

Background: The degree of KCNQ1/KCNE1 (IKs components) colocalization in cardiomyocytes is unclear.
Results: In ventricular myocytes, KCNE1 is mainly on the cell surface, whereas KCNQ1 is mainly in the intracellular compartment. [Ca2+]i elevation or oxidative stress triggers KCNQ1 trafficking to the cell surface.
Conclusion: Stress-induced KCNQ1 trafficking influences IKs amplitude.
Significance: This is a novel mechanism for IKs function as a “ventricular repolarization reserve.”
Keywords: Confocal Microscopy, Endoplasmic Reticulum (ER), Potassium Channels, Stress Response, Trafficking, Confocal Microscopy, Stress Response, Trafficking
Abstract
Our goals are to simultaneously determine the three-dimensional distribution patterns of KCNQ1 and KCNE1 in cardiac myocytes and to study the mechanism and functional implications for variations in KCNQ1/KCNE1 colocalization in myocytes. We monitored the distribution patterns of KCNQ1, KCNE1, and markers for subcellular compartments/organelles using immunofluorescence/confocal microscopy and confirmed the findings in ventricular myocytes by directly observing fluorescently tagged KCNQ1-GFP and KCNE1-dsRed expressed in these cells. We also monitored the effects of stress on KCNQ1-GFP and endoplasmic reticulum (ER) remodeling during live cell imaging. The data showed that 1) KCNE1 maintained a stable cell surface localization, whereas KCNQ1 exhibited variations in the cytosolic compartment (striations versus vesicles) and the degree of presence on the cell surface; 2) the degree of cell surface KCNQ1/KCNE1 colocalization was positively correlated with slow delayed rectifier (IKs) current density; 3) KCNQ1 and calnexin (an ER marker) shared a cytosolic compartment; and 4) in response to stress ([Ca2+]i elevation, oxidative overload, or AT1R stimulation), KCNQ1 exited the cytosolic compartment and trafficked to the cell periphery in vesicles. This was accompanied by partial ER fragmentation. We conclude that the cellular milieu regulates KCNQ1 distribution in cardiac myocytes and that stressful conditions can increase IKs by inducing KCNQ1 movement to the cell surface. This represents a hitherto unrecognized mechanism by which IKs fulfills its function as a repolarization reserve in ventricular myocytes.
Introduction
KCNQ1 and KCNE1 associate to form slow delayed rectifier (IKs) channels (1). IKs functions as a “repolarization reserve” in human ventricles by contributing outward currents to prevent undue action potential prolongation when the β-adrenergic tone is high or when other repolarizing currents are diminished (2). Loss-of-function mutations in KCNQ1 or KCNE1 have been linked to long QT syndrome (LQT1 and LQT5) (3), whereas gain-of-function mutations have been linked to familial atrial fibrillation or short QT syndrome (SQT2) (4, 5). These observations indicate that proper control of IKs amplitude is required to maintain the electrical stability of both ventricles and atria. Situations of too little or too much IKs are equally dangerous.
We have shown that, in COS-7 cells, the partnership between KCNQ1 and KCNE1 is dynamic (6). KCNE1 can dissociate from KCNQ1, leaving room in the KCNQ1 channel to pick up new KCNE partners. Whether these observations can be extrapolated to native KCNQ1 and KCNE1 in cardiac myocytes is an open and important question. Other members of the KCNE family (KCNE2-KCNE4) have been detected in the human heart (7–9). In heterologous expression systems, these KCNE subunits can each associate with KCNQ1 to alter its current amplitude and gating kinetics (10). The degree and stability of KCNQ1/KCNE1 assembly in cardiac myocytes will influence how likely it is that the other KCNE subunits can associate with KCNQ1 and modulate its function.
It is not clear how well and where native KCNQ1 and KCNE1 are colocalized in cardiac myocytes. The major distribution pattern of KCNQ1 in atrial and ventricular myocytes of several animal species is transverse striations (11–15). On the other hand, the distribution pattern of native KCNE1 in cardiac myocytes has been described as “diffuse” (16), clustered to the intercalated disc region (13) or localized in the z-line region (15, 16). In this study, we used multiple approaches to simultaneously determine the distribution patterns of native KCNQ1 and KCNE1 in cardiac myocytes. We also explored the mechanism and functional implications for variations in the degree of KCNQ1/KCNE1 colocalization in cardiac myocytes. We used the guinea pig model because its atrial and ventricular myocytes express a robust IKs.
EXPERIMENTAL PROCEDURES
Molecular Biology
Human KCNE1 (NP_001121140, abbreviated as “E1”) was subcloned in-frame into pDsRed-monomer_Hyg_N1 (Clontech) to create E1-dsR. The HA epitope was attached to the C terminus of E1 to create E1-HA. The V5 epitope was attached to the C terminus of human KCNQ1 (NP_000209, 676 amino acids long, abbreviated as “Q1”) to create Q1-V5. Human KCNQ1 with enhanced GFP attached to the C terminus (Q1-GFP) was a kind gift from Dr. Andrew Tinker (University College London, UK) (17). To create the adenovirus carrying Q1-GFP and E1-dsR, the genes of interest were subcloned into the Adeno_shuttle vector, and, after confirming gene function (by patch clamp, immunoblotting, and confocal experiments), they were sent to Vector Biolabs for adenoviral production.
Animals
The investigation conformed to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (Publication No. 85-23, revised in 1996). The animal protocol (AM10294) has been reviewed and approved annually by the Institutional Animal Care and Use Committee of Virginia Commonwealth University. For euthanasia, animals were given xylazine (10 mg/kg) and acepromazine (1 mg/kg) by intramuscular injection. When the animal had reached the stage of general anesthesia (monitored by the total lack of response to toe pinch and cornea touching), ketamine (100 mg/kg) was administered by intramuscular injection. All three reagents were given only once. Euthanasia was accomplished by removing the heart.
Myocyte Isolation, Culture, and Adenoviral Infection
Guinea pig atrial and ventricular (GPA2 and GPV) myocytes were isolated from 2- to 3-month-old adult animals using enzymatic (collagenase, type II, Worthington) digestion, followed by mechanical trituration as described previously (6). Isolated myocytes were used in three types of experiments. For patch clamp experiments, myocytes were stored in Kraftbruhe (KB) medium at 4 °C and used within 10 h after isolation. For immunofluorescence/confocal experiments, myocytes were plated on poly-L-lysine-coated coverslips, fixed by 2% paraformaldehyde in PBS (room temperature, 15–30 min), rinsed, and stored in non-permeabilizing blocking buffer (5% FBS and 0.1% NaN3 in PBS) at 4 °C until experiments. For adenovirus-mediated gene expression experiments, adult GPV myocytes were isolated under sterile conditions. Cells were allowed to recover in KB for 2 h at room temperature. Then, the medium was changed to nominally calcium-free normal Tyrode's (147 mm NaCl, 4 mm KCl, 5.5 mm dextrose, and 5 mm HEPES (pH 7.3)), and the calcium concentration was raised stepwise to 1 mm during 2 h. Cells were plated on mouse laminin-coated coverslips, allowed to recover for ∼ 4 h in a serum-free medium (medium 199, Invitrogen), supplemented with BSA, l-carnitine, creatine, taurine, and penicillin/streptomycin as described previously (18) at 36 °C in a 5% CO2 moist incubator. Cells were incubated with adenoviruses harboring enhanced GFP (0.5 × 107 plaque-forming units/ml) or Q1-GFP and E1-dsR (0.25 × 107 plaque-forming units/ml each) overnight. Where noted, cells were incubated with Q1-GFP (0.25 × 107 plaque-forming units/ml) and a red fluorescent protein-tagged ER marker (ER-RFP, in bacuolovirus) according to the instructions of the manufacturer. After removing the viruses, cells were incubated for another 24 h before fixation for confocal experiments.
Neonatal rat ventricular myocytes (NRVMs) were isolated from 1- or 2-day-old Wistar-Kyoto rats under sterile conditions using mild enzymatic (0.1% trypsin) digestion and gentle mechanical trituration as described previously (19). After preplating (36 °C for 30 min) to remove contaminating fibroblasts, myocytes were plated on laminin- or fibronectin-coated coverslips in minimum Eagle's medium containing 10% FBS and cultured at 36 °C in 5% CO2 moist incubator. After being cultured for 3 days, cells were incubated with adenoviruses harboring Q1-GFP and E1-dsR (0.25 × 107 plaque-forming units/ml each) overnight. After removing the viruses, culture continued for the specified amounts of time (1, 3, 6, and 12 h). At the end of the culture, cells were used for patch clamp experiments or live- or fixed-cell confocal imaging.
Patch Clamp Experiments
Currents were measured using the whole-cell recording configuration of the patch clamp technique with EPC-8 (HEKA, Germany) or Axiopatch 200B (Molecular Devices) amplifiers. Patch pipettes had tip resistances of 2.5–4.0 MΩ (atrial myocytes, COS-7, and NRVM) or 1.5–2.5 MΩ (ventricular myocytes). After forming the whole-cell recording configuration, series resistance was compensated up to 95%. The pipette tip potential was zeroed before forming the whole-cell recording configuration, and the liquid junction potential (pipette versus bath solutions, estimated to be −10 mV) was subtracted during data analysis. To record the IKs from GPA and GPV myocytes, after establishing the whole-cell recording configuration in normal Tyrode's, the superfusate was switched to sodium-, potassium-, and calcium-free solution with 3 μm E-4031. These conditions isolate IKs from overlapping INa, ICaL, IK1, IKr, and INCX. In all experiments, the bath solution was maintained at 35 ± 1 °C. The program suite pClamp10 was used for data acquisition and analysis. Specific voltage clamp protocols and data analysis are described in the figure legends.
COS-7 Cell Culture and cDNA Transfection
COS-7 cells were maintained in DMEM (Invitrogen) supplemented with 10% FCS and penicillin/streptomycin in a 5% CO2 moist incubator at 36 °C. Cells were plated at a subconfluence level the day before transfection. Cells were transfected with cDNAs encoding Q1-GFP and E1-dsR (cDNA molar ratio 1:1) and, where noted, angiotensin type 1 receptor (AT1R) or ER-FRP. Transfection was facilitated by Lipofectamine 2000.
Immunofluorescence, Confocal Microscopy, and Image Analysis
Fixed cells attached to coverslips were permeabilized by 0.2% saponin (at 4 °C overnight or at room temperature for 2 h). Cells were incubated with primary/Alexa Fluor-conjugated secondary antibody pairs (see below). Where specified, cells were incubated with Alexa Fluor-conjugated wheat germ agglutinin (WGA). Nuclei were stained with DAPI. Fluorescence images were obtained with a Zeiss 510 Meta or Zeiss 710 confocal microscope. In most experiments, we collected data from four channels with uniform 0.7-μm optic slice thickness (excitation/emission): DAPI (405 nm/band pass (BP) 420–480 nm), Alexa Fluor 488 or GFP (488 nm/BP 493–530 nm), Alexa Fluor 568 or dsRed (561 nm/BP 570–630 nm), and Alexa Fluor 633 or Alexa Fluor 647 (633 nm/long pass (LP) 657 nm). Z-stack images were obtained at a frequency of 0.37 μm/optic slice.
For live cell imaging, COS-7 cells were plated on fibronectin-coated glass-bottom (glass thickness, 0.17 mm/#1.5) 35-mm dishes. Before imaging, nuclei were stained with Hoechst 33342, and, where noted, the endoplasmic reticulum was stained with ER Tracker blue/white dye (Molecular Probes). Cells were bathed in phenol red-free, HEPES-buffered medium supplemented with vitamin C (50 μm) at 37 °C and viewed with Zeiss 710.
To quantify KCNQ1 immunofluorescence signals in the cell periphery (Figs. 1F and 9C), the z-stack images of each myocyte were projected into a summed image that was used to calculate the total KCNQ1 signals, Qtotal (within the cell contour), and periphery KCNQ1 signals, Qperiphery (within a pace of ≤ 2 μm width from the cell contour). The percentage of KCNQ1 in the cell periphery was calculated as (Qperiphery/Qtotal) × 100%.
FIGURE 1.

Native KCNQ1 and KCNE1 (abbreviated as Q1 and E1) in cardiac myocytes are often not colocalized. A, validating KCNQ1 pAbs (goat and rabbit) and KCNE1 mAb (mouse) for detecting native proteins in GPA and GPV myocytes, respectively, using immunofluorescence (IF). +, Ab preincubated with antigen (Ag) overnight; −, Ab preincubated overnight in buffer without antigen. B and C, simultaneously detecting three-dimensional distribution patterns of KCNQ1 and KCNE1 in ventricular and atrial myocytes using KCNQ1 goat pAb and KCNE1 mouse mAb. The main panels depict x-y plane images of KCNQ1 and KCNE1 immunofluorescence signals taken from the central z slice, with x-z plane images reconstructed from the z-stacks shown on top. In B, enlarged views of the areas boxed in yellow frames are shown below. D, KCNQ1 rabbit pAb detected similar variations in KCNQ1 distribution patterns in ventricular and atrial myocytes. In each panel, a cell or cell cluster is identified as #1 or #2, and the KCNQ1 distribution patterns are noted (the definition of KCNQ1 distribution patterns is described in the text). Scale bars in B–D = 5 μm. E, percentages of atrial (A) and ventricular (V) myocytes manifesting KCNQ1 distribution patterns 1, 2, and 3. F, degrees of KCNQ1 presence in periphery of atrial and ventricular myocytes of KCNQ1 distribution pattern 1 versus 2 or 3. ***, t test, p < 0.001.
FIGURE 9.

In response to stress ([Ca2+]i elevation and/or oxidative stress), KCNQ1 exits the ER/SR in vesicles and travels to the cell periphery. A, Q1-GFP redistribution in a beating NRVM after repeated laser scans. Left panel, first scan. Center panel, after 60 scans applied once every 5 s. Right panel, scan obtained after resting (stopping laser scans) for 3 min. B, Q1-GFP redistribution in live COS-7 cells in response to stress. a, before and after ionomycin (10 μm) application to a cell expressing Q1-GFP and ER-RFP. b, before and after H2O2 (0.1%) application to a cell expressing Q1-GFP. c, images of a cell expressing Q1-GFP with the ER stained by ER Tracker in the presence of 0.1% H2O2. d, before and after Ang II (1 μm) application to a cell expressing Q1-GFP and angiotensin type 1 receptor (AT1R). e, images of two cells expressing Q1-GFP, AT1R, and ER-RFP under control conditions or after incubation with Ang II (1 μm, 60 min). C, the effect of incubation with Ang II (1 μm, 60 min) on native KCNQ1 distribution in guinea pig atrial and ventricular myocytes. The same batch of myocytes was incubated under the same conditions without Ang II as a control. Left panel, representative KCNQ1 immunofluorescence images. Right panel, quantification of the percentage of KCNQ1 signal in the cell periphery. Numbers of myocytes analyzed are shown (from one animal). *, p < 0.05. Live cell imaging and incubation with Ang II were all carried out at 37 °C. Scale bars = 5 μm. Videos of time-lapse live cell imaging are available in the supplemental information.
For colocalization analysis (Fig. 5B), confocal data were analyzed using the Volocity program (PerkinElmer Life Sciences). We focused on the central z-optical slice. After image deconvolution to reduce out-of-focus pixels from adjacent z-sections, the region of interest (the lateral cell surface defined as a ≤ 2 μm-wide space within the cell contour) was specified. The threshold value for each fluorescence channel was set by the average pixel intensity in the cell-free area, and the degree of colocalization of KCNQ1/KCNE1 immunofluorescence signals was evaluated by the Pearson's correlation coefficient.
FIGURE 5.

KCNQ1 and KCNE1 are colocalized to a higher degree in guinea pig atrial than in ventricular myocytes. In all cases, KCNQ1 and KCNE1 were detected by Alexa Fluor 488 and Alexa Fluor 568, respectively. A, x-y plane (large panels) and x-z plane (small panels) images of separate and merged KCNQ1 and KCNE1 immunofluorescence signals. Scale bars = 5 μm. B, quantification of the colocalization of KCNQ1 and KCNE1 immunofluorescence signals in the cell periphery. ***, p < 0.001; Student's t test. C, detecting KCNQ1 and KCNE1 functional assembly by apFRET. KCNQ1/Alexa Fluor 488 served as the FRET donor and KCNE1/Alexa Fluor 568 as the FRET acceptor. Shown are representative fluorescence images of KCNQ1 and KCNE1 in atrial and ventricular myocytes before and after photobleaching Alexa Fluor 568 (Pre-bleach and Post-bleach). The FRET map images depict the calculated FRET efficiency, as [(donorpost − donorpre)/donorpre] × 100%, using the color scale shown on the right. D, time courses of changes in Alexa Fluor 488 fluorescence intensity in guinea pig atrial and ventricular myocytes before and after photobleaching and in reference myocytes (REF, combined atrial and ventricular myocytes that were in the same microscopic field whose Alexa Fluor 568 signals were not bleached). The dashed line denotes the putative time course of intrinsic donor photobleaching during the five scans. Analysis of variance, p < 0.001. Pair-wise comparison, #, p < 0.01, GPV versus REF; *, p < 0.05, GPA versus GPV. The numbers in parentheses denote the number of myocytes studied/number of animals used.
We used acceptor photobleach fluorescence resonance energy transfer (apFRET) to detect and quantify the functional assembly between native KCNQ1 and KCNE1 in GPA and GPV myocytes (Fig. 5, C and D) or between native ether-a-go-go related gene (ERG) and KCNE1 in GPV myocytes (Fig. 11). KCNQ1 or ERG was labeled by rabbit Ab/Alexa Fluor 488 anti-rabbit (as FRET donor), and KCNE1 was labeled by mouse Ab/Alexa568 anti-mouse (FRET acceptor). Each apFRET experiment had five scans, applied once every 16 s. Each scan contained sequential scans of Alexa Fluor 488 and Alexa Fluor 568 using 1% laser power (to minimize unintentional photobleaching). An optic thickness of 4 μm was used to catch most of the fluorescence signals from cells. Scans 1 and 2 were prebleach scans. Between scans 2 and 3, laser 561 at 100% power was applied 100 times to selected regions of interest to bleach Alexa Fluor 568 to < 20% of the control. Cells in the same microscopic field whose Alexa Fluor 568 fluorescence was not bleached served as a reference, “REF”, reporting changes in Alexa Fluor 488 intensity unrelated to apFRET. Scans 3–5 were post-bleach scans. The pixel contents of donor fluorescence from the whole cell contour were measured for all five scans, background-subtracted, and normalized by the scan 2 data. The degree of apFRET was calculated as [(Donorpost − Donorpre)/Donorpre] × 100%. A FRET map was generated using the ImageJ plug-in “FRETcalc.”
FIGURE 11.
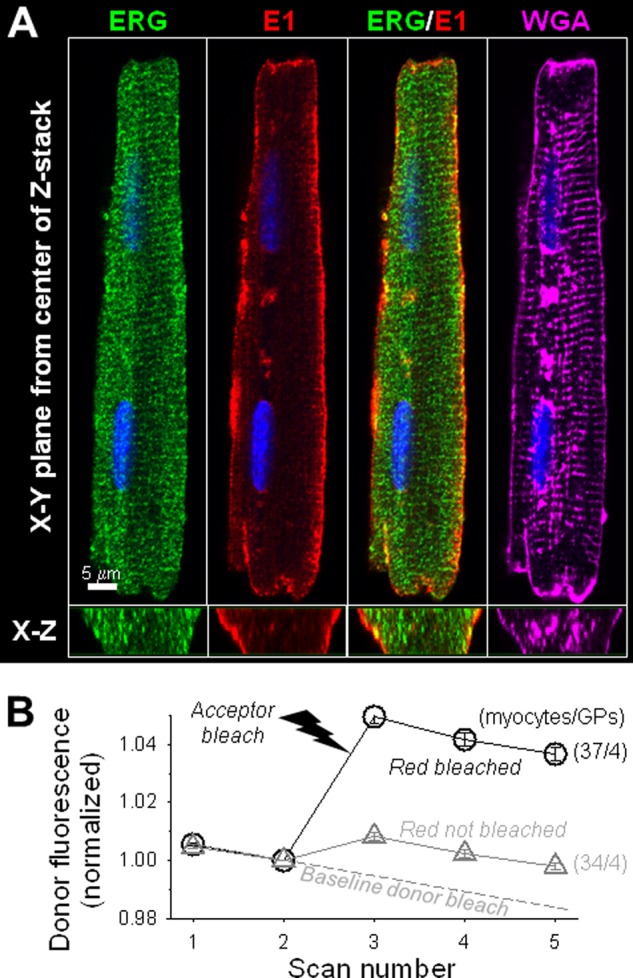
Three-dimensional patterns of ERG and KCNE1 immunofluorescence in guinea pig ventricular myocytes. A, fluorescence signals from a GPV myocyte taken from the central plane of z-stack (x-y) and cross section reconstructed from the z-stack (x-z) of ERG (rabbit pAb/Alexa Fluor 488 anti-rabbit), KCNE1 (mouse mAb/Alexa Fluor 568 anti-mouse), ERG/KCNE1 merge, and WGA (Alexa Fluor 647). B, degree of apFRET between Alexa Fluor 488-labeled ERG (donor) and Alexa Fluor 568-labeled KCNE1 (acceptor). The imaging protocol and data analysis are the same as described for Fig. 5, C and D. The number of myocytes examined/number of guinea pigs used are marked.
Immunoblot Experiments
Whole-tissue or whole-cell lysate (WTL or WCL, respectively) was prepared from guinea pig heart or myocytes using the protocol described by O'Rourke et al. (20). Whole cell lysate was prepared from COS-7 cells using a method described previously (6). Protein concentration was measured with a Micro BCATM protein assay kit (Pierce). Samples were loaded onto 10% (KCNQ1) or 16% (KCNE) denaturing (SDS)/reducing (mercaptoethanol) polyacrylamide gels. After electrophoresis, the proteins were blotted onto PVDF membranes and probed with suitable antibodies. Immunoreactivity was visualized using an ECL detection kit (Amersham Biosciences). To check loading variations, the same membranes were stripped and reprobed with actin mAb, or the remaining proteins in the gels were stained with Coomassie Blue. Immunoreactive band or Coomassie Blue intensities were measured by densitometry (ChemiImager model 4400, α-Innotech).
Antibodies and Reagents
The following primary antibodies were used: KCNQ1 goat (Santa Cruz Biotechnology) and rabbit (Alomone) pAbs, ERG rabbit pAb (Alomone), KCNE1 mouse mAb (AbNova), V5 mouse mAb (Invitrogen), HA mouse mAb (Covance), calnexin rabbit (Sigma) and goat (Santa Cruz Biotechnology) pAbs, ryanodine receptor type 2 (RyR2) mouse mAb (Abcam), dystrophin mouse mAb (Sigma), telethonin mouse mAb (Santa Cruz Biotechnology), α-actinin mouse mAb (Sigma), early endosome antigen 1 (EEA1) mouse mAb (BD Biosciences), mannose 6-phosphate receptor (M6PR) mouse mAb (Abcam), lysosome-associated membrane protein 1 (LAMP1) mouse mAb (Abcam), Rab5 mouse mAb (BD Biosciences), and Rab11 mouse mAb (BD Biosciences). Alexa Fluor-conjugated secondary antibodies, WGA, and ER Tracker dye were from Molecular Probes.
Ionomycin was dissolved in dimethyl sulfoxide to make 10 mm stock solutions, stored in aliquots at −20 °C and diluted to 10 μm in bath solution for experiments. Angiotensin II (Sigma) was dissolved in Millipore water to make 1 mm stock solutions, stored in aliquots at −20 °C and diluted to 1 μm in bath solution for experiments. H2O2 (30% solution, Sigma) was diluted to 0.01–0.1% with bath solution for experiments.
Statistics
Data are presented as mean ± S.E. Comparison between two groups was done by two-tailed Student's t test. Multiple group comparisons were assessed by one-way analysis of variance, followed by Dunn's test versus reference. SigmaStat (version 2) was used for statistical analysis.
RESULTS
Three-dimensional Distribution Patterns of Native KCNQ1 and KCNE1 in Adult Guinea Pig Atrial and Ventricular Myocytes
We simultaneously monitored the immunofluorescence signals of KCNQ1 and KCNE1 and used z-stack images to reconstruct their three-dimensional patterns. This allowed us to “see” where they were, both inside of the cells and on the cell surface. A prerequisite for the success of this approach was reliable Abs for immunofluorescence detection. Fig. 1A shows that preincubating primary Abs (anti-KCNQ1 goat and rabbit pAbs and anti-KCNE1 mouse mAb) with respective antigens abolished the immunofluorescence signals, supporting their specificity.
In both guinea pig atrial and ventricular myocytes, KCNE1 maintained a stable cell surface localization. This is best seen in the cross-section images reconstructed from the z-stacks (Fig. 1, B and C, top panels). On the other hand, the KCNQ1 immunofluorescence signals showed clear variations. There were three main patterns. In pattern 1, KCNQ1 was distributed as prominent transverse striations with a spacing of 1.9 ± 0.1 μm (Fig. 1, B (GPV #1) and C (GPA #1)). The cross-section images show that these striations were deep in the cytosol. In pattern 2, KCNQ1 was in prominent vesicles as well as in striations (Fig. 1D, GPV #1 and GPA #1). In pattern 3, KCNQ1 was mainly in vesicles, with no or only dim/incomplete striations (Fig. 1D, GPV #2 and GPA #2).
On the basis of data from 172 atrial and 125 ventricular myocytes isolated from six guinea pigs and analyzed in seven independent confocal experiments, 53, 32, and 15% of atrial myocytes manifested KCNQ1 distribution patterns 1, 2, and 3, respectively (Fig. 1E). For ventricular myocytes, the numbers were comparable (62, 28, and 10%). Most atrial myocytes had a strong KCNQ1 presence on the lateral surface, irrespective of the cytosolic KCNQ1 distribution pattern (Fig. 1F). In “pattern 1” ventricular myocytes, KCNQ1 signals on the lateral surface were weak and uneven. Reconstructed three-dimensional images suggest a patchy KCNQ1 distribution pattern on the lateral surface. “Pattern 2” or “pattern 3” ventricular myocytes had a significantly higher KCNQ1 presence in the cell periphery (Fig. 1F). The nature of these variations in the KCNQ1 distribution pattern and their functional significance were explored in the following experiments.
Location of KCNQ1 Striations in Guinea Pig Atrial and Ventricular Myocytes
We compared the KCNQ1 striation pattern with those of markers for subcellular compartments or organelles. Alexa Fluor-conjugated WGA was used to stain surface membrane and t-tubules. In most ventricular myocytes, the KCNQ1 striations coincided with those of WGA, consistent with the notion that KCNQ1 is in t-tubules (Fig. 2A, a) (21). However, in ventricular myocytes that had been partially detubulated (incubation for 30 min in 1.5 m formamide), we still saw clear KCNQ1 striations in areas where t-tubules were missing (Fig. 2A, b). Guinea pig atrial myocytes did not have t-tubules (WGA stained the lateral surface without striations), yet KCNQ1 was in clear striations (Fig. 2A, c). Finally, Fig. 2A, d, illustrates a ventricular myocyte in which KCNQ1 striations alternated with WGA striations. There was a minor KCNQ1 band between the major striation bands that coincided with the WGA band.
FIGURE 2.

Probing the location of KCNQ1 striations in ventricular and atrial myocytes. A, varying relationships between KCNQ1 and t-tubules (detected by Alexa Fluor-conjugated WGA). Each row shows fluorescence images of KCNQ1 (left panels), WGA (center panels) and merged images (right panels). a, KCNQ1 and WGA striations overlapped (arrows). b, in partially detubulated ventricular myocytes clear KCNQ1 striations (arrows) were seen in areas missing t-tubules. c, atrial myocytes did not have t-tubules (no WGA striations), although KCNQ1 showed clear striations (arrows). d, KCNQ1 major striations (solid arrowheads) alternated with WGA striations (open arrowhead), whereas minor KCNQ1 bands coincided with WGA striations. Scale bars = 5 μm. B, in both ventricular and atrial myocytes, KCNQ1 striations registered with three cytosolic markers: a, ryanodine receptor (RyR2, marking the junctional SR); b, α-actinin (z-lines); and c, calnexin (SR). For each myocyte, the x-y plane images of separate KCNQ1 and marker immunofluorescence signals (left and center panels) and the reconstructed y-z plane image of merged immunofluorescence signals (right panel) are shown. Scale bars = 5 μm.
We further compared KCNQ1 striation patterns with the following cytosolic markers: ryanodine receptor (RyR2, marking the junctional sarcoplasmic reticulum (SR)), α-actinin (z-lines), and calnexin (ER/SR). Fig. 2B shows that, in both atrial and ventricular myocytes, KCNQ1 striations registered with those of RyR2, α-actinin, and calnexin. The cross-section images of merged immunofluorescence signals clearly show that KCNQ1 was distributed deep into the cytosol along with these cytosolic markers. Together, these data suggest that KCNQ1 striations do not necessarily represent t-tubule localization. Instead, they suggest that KCNQ1 resides in the z-line region, probably in an intracellular compartment.
Verification of KCNQ1 and KCNE1 Immunofluorescence Patterns in Guinea Pig Ventricular Myocytes
The above data suggest that, in guinea pig ventricular myocytes, KCNQ1 and KCNE1 are often not colocalized. If confirmed and extrapolated to human ventricular myocytes, this observation has far-reaching implications in terms of the control of IKs current amplitude and KCNQ1 channel function. Although we have validated our Abs for immunofluorescence application (Fig. 1A), one remaining concern was that Abs might bind to proteins that were unrelated to KCNQ1 or KCNE1 but had three-dimensional conformations similar to the epitopes. Therefore, we further tested the distribution patterns of KCNQ1 and KCNE1 in guinea pig ventricular myocytes without the need for Abs. We used a KCNQ1 construct with enhanced GFP ligated to its C terminus (Q1-GFP (17)) and a KCNE1 construct with mono-dsRed ligated to its C terminus (E1-dsR). By using adenoviruses to introduce these constructs into guinea pig ventricular myocytes, we could directly and simultaneously monitor their distribution patterns with fluorescence microscopy.
Validating Fluorescent Protein-tagged KCNQ1 and KCNE1
Q1-GFP and E1-dsR expressed in both COS-7 cells (Fig. 3A) and NRVMs (data not shown) could assemble into functional IKs channels. To test whether the attached enhanced GFP and mono-dsRed (225 and 239 amino acids, respectively) might interfere with their ability of trafficking, we compared the distribution patterns of E1-dsR and E1-HA (HA epitope attached to the C terminus of KCNE1) in the presence of KCNQ1. We also compared the distribution patterns of Q1-GFP and Q1-V5 (V5 epitope attached to the C terminus of KCNQ1) in the presence KCNE1. The small HA and V5 epitopes (9 and 14 amino acids) should have a minimal impact on their trafficking ability. In the COS-7 expression system, E1-dsR and E1-HA had the same distribution pattern. They were colocalized in vesicles and on the cell surface. Q1-GFP and Q1-V5 also had the same distribution pattern. They were colocalized in the ER and on the cell surface. These data confirm that attaching fluorescent proteins to the C termini of KCNQ1 and KCNE1 did not interfere with their ability to traffic in COS-7 cells.
FIGURE 3.

Validating KCNQ1-GFP and KCNE1-dsR for their channel formation function and subcellular distribution patterns. A, COS-7 expression. Top panel, patch clamp recording from COS-7 cells expressing Q1-GFP alone or with E1-dsR. Currents were elicited by 2-s test pulses from −80 mV holding voltage to −70 to + 100 mV in 10-mV increments, and tail currents were recorded after repolarization to −60 mV. The relationship between tail current amplitude (Itail) and test pulse voltage (Vt) was fit with a simple Boltzmann function: Itail = Imax/(1 + exp[(V0.5-Vt)/k]) to estimate the maximal Itail (Imax), half-maximum activation voltage (V0.5) and slope factor (k). Itail values were normalized by Imax (fraction activated) and plotted against Vt on the right. The V0.5 and k values are −8.3 ± 5 and 15.1 ± 1.8 mV for Q1-GFP alone (n = 6) and 41.0 and 20.4 mV for Q1-GFP/E1-dsR. Bottom panel, fluorescence images of COS-7 cells coexpressing E1-HA and E1-dsR (with KCNQ1, top row) or Q1-GFP and Q1-V5 (with KCNE1, bottom row). The HA and V5 epitopes were detected by mouse primary mAbs/Alexa Fluor 488- or Alexa Fluor 568-conjugated anti-mouse after cell membrane permeabilization. In the bottom row, the ER was stained by ER blue-white tracker dye. The arrows point to cell surface distribution. Distribution in the ER or vesicles is also noted. Scale bars = 5 μm. B, NRVM expression. Shown are Q1-GFP and E1-dsR fluorescence images from myocytes cultured for 3 or 12 h after overnight incubation with adenoviruses carrying these constructs. Native calnexin immunofluorescence was detected by Alexa Fluor 647. The arrows and open arrowheads point to cell surface and striation distribution, respectively.
However, the distribution patterns of KCNQ1 and KCNE1 in COS-7 cells were distinctly different from those in cardiac myocytes. To test whether this was due to differences in the cellular environment, e.g. sarcomere organization in cardiac myocytes but not in COS-7 cells, we further examined the distribution patterns of Q1-GFP and E1-dsR in NRVMs. NRVMs in culture gradually develop ordered sarcomeres (22), allowing us to compare the distribution patterns of Q1-GFP and E1-dsR in a myocyte environment before and after the development of sarcomere organization. NRVMs were incubated overnight with adenoviruses carrying Q1-GFP and E1-dsR. After removing the viruses, the cells were cultured for varying lengths of time before fixation for confocal experiments. In NRVMs without clear sarcomere organization (e.g. 3-h myocyte in Fig. 3B), Q1-GFP was detected in the ER tubular network (colocalization with the ER marker calnexin), whereas E1-dsR was detected in vesicles. Their patterns were similar to those of KCNQ1 and KCNE1 in COS-7 cells. In NRVMs that had developed ordered sarcomeres (e.g. 12-h myocyte in Fig. 3B), the Q1-GFP along with calnexin was partially organized into striations. E1-dsR and Q1-GFP were both detected on the surface of the 12-h myocyte. These observations suggest that the KCNQ1 distribution pattern depended on cellular architecture; i.e. it manifested striations in cardiac myocytes that had organized sarcomeres.
Confirming the KCNQ1 and KCNE1 Distribution Patterns in Guinea Pig Ventricular Myocytes
To reach a suitable expression level of Q1-GFP and E1-dsR in adult guinea pig ventricular myocytes, we needed to culture the myocytes for a total of 3 days. Many, although not all, cultured guinea pig ventricular myocytes maintained their architecture, as shown by the similar distribution patterns of subcellular compartment markers to those in freshly isolated myocytes (data not shown).
We examined myocytes from six animals (independent procedures of cell isolation, culture, and adenoviral infection), focusing on myocytes that had an elongated shape with striations. Similar results were obtained in a total of 65 myocytes, and representative images are shown in Fig. 4. Q1-GFP expressed in guinea pig ventricular myocytes clearly exhibited a transverse striation pattern with a spacing of 1.7 ± 0.2 μm, whereas E1-dsR was present on the lateral cell surface as well as in vesicles (Fig. 4A). The Q1-GFP striations registered with those of RyR2, α-actinin, and calnexin (Fig. 4B, a–c).
FIGURE 4.

Distribution patterns of Q1-GFP and E1-dsR in cultured guinea pig ventricular myocytes. A, representative fluorescence images of Q1-GFP and E1-dsR. Main panel, x-y plane images. Lower left inset, reconstructed X-Z plane images. B, Q1-GFP striations registered with those of cytosolic markers: RyR2, α-actinin, and calnexin. The arrows point to striations.
The distribution patterns of Q1-GFP and E1-dsR were reminiscent of the main features of native KCNQ1 and KCNE1 in guinea pig ventricular myocytes detected by immunofluorescence (Figs. 1 and 2B). Q1-GFP and E1-dsR also showed a strong presence in the perinuclear region, where the rough ER and Golgi apparatus were. In addition, E1-dsR was detected in prominent vesicular compartments, similar to what was seen in COS-7 and NRVM expression (Fig. 3). These reflected the high level of protein translation and trafficking following adenovirus-mediated gene expression. We conclude that the antibodies faithfully detected native KCNQ1 and KCNE1 in guinea pig cardiac myocytes, and, indeed, KCNQ1 and KCNE1 are not well colocalized in the ventricular myocytes.
Functional Consequence of Variations in KCNQ1/KCNE1 Colocalization in Cardiac Myocytes
The degree of KCNQ1/KCNE1 colocalization in the cell periphery was higher in guinea pig atrial than ventricular myocytes (Fig. 5, A and B). To further test whether KCNQ1/KCNE1 colocalization was correlated with assembly into IKs channels, we tested whether fluorescently labeled KCNQ1 and KCNE1 could engage in FRET. FRET between a fluorescent donor and acceptor occurs when the fluorophores are within 20–140 Å from each other with proper orientation. The degree of FRET declines as the sixth power of the distance between the fluorophores (23). It has been reported that the fluorescent donor and acceptor attached to the C termini of KCNQ1 and KCNE1, respectively, can engage in FRET when the tagged KCNQ1 and KCNE1 form a functional IKs channel (24). We labeled native KCNQ1 with Alexa Fluor 488 (as FRET donor) and native KCNE1 with Alexa Fluor 568 (FRET acceptor) and compared the Alexa Fluor 488 signal intensities before and after photobleaching Alexa Fluor 568. If there was FRET between the two, the Alexa Fluor 488 signal would increase after Alexa Fluor 568 had been bleached. Fig. 5C depicts examples of Alexa Fluor 488 and Alexa Fluor 568 fluorescence images from guinea pig atrial and ventricular myocytes before and after photobleaching. The time courses of changes in Alexa Fluor 488 fluorescence signals before and after photobleaching are shown in Fig. 5D. Photobleaching Alexa Fluor 568 led to an increase in Alexa Fluor 488 fluorescence intensity, and the degree of increase was significantly higher in atrial than ventricular myocytes (5.2 ± 0.2% versus 2.9 ± 0.2%, calculated for scan 3). The latter was higher than that seen in reference (REF, reporting changes in the Alexa Fluor 488 fluorescence signal without photobleaching Alexa Fluor 568, 0.8 ± 0.2%). We conclude that a higher degree of cell periphery KCNQ1/KCNE1 colocalization corresponds to a higher degree of KCNQ1/KCNE1 assembly into IKs channels in atrial than in ventricular myocytes.
We patch-clamped 24 atrial and 21 ventricular myocytes isolated from seven guinea pigs. To isolate IKs from other overlapping currents, the cells were bathed in sodium-, potassium-, and calcium-free Tyrode's solution containing 3 μm E4031 (Fig. 6A). The IKs current density in atrial myocytes was significantly higher than that in ventricular myocytes (estimated maximal tail currents recorded at −50 mV, 5.4 ± 0.4 versus 2.2 ± 0.2 pA/pF, p < 0.05). Although the IKs current densities differed markedly between atrial and ventricular myocytes, the IKs gating kinetics was similar in terms of voltage-dependence of activation, rates of activation, and deactivation (Fig. 6, B–E).
FIGURE 6.

Comparison of IKs current density and gating kinetics in guinea pig atrial and ventricular myocytes. Whole cell currents recorded in sodium-, potassium-, and calcium-free solution at 35 °C in the presence of 3 μm E4031. A, representative current traces elicited by the diagrammed voltage clamp protocol applied once per 10 s. Currents were normalized by cell capacitance (calibrations applied to both panels). Horizontal bars denote zero current. pA, picoampere; pF, picofarad. B, left panel, tail current density plotted against test pulse voltage. Right panel, voltage-dependence of IKs activation. For each myocyte, the relationship between tail current amplitude (Itail) and test pulse voltage (Vt) was fit with a simple Boltzmann function as described in Fig. 3A. The fraction of channel activated (Itail/Imax) was plotted against Vt. C, current traces elicited by an “envelope test” protocol. Test pulse durations ranged from 50–3000 ms, and the interpulse interval was 10 s. nA, nanoampere. D, time course of IKs activation constructed from the growth of tail currents during the envelope test protocol as shown in C. For each myocyte, the relationship between tail current amplitude (Itail) and test pulse duration (t) was fit with a single exponential function with a delay, d: Itail = Iss(1-exp[-(t-d)/τ]), where Iss and τ are the plateau Itail amplitude and time constant of IKs activation. E, rate of IKs deactivation. Inset, representative IKs tail currents from atrial and ventricular myocytes recorded at repolarization voltages (Vr) of −100 to −40 mV in 20-mV increments. The time courses were fit with a double (−40 and −60 mV) or single (-80 and −100 mV) exponential function: Itail = Σ Iiexp(-t/τd), where i = 1 or 2 and τd was the deactivation time constant of component i. The τd values (of the major component, for double exponential fit) were pooled and plotted against repolarization voltages. In B, left panel, and D, the numbers in parentheses denote the number of myocytes studied/number of animals used.
We further quantified the KCNQ1 and KCNE1 protein levels in whole tissue lysates prepared from five regions of guinea pig hearts. Fig. 7 shows that the KCNQ1 protein levels were modestly lower in ventricles than in atria. There were no statistically significant differences in the KCNE1 protein levels among the five regions. Together, these data suggest that IKs current amplitude is determined not only by the expression levels of KCNQ1 and KCNE1 but also by the degree of their colocalization on the cell surface.
FIGURE 7.
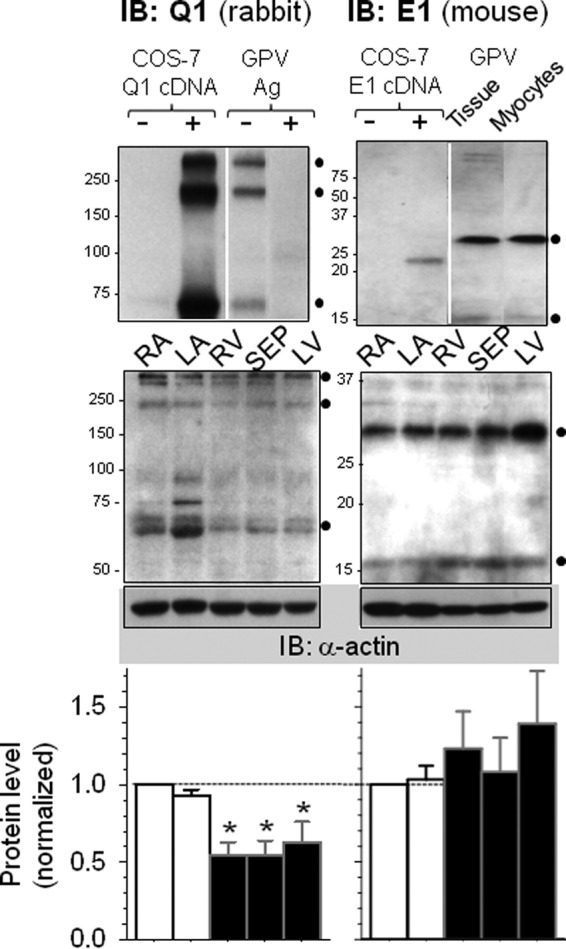
Quantification of KCNQ1 and KCNE1 protein levels in guinea pig heart. Left panels, quantification of KCNQ1 in five regions of guinea pig hearts: right (RA) and left (LA) atrial appendages, interventricular septum (SEP), and right ventricular (RV) and left ventricular (LV) free walls. Left top panel, validating KCNQ1 rabbit pAb for immunoblot (IB) application. The KCNQ1 Ab detected 75, 225- and 300-kDa bands in COS-7 cells transfected with KCNQ1 cDNA but none in untransfected cells (+ and − Q1 cDNA, respectively). The Ab detected three similar bands in whole tissue lysate (WTL) from guinea pig ventricles, and the bands were abolished by preabsorbing the Ab with excess Ag (− and + Ag, respectively). Left center panel, a representative KCNQ1 immunoblot image. WTLs from five regions were fractionated by SDS-PAGE. After probing for KCNQ1, the membrane was stripped and reprobed for α-actin (bottom, as loading control). Size marker positions are marked on the left. ●, KCNQ1-specific bands. Left bottom panel, densitometry data summarized from six guinea pig hearts. The combined intensities of KCNQ1-specific bands were divided by the α-actin band intensity of the same lane and then normalized by that of the RA lane of the same membrane. One-way analysis of variance, p = 0.013. Dunn's test versus RA, *, p < 0.05. Right bottom panel, quantification of KCNE1 in the same five regions. Right top panel, validating the KCNE1 mouse mAb for IB application. The KCNE1 Ab detected a 25-kDa band in COS-7 cells transfected with KCNE1 but not in untransfected cells. The Ab detected a 30-kDa and a faint 15-kDa band in WTL of guinea pig ventricle and whole cell lysate prepared from isolated guinea pig ventricular myocytes (● on the right). These are interpreted as KCNE1 core protein (15 kDa) and the glycosylated/dimer form (30 kDa). Right center panel, a representative KCNE1 immunoblot image. Right bottom panel, densitometry data summarized from six hearts. Data analysis was the same as that described for KCNQ1. One-way analysis of variance, p = 0.778.
Mechanism for Dynamic KCNQ1 Distribution Patterns in Cardiac Myocytes
Ventricular myocytes isolated from the same guinea pig heart were incubated in KB (containing 0.5 mm EGTA without added calcium, essentially calcium-free) or medium 199 (containing 1.8 mm calcium) for 8 h before fixation and processing for confocal microscopy. Although KCNE1 maintained stable lateral surface localization under both conditions, KCNQ1 distribution switched from mainly pattern 1 in KB to mainly pattern 2 in medium 199 (Fig. 8A). In pattern 2 ventricular myocytes, most of the KCNQ1 vesicles appeared attached to the striations or in their vicinity, whereas in pattern 3 ventricular myocytes, the KCNQ1 vesicles were clustered to the cell periphery (Fig. 8B). To understand what might have caused the dynamic distribution patterns of KCNQ1, we probed the origin and destination of KCNQ1 vesicles in cardiac myocytes.
FIGURE 8.

Probing the nature of KCNQ1 vesicles in guinea pig ventricular myocytes. A, effects of changing [Ca2+]o on KCNQ1 and KCNE1 distribution patterns in guinea pig ventricular myocytes. Myocytes isolated from the same heart were incubated in a calcium-free medium (KB, containing 0.5 mm EGTA) or in medium 199 ([Ca] = 1.8 mm) for 8 h, fixed, and subjected to immunofluorescence detection. The numbers of KCNQ1 vesicles per 1-μm optic slice were 114 ± 12 and 180 ± 21 in myocytes incubated in KB and medium 199, respectively (n = 3 and 7). B, KCNQ1 vesicle distribution in pattern 2 and 3 ventricular myocytes. C, comparison of KCNQ1 vesicles with vesicles bearing organelle markers: early endosome associated protein-1 (EEA1), M6PR, lysosome-associated membrane protein 1 (LAMP1), Rab5, and Rab11. Each row shows immunofluorescence images of KCNQ1 (left column), marker (center column), and merge (right column). Circles highlight KCNQ1 vesicles positive for EEA1 or M6PR. D, comparison of KCNQ1 distribution pattern with that of calnexin in KCNQ1 pattern 2 and 3 ventricular myocytes. Each graph in B and D depicts the x-y (main panel), x-z (top panels) and y-z (right panels) views of immunofluorescence signals.
KCNQ1 and Endocytic/Recycling Vesicles in Cardiac Myocytes
We compared the immunofluorescence signals of KCNQ1 with those of markers for vesicles in the endocytic/recycling pathways: early endosome antigen 1 (EEA1, a marker of early endosomes), M6PR (late endosomes), lysosome-associated membrane protein 1 (LAMP1, lysosomes), Rab5 (early endosomes), and Rab11 (recycling endosomes) (Fig. 8C). On the basis of analysis of the central z-plane images, we detected 4.1 ± 0.7 KCNQ1 vesicles/guinea pig ventricular myocyte that were positive for EEA1, 2.2 ± 0.3 KCNQ1 vesicles positive for M6PR, and 0.8 ± 0.2 KCNQ1 vesicles positive for LAMP1 (n = 29, 30, and 30 myocytes, respectively, from two animals). We could not detect any overlap between KCNQ1 vesicles and Rab5 or Rab11. Therefore, only a minute fraction of KCNQ1 vesicles represented endocytosed KCNQ1 in the early or late endosomal compartment. The majority of KCNQ1 vesicles originated from somewhere else.
KCNQ1 and SR Vesiculation in Cardiac Myocytes
In pattern 1 myocytes, KCNQ1 striations registered with those of the ER/SR marker calnexin (Fig. 2B, c). In pattern 2 ventricular myocytes, both KCNQ1 and calnexin were in prominent vesicles and dim in striations (Fig. 8D, left panel). In pattern 3 ventricular myocytes, both KCNQ1 and calnexin clustered to the cell periphery (Fig. 8D, right panel). It has been reported that, in rat basophilic leukemia (2H3) and mouse fibroblast (3T3) cells, elevating [Ca2+]i with ionomycin causes reversible ER fragmentation (25). The striking correlation between KCNQ1 and calnexin in guinea pig ventricular myocytes provided a clue that KCNQ1 vesicles might originate from SR vesiculation and [Ca2+]i elevation might be the trigger. It is possible that, during myocyte isolation, the cells experienced different degrees of “assault” that led to [Ca2+]i elevation, producing KCNQ1 distribution patterns 1, 2, and 3. This scenario is consistent with the data shown in Fig. 8A.
[Ca2+]i elevation might not be the only trigger of KCNQ1/SR vesiculation, as was suggested by live cell imaging of NRVMs expressing Q1-GFP. Fig. 9A and supplemental video 1 show that, in a beating NRVM (cyclic rise in [Ca2+]i), Q1-GFP was in the ER tubular network (first scan). However, Q1-GFP vesiculated after repeated laser scans. This process was readily reversible (Q1-GFP resumed ER tubular network morphology) after stopping laser scanning for 3 min. Thus, in NRVMs, the cyclic rise in [Ca2+]i was not sufficient to induce Q1-GFP vesiculation. Q1-GFP vesiculation occurred when the cell was further stressed by laser scanning, which produced reactive oxygen species.
Direct Observation of Simultaneous Q1-GFP and ER Vesiculation in Live COS-7 Cells
To test whether Q1-GFP and ER simultaneously vesiculated in response to stress ([Ca2+]i elevation or oxidative stress), we switched to the COS-7 expression system because these robust cells could sustain repeated laser scans for long durations. Fig. 9B, a–c, and supplemental videos 2 and 3) confirm that elevating [Ca2+]i (by ionomycin) or oxidative stress (by H2O2) induced simultaneous vesiculation of Q1-GFP and ER. Similar observations were obtained in nine (ionomycin treatment) and three (H2O2 treatment) cells. On the other hand, two other stressors that have been shown to increase IKs (11) did not induce Q1-GFP or ER vesiculation: hypo-osmotic cell swelling (50% reduction of bath solution osmolarity) and β-adrenergic stimulation (isoproterenol, 1 μm, on the coexpressed β1-adrenergic receptor).
KCNQ1 Vesiculation and Cell Surface Expression in Response to Angiotensin II Receptor (AT1R) Stimulation
AT1R stimulation leads to [Ca2+]i elevation (through the inositol 1,4,5-trisphosphate receptor) and production of reactive oxygen species (through NADPH oxidase) (26). Fig. 9B, d, and supplemental video 4 show that stimulating AT1R coexpressed with Q1-GFP in COS-7 cells induced Q1-GFP vesiculation. Similar observations were obtained from four cells. After incubating COS-7 cells coexpressing Q1-GFP, AT1R, and ER-RFP with 1 μm angiotensin II for 60 min, Q1-GFP was redistributed from the ER compartment to the cell surface (Fig. 9B, e, representative of 10 cells under the control conditions and six cells after Ang II incubation).
To test whether this occurred with native KCNQ1, we incubated guinea pig cardiac myocytes with angiotensin II (1 μm, 60 min). This induced a translocation of KCNQ1 from cytosolic striations to the cell periphery in ventricular myocytes (Fig. 9C). On the other hand, in guinea pig atrial myocytes where native KCNQ1 was already high on the cell surface, incubation with angiotensin II did not induce a further increase.
DISCUSSION
KCNQ1 Striations and Endo(sarco)plasmic Reticulum
We propose that, in most myocytes, KCNQ1 striations represent an intracellular compartment close to the Z-lines. On the basis of the tight correlation between KCNQ1 and calnexin, we further propose that this KCNQ1 striation compartment is part of the ER/SR. Calnexin has been used as a specific marker for the ER/SR. Colocalization between membrane channels and calnexin has been interpreted as channel retention in the ER because of misfolding or trafficking defects. However, the ER/SR is an extensive, highly dynamic structure and is compartmentalized to fulfill its many functions (27, 28). In adult guinea pig ventricular myocytes, the striation pattern of calnexin likely reflects intimate contacts between the calnexin-positive ER/SR domain and the t-tubule membranes (27).
KCNQ1 Vesicles and Trafficking in Response to Stress
On the basis of the data presented in Figs. 8 and 9 (and in supplemental videos 1–4), we suggest the following scenario (schematic in Fig. 10). In unstressed guinea pig ventricular myocytes, KCNQ1 is mainly in the cytosolic ER/SR compartment. When ventricular myocytes are stressed ([Ca2+]i elevation and/or oxidative overload), KCNQ1 vesiculates, along with partial and reversible ER/SR fragmentation. The KCNQ1 vesicles travel to the cell periphery, where KCNQ1 can be delivered to the cell surface and assemble with KCNE1 to form functional IKs channels. Cell surface KCNQ1 can reenter the cytosolic compartment via vesicles, consistent with the observation that some of the KCNQ1 vesicles carried markers for early and late endosomes (EEA1 and M6PR).
FIGURE 10.
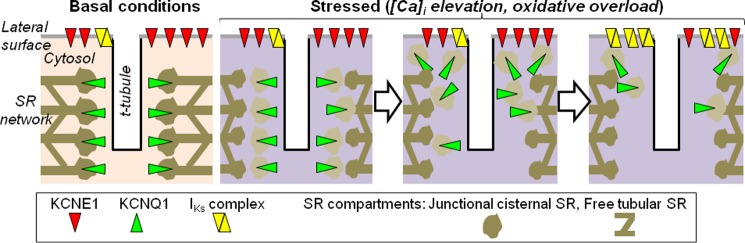
Schematic depiction of KCNQ1 translocation and regulation of IKs current density in ventricular myocytes in response to stress. Under basal conditions, KCNE1 is mainly on the lateral cell surface, whereas KCNQ1 is mainly in a cytosolic compartment, associated with or as part of the junctional SR adjacent to t-tubules/z-lines. The IKs current is mediated by a small fraction of available KCNQ1 channels and KCNE1 subunits that are colocalized/assembled on the cell surface. When myocytes are stressed by [Ca2+]i elevation or oxidative overload, junctional SR is partially fragmented. KCNQ1 vesicles exiting the cytosolic compartment travel to the cell periphery, where KCNQ1 is delivered to the cell surface. This leads to an increase in the IKs current density by increased KCNQ1/KCNE1 colocalization/assembly.
Differential Response to Stress between Guinea Pig Atrial and Ventricular Myocytes
Although we observed similar variations in KCNQ1 distribution patterns in guinea pig atrial and ventricular myocytes (Fig. 1E), there was a much higher KCNQ1 presence on the surface of atrial than ventricular myocytes (F). In situ atrial myocytes may be chronically stressed by mechanical stretch (because of the thin atrial wall) and by oxidative overload (because of high oxygen tension in the left atrium). This may lead to a situation similar to pattern 3 ventricular myocytes. This is consistent with the observations that KCNQ1 distribution in atrial myocytes was relatively insensitive to Ang II incubation (Fig. 9C).
Comparison with Previous Studies
The major distribution pattern of KCNQ1 in atrial and ventricular myocytes is transverse striations (11–15), interpreted as KCNQ1 localization in t-tubules (21). In the majority (50–60%) of guinea pig atrial and ventricular myocytes, we observed a KCNQ1 distribution pattern as transverse striations, with a spacing of 1.9 ± 0.1 μm. Our observation is consistent with the literature. However, the interpretation differs. Fig. 2A shows that KCNQ1 was in clear striations even when there was no t-tubule, indicating that the striation pattern does not necessarily mean t-tubule localization.
It has been shown that stress-induced activation of serum- and glucocorticoid-inducible kinase 1 (SGK-1) can increase the IKs amplitude by facilitating exocytosis of KCNQ1 vesicles (29), although such KCNQ1 vesicles have not been described for cardiac myocytes. Furthermore, elevating [Ca2+]i can increase IKs in guinea pig ventricular myocytes (30). One of the mechanisms is calcium binding to the calmodulin molecules associated with the cytoplasmic C-terminal domain of KCNQ1, facilitating KCNQ1 trafficking to the cell surface (31). The stress-induced transition of KCNQ1 distribution from pattern 1 to pattern 3 reported here may underlie the above observations.
There is a precedent for KCNQ1 redistribution in response to stimulus. In resting gastric parietal cells, KCNQ1 is mainly in an intracellular compartment. Cell activation leads to KCNQ1 translocation to the cell surface (32).
Is Cell Surface KCNE1 Partnered with ERG?
It has been shown in heterologous expression experiments that KCNE1 can associate with human ERG to modulate its current amplitude (33). To test this possibility in guinea pig ventricular myocytes, we simultaneously monitored the three-dimensional distribution patterns of guinea pig ERG and KCNE1 immunofluorescence along with WGA (surface and t-tubule marker). There was a partial overlap between ERG and KCNE1 on the lateral cell surface (Fig. 11A). Furthermore, Alexa Fluor 488-labeled ERG and Alexa Fluor 568-labeled KCNE1 could engage in apFRET (Fig. 11B). These observations suggest that, in guinea pig ventricular myocytes, cell surface KCNE1 can assemble with KCNQ1 or ERG. Because KCNQ1 and ERG may be colocalized in cardiac myocytes (34), it is possible that KCNE1 may switch its partnership between these two K channels. Whether, and under what conditions, this happens in cardiac myocytes requires further investigation.
KCNQ1, KCNE1, other KCNE Subunits, and IKs Function
IKs functions as a repolarization reserve in human ventricular myocytes. Our study suggests a previously unrecognized mechanism whereby the IKs amplitude in ventricular myocytes is maintained at a low level until it is needed. The IKs function in atrial myocytes is less clear. Because of the atria-specific expression of ultra-rapid delayed rectifier (IKur) channels, atrial myocytes do not need a repolarization reserve. We observed a high cell surface KCNQ1/KCNE1 colocalization and high IKs density in guinea pig atrial myocytes under basal conditions. If this can be extrapolated to human atrial myocytes, IKs may be a risk factor for atrial fibrillation. This is in keeping with the observations that genetic variations leading to “gain of function” in IKs and atrial action potential shortening are a common mechanism underlying diverse familial atrial fibrillation syndromes (35).
We propose a scenario of a dynamic relationship between native KCNQ1 and KCNE1 in ventricular myocytes. This is consistent with the transient nature of the KCNQ1/KCNE1 partnership observed in COS-7 experiments (6). This raises the possibility that KCNQ1 may associate with other members of the KCNE family in ventricular myocytes. In particular, KCNE2 is expressed in ventricles of humans and guinea pigs (7). In guinea pig ventricular myocytes, KCNE2 is distributed in cytosolic striations as well as on the lateral surface, i.e. it is better colocalized with KCNQ1 than KCNE1. It is possible that, in ventricular myocytes, KCNE2 can associate with KCNQ1 to suppress the IKs amplitude (15).
Acknowledgments
We thank Dr. Andrew Tinker for providing the KCNQ1-GFP construct and Drs. Minoru Horie, Hiroshi Matsuura, and Futoshi Toyoda for providing guinea pig myocytes (data presented in Fig. 6).
This study was supported, in whole or in part, by National Institutes of Health Grant RO1 HL107294 (to G. N. T.) and NINDS National Institutes of Health Center Core Grant 5P30NS047463 (for microscopy performed at the Virginia Commonwealth University Department of Neurobiology and Anatomy Microscopy Facility). This work was also supported by an American Heart Association/Mid-Atlantic Affiliate Postdoctoral Fellowship (to D. P. Z.).

This article contains supplemental videos 1–4.
- GPA
- guinea pig atrial
- GPV
- guinea pig ventricular
- RFP
- red fluorescent protein
- NRVM
- neonatal rat ventricular myocite
- apFRET
- acceptor photobleach FRET
- M6PR
- mannose 6-phosphate receptor
- SR
- sarcoplastic reticulum
- ER
- endoplasmic reticulum
- Ang II
- angiotensin II
- WGA
- wheat germ agglutinin
- IKs
- slow delayed rectifier channel or current
- DsR
- DsRed
- INa
- Na current
- ICaL
- L-type Ca current
- IK1
- inward rectifier current
- IKr
- rapid delayed rectifier
- INCX
- Na/Ca exchanger current
- pAb
- polyclonal antibody.
REFERENCES
- 1. Sanguinetti M. C., Curran M. E., Zou A., Shen J., Spector P. S., Atkinson D. L., Keating M. T. (1996) Coassembly of KvLQT1 and minK (IsK) proteins to form cardiac IKs potassium channel. Nature 384, 80–83 [DOI] [PubMed] [Google Scholar]
- 2. Jost N., Virág L., Bitay M., Takács J., Lengyel C., Biliczki P., Nagy Z., Bogáts G., Lathrop D. A., Papp J. G., Varró A. (2005) Restricting excessive cardiac action potential and QT prolongation. A vital role for IKs in human ventricular muscle. Circulation 112, 1392–1399 [DOI] [PubMed] [Google Scholar]
- 3. Splawski I., Shen J., Timothy K. W., Lehmann M. H., Priori S., Robinson J. L., Moss A. J., Schwartz P. J., Towbin J. A., Vincent G. M., Keating M. T. (2000) Spectrum of mutations in long-QT syndrome genes KvLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation 102, 1178–1185 [DOI] [PubMed] [Google Scholar]
- 4. Borggrefe M., Wolpert C., Antzelevitch C., Veltmann C., Giustetto C., Gaita F., Schimpf R. (2005) Short QT syndrome genotype-phenotype correlations. J. Electrocardiol, 38, 75–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Olesen M. S., Bentzen B. H., Nielsen J. B., Steffensen A. B., David J.-P., Jabbari J., Jensen H. K., Haunsø S., Svendsen J. H., Schmitt N. (2012) Mutations in the potassium channel subunit KCNE1 are associated with early-onset familial atrial fibrillation. BMC Med. Genet. 13, 24–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jiang M., Xu X., Wang Y., Toyoda F., Liu X.-S., Zhang M., Robinson R. B., Tseng G.-N. (2009) Dynamic partnership between KCNQ1 and KCNE1 and influence on cardiac IKs current amplitude by KCNE2. J. Biol. Chem. 284, 16452–16462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang M., Wang Y.-H., Jiang M., Zankov D. P., Chowdhury S. R., Kasirajan V., Tseng G.-N. (2012) KCNE2 protein is more abundant in ventricles than in atria and can accelerate hERG protein degradation in a phosphorylation-dependent manner. Am. J. Physiol. Heart Circ. Physiol. 302, H910–H922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Delpón E., Cordeiro J. M., Núñez L., Thomsen P. E., Guerchicoff A., Pollevick G. D., Wu Y., Kanters J. K., Larsen C. T., Hofman-Bang J., Burashnikov E., Christiansen M., Antzelevitch C. (2008) Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome. Circ. Arrhythm. Electrophysiol. 1, 209–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Manderfield L. J., George A. L., Jr. (2008) KCNE4 can co-associate with the IKs (KCNQ1-KCNE1) channel complex. FEBS J. 275, 1336–1349 [DOI] [PubMed] [Google Scholar]
- 10. Bendