be present on the Synechocystis H-protein. In addition to the effect from several conserved residues, a large contribution to the charge seems to originate from exposed backbone carbonyl oxygens, in particular in the area closest to the lipoyl arm. The P-protein from Synechocystis has a lower affinity for the H-protein at pH 8 than at pH 6 (69); this may be caused by pH effects on charged residues on the protein surface. Presumably, the switch from the enzymatically inactive form to the enzymatically active form could also improve the H-protein docking via the observed change of surface charge.
Based on this, we envision the following scenario for regulation and catalysis in the homodimeric enzyme; formation of the 353–972 disulfide is subject to regulation by the redox status of the cell (enabled by a thioredoxin-linked system) and restricts access of the substrate to the active site (and possibly also of PLP). During conditions favoring catalysis, the disulfide bond is broken, and the C terminus is released and moves to its open unlocked position. The combined effect of the changes in conformation and surface charge enables passage of the two substrates, glycine and H-protein, to the active site. The concerted action of the entry of the H-protein's lipoyl arm and the conformational change of the mobile loop provides an efficient closure of the active site necessary for catalysis.
Acknowledgments
We thank the ESRF and the EMBL Outstation in Grenoble for providing beam time and data collection facilities and the beam line staff of ESRF for outstanding support.
This work was supported by the Deutscher Akademischer Austauschdienst and Landesgraduiertenstipendium Mecklenburg-Vorpommern and grants from the Swedish Research Council FORMAS.
The atomic coordinates and structure factors (codes 4LGL, 4LHC, and 4LHD) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- GCS
- glycine cleavage system
- PLP
- pyridoxal 5′-phosphate
- ESRF
- European Synchrotron Radiation Facility
- PDB
- Protein Data Bank
- TCEP
- tris(2-carboxyethyl)phosphine
- r.m.s.
- root mean square
- Bicine
- N,N-bis(2-hydroxyethyl)glycine.
REFERENCES
- 1. Falb M., Müller K., Koenigsmaier L., Oberwinkler T., Horn P., von Gronau S., Gonzalez O., Pfeiffer F., Bornberg-Bauer E., Oesterhelt D. (2008) Metabolism of halophilic archaea. Extremophiles 12, 177–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Klein S. M., Sagers R. D. (1967) Glycine metabolism. III. A flavin-linked dehydrogenase associated with the glycine cleavage system in Peptococcus glycinophilus. J. Biol. Chem. 242, 297–300 [PubMed] [Google Scholar]
- 3. Lamers Y., Williamson J., Theriaque D. W., Shuster J. J., Gilbert L. R., Keeling C., Stacpoole P. W., Gregory J. F. (2009) Production of 1-carbon units from glycine is extensive in healthy men and women. J. Nutr. 139, 666–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McNeil J. B., Zhang F., Taylor B. V., Sinclair D. A., Pearlman R. E., Bognar A. L. (1997) Cloning, and molecular characterization of the GCV1 gene encoding the glycine cleavage T-protein from Saccharomyces cerevisiae. Gene 186, 13–20 [DOI] [PubMed] [Google Scholar]
- 5. Motokawa Y., Kikuchi G. (1971) Glycine metabolism in rat liver mitochondria. V. Intramitochondrial localization of the reversible glycine cleavage system and serine hydroxymethyltransferase. Arch. Biochem. Biophys. 146, 461–464 [DOI] [PubMed] [Google Scholar]
- 6. Neuburger M., Bourguignon J., Douce R. (1986) Isolation of a large complex from the matrix of pea leaf mitochondria involved in the rapid transformation of glycine into serine. FEBS Lett. 207, 18–22 [Google Scholar]
- 7. Hiraga K., Kochi H., Motokawa Y., Kikuchi G. (1972) Enzyme complex nature of the reversible glycine cleavage system of cock liver mitochondria. J. Biochem. 72, 1285–1289 [DOI] [PubMed] [Google Scholar]
- 8. Kawasaki H., Sato T., Kikuchi G. (1966) A new reaction for glycine biosynthesis. Biochem. Biophys. Res. Commun. 23, 227–233 [DOI] [PubMed] [Google Scholar]
- 9. Maaheimo H., Fiaux J., Cakar Z. P., Bailey J. E., Sauer U., Szyperski T. (2001) Central carbon metabolism of Saccharomyces cerevisiae explored by biosynthetic fractional 13C labeling of common amino acids. Eur. J. Biochem. 268, 2464–2479 [DOI] [PubMed] [Google Scholar]
- 10. Pasternack L. B., Laude D. A., Jr., Appling D. R. (1992) 13C NMR detection of folate-mediated serine and glycine synthesis in vivo in Saccharomyces cerevisiae. Biochemistry 31, 8713–8719 [DOI] [PubMed] [Google Scholar]
- 11. Eliot A. C., Kirsch J. F. (2004) Pyridoxal phosphate enzymes. Mechanistic, structural, and evolutionary considerations. Annu. Rev. Biochem. 73, 383–415 [DOI] [PubMed] [Google Scholar]
- 12. Douce R., Bourguignon J., Neuburger M., Rébeillé F. (2001) The glycine decarboxylase system. A fascinating complex. Trends Plant Sci. 6, 167–176 [DOI] [PubMed] [Google Scholar]
- 13. Kikuchi G., Motokawa Y., Yoshida T., Hiraga K. (2008) Glycine cleavage system. Reaction mechanism, physiological significance, and hyperglycinemia. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 84, 246–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hanson A. D., Roje S. (2001) One-carbon metabolism in higher plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 52, 119–137 [DOI] [PubMed] [Google Scholar]
- 15. Bykova N. V., Keerberg O., Pärnik T., Bauwe H., Gardeström P. (2005) Interaction between photorespiration and respiration in transgenic potato plants with antisense reduction in glycine decarboxylase. Planta 222, 130–140 [DOI] [PubMed] [Google Scholar]
- 16. Kanno J., Hutchin T., Kamada F., Narisawa A., Aoki Y., Matsubara Y., Kure S. (2007) Genomic deletion within GLDC is a major cause of non-ketotic hyperglycinaemia. J. Med. Genet. 44, e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kure S., Mandel H., Rolland M. O., Sakata Y., Shinka T., Drugan A., Boneh A., Tada K., Matsubara Y., Narisawa K. (1998) A missense mutation (His42Arg) in the T-protein gene from a large Israeli-Arab kindred with non-ketotic hyperglycinemia. Hum. Genet. 102, 430–434 [DOI] [PubMed] [Google Scholar]
- 18. Engel N., van den Daele K., Kolukisaoglu U., Morgenthal K., Weckwerth W., Pärnik T., Keerberg O., Bauwe H. (2007) Deletion of glycine decarboxylase in Arabidopsis is lethal under non-photorespiratory conditions. Plant Physiol. 144, 1328–1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bauwe H., Hagemann M., Fernie A. R. (2010) Photorespiration. Players, partners and origin. Trends Plant Sci. 15, 330–336 [DOI] [PubMed] [Google Scholar]
- 20. Oliver D. J., Neuburger M., Bourguignon J., Douce R. (1990) Interaction between the component enzymes of the glycine decarboxylase multienzyme complex. Plant Physiol. 94, 833–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Heineke D., Bykova N., Gardeström P., Bauwe H. (2001) Metabolic response of potato plants to an antisense reduction of the P-protein of glycine decarboxylase. Planta 212, 880–887 [DOI] [PubMed] [Google Scholar]
- 22. Timm S., Florian A., Arrivault S., Stitt M., Fernie A. R., Bauwe H. (2012) Glycine decarboxylase controls photosynthesis and plant growth. FEBS Lett. 586, 3692–3697 [DOI] [PubMed] [Google Scholar]
- 23. Hagemann M., Vinnemeier J., Oberpichler I., Boldt R., Bauwe H. (2005) The glycine decarboxylase complex is not essential for the cyanobacterium Synechocystis sp. strain PCC 6803. Plant Biol. 7, 15–22 [DOI] [PubMed] [Google Scholar]
- 24. Eisenhut M., Bauwe H., Hagemann M. (2007) Glycine accumulation is toxic for the cyanobacterium Synechocystis sp. strain PCC 6803, but can be compensated by supplementation with magnesium ions. FEMS Microbiol. Lett. 277, 232–237 [DOI] [PubMed] [Google Scholar]
- 25. Eisenhut M., Ruth W., Haimovich M., Bauwe H., Kaplan A., Hagemann M. (2008) The photorespiratory glycolate metabolism is essential for cyanobacteria and might have been conveyed endosymbiontically to plants. Proc. Natl. Acad. Sci. U.S.A. 105, 17199–17204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Oliver D. J. (1994) The glycine decarboxylase complex from plant mitochondria. Annu. Rev. Plant Physiol. Plant Mol. Biol. 45, 323–337 [Google Scholar]
- 27. Kikuchi G., Hiraga K. (1982) The mitochondrial glycine cleavage system. Unique features of the glycine decarboxylation. Mol. Cell. Biochem. 45, 137–149 [DOI] [PubMed] [Google Scholar]
- 28. Guilhaudis L., Simorre J. P., Blackledge M., Marion D., Gans P., Neuburger M., Douce R. (2000) Combined structural and biochemical analysis of the H-T complex in the glycine decarboxylase cycle. Evidence for a destabilization mechanism of the H-protein. Biochemistry 39, 4259–4266 [DOI] [PubMed] [Google Scholar]
- 29. Faure M., Bourguignon J., Neuburger M., MacHerel D., Sieker L., Ober R., Kahn R., Cohen-Addad C., Douce R. (2000) Interaction between the lipoamide-containing H-protein and the lipoamide dehydrogenase (L-protein) of the glycine decarboxylase multienzyme system. 2. Crystal structures of H- and L-proteins. Eur. J. Biochem. 267, 2890–2898 [DOI] [PubMed] [Google Scholar]
- 30. Lee H. H., Kim D. J., Ahn H. J., Ha J. Y., Suh S. W. (2004) Crystal structure of T-protein of the glycine cleavage system. Cofactor binding, insights into H-protein recognition, and molecular basis for understanding nonketotic hyperglycinemia. J. Biol. Chem. 279, 50514–50523 [DOI] [PubMed] [Google Scholar]
- 31. Nakai T., Ishijima J., Masui R., Kuramitsu S., Kamiya N. (2003) Structure of Thermus thermophilus HB8 H-protein of the glycine-cleavage system, resolved by a six-dimensional molecular-replacement method. Acta Crystallogr. D Biol. Crystallogr. 59, 1610–1618 [DOI] [PubMed] [Google Scholar]
- 32. Nakai T., Nakagawa N., Maoka N., Masui R., Kuramitsu S., Kamiya N. (2005) Structure of P-protein of the glycine cleavage system. Implications for nonketotic hyperglycinemia. EMBO J. 24, 1523–1536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bauwe H., Chu C. C., Kopriva S., Nan Q. (1995) Structure and expression analysis of the gdcsPA and gdcsPB genes encoding two P-isoproteins of the glycine cleavage system from Flaveria pringlei. Eur. J. Biochem. 234, 116–124 [DOI] [PubMed] [Google Scholar]
- 34. Kume A., Koyata H., Sakakibara T., Ishiguro Y., Kure S., Hiraga K. (1991) The glycine cleavage system. Molecular cloning of the chicken and human glycine decarboxylase cDNAs and some characteristics involved in the deduced protein structures. J. Biol. Chem. 266, 3323–3329 [PubMed] [Google Scholar]
- 35. Turner S. R., Ireland R., Rawsthorne S. (1992) Cloning and characterization of the P subunit of glycine decarboxylase from pea (Pisum sativum). J. Biol. Chem. 267, 5355–5360 [PubMed] [Google Scholar]
- 36. Freudenberg W., Andreesen J. R. (1989) Purification and partial characterization of the glycine decarboxylase multienzyme complex from Eubacterium acidaminophilum. J. Bacteriol. 171, 2209–2215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Okamura-Ikeda K., Ohmura Y., Fujiwara K., Motokawa Y. (1993) Cloning and nucleotide sequence of the gcv operon encoding the Escherichia coli glycine-cleavage system. Eur. J. Biochem. 216, 539–548 [DOI] [PubMed] [Google Scholar]
- 38. Hasse D., Mikkat S., Thrun H. A., Hagemann M., Bauwe H. (2007) Properties of recombinant glycine decarboxylase P- and H-protein subunits from the cyanobacterium Synechocystis sp. strain PCC 6803. FEBS Lett. 581, 1297–1301 [DOI] [PubMed] [Google Scholar]
- 39. Kern R., Bauwe H., Hagemann M. (2011) Evolution of enzymes involved in the photorespiratory 2-phosphoglycolate cycle from cyanobacteria via algae toward plants. Photosynth. Res. 109, 103–114 [DOI] [PubMed] [Google Scholar]
- 40. Hasse D., Hagemann M., Andersson I., Bauwe H. (2010) Crystallization and preliminary X-ray diffraction analyses of the homodimeric glycine decarboxylase (P-protein) from the cyanobacterium Synechocystis sp. PCC 6803. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 66, 187–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 42. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 43. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lamzin V. S., Perrakis A., Wilson K. S. (2001) The ARP/WARP suite for automated construction and refinement of protein models. in International Tables for Crystallography, Vol. F (Rossman M. G., Arnold E., eds) pp. 720–722, Kluwer Academic Publishers, Dordrecht, The Netherlands [Google Scholar]
- 45. Jones T. A., Zou J. Y., Cowan S. W., Kjeldgaard M. (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47, 110–119 [DOI] [PubMed] [Google Scholar]
- 46. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 47. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX. A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Perrakis A., Morris R., Lamzin V. S. (1999) Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol. 6, 458–463 [DOI] [PubMed] [Google Scholar]
- 49. Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. (1993) Procheck. A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 50. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity. All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 52. Kabsch W., Sander C. (1983) Dictionary of protein secondary structure. Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 [DOI] [PubMed] [Google Scholar]
- 53. DeLano W. L. (2010) The PyMOL Molecular Graphics System, version 1.3r1, Schrodinger, LLC, New York [Google Scholar]
- 54. Lee B., Richards F. M. (1971) The interpretation of protein structures. Estimation of static accessibility. J. Mol. Biol. 55, 379–400 [DOI] [PubMed] [Google Scholar]
- 55. Dunathan H. C. (1966) Conformation and reaction specificity in pyridoxal phosphate enzymes. Proc. Natl. Acad. Sci. U.S.A. 55, 712–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Walker J. L., Oliver D. J. (1986) Glycine decarboxylase multienzyme complex. Purification and partial characterization from pea leaf mitochondria. J. Biol. Chem. 261, 2214–2221 [PubMed] [Google Scholar]
- 57. Buchanan B. B. (1980) Role of light in the regulation of chloroplast enzymes. Annu. Rev. Plant Physiol. 31, 341–374 [Google Scholar]
- 58. Schürmann P., Buchanan B. (2008) The ferredoxin/thioredoxin system of oxygenic photosynthesis. Antioxid. Redox Signal. 10, 1235–1274 [DOI] [PubMed] [Google Scholar]
- 59. Balmer Y., Vensel W. H., Tanaka C. K., Hurkman W. J., Gelhaye E., Rouhier N., Jacquot J. P., Manieri W., Schürmann P., Droux M., Buchanan B. B. (2004) Thioredoxin links redox to the regulation of fundamental processes of plant mitochondria. Proc. Natl. Acad. Sci. U.S.A. 101, 2642–2647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Marchand C., Le Maréchal P., Meyer Y., Miginiac-Maslow M., Issakidis-Bourguet E., Decottignies P. (2004) New targets of Arabidopsis thioredoxins revealed by proteomic analysis. Proteomics 4, 2696–2706 [DOI] [PubMed] [Google Scholar]
- 61. Martí M. C., Olmos E., Calvete J. J., Díaz I., Barranco-Medina S., Whelan J., Lázaro J. J., Sevilla F., Jiménez A. (2009) Mitochondrial and nuclear localization of a novel pea thioredoxin. Identification of its mitochondrial target proteins. Plant Physiol. 150, 646–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yoshida K., Noguchi K., Motohashi K., Hisabori T. (2013) Systematic exploration of thioredoxin target proteins in plant mitochondria. Plant Cell Physiol. 54, 875–892 [DOI] [PubMed] [Google Scholar]
- 63. Michelet L., Zaffagnini M., Massot V., Keryer E., Vanacker H., Miginiac-Maslow M., Issakidis-Bourguet E., Lemaire S. D. (2006) Thioredoxins, glutaredoxins, and glutathionylation. New cross-talks to explore. Photosynth. Res. 89, 225–245 [DOI] [PubMed] [Google Scholar]
- 64. Lindahl M., Florencio F. J. (2003) Thioredoxin-linked processes in cyanobacteria are as numerous as in chloroplasts, but targets are different. Proc. Natl. Acad. Sci. U.S.A. 100, 16107–16112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Li M., Yang Q., Zhang L., Li H., Cui Y., Wu Q. (2007) Identification of novel targets of cyanobacterial glutaredoxin. Arch. Biochem. Biophys. 458, 220–228 [DOI] [PubMed] [Google Scholar]
- 66. Lindahl M., Kieselbach T. (2009) Disulphide proteomes and interactions with thioredoxin on the track towards understanding redox regulation in chloroplasts and cyanobacteria. J. Proteomics 72, 416–438 [DOI] [PubMed] [Google Scholar]
- 67. Beffa T., Blanc M., Lyon P. F., Vogt G., Marchiani M., Fischer J. L., Aragno M. (1996) Isolation of Thermus strains from hot composts (60 °C–80 °C). Appl. Environ. Microbiol. 62, 1723–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Jorda J., Yeates T. O. (2011) Widespread disulfide bonding in proteins from thermophilic archaea. Archaea 2011, 409156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hasse D., Mikkat S., Hagemann M., Bauwe H. (2009) Alternative splicing produces an H-protein with better substrate properties for the P-protein of glycine decarboxylase. FEBS J. 276, 6985–6991 [DOI] [PubMed] [Google Scholar]
- 70. Gouet P., Courcelle E., Stuart D. I., Métoz F. (1999) ESPript. Analysis of multiple sequence alignments in PostScript. Bioinformatics 15, 305–308 [DOI] [PubMed] [Google Scholar]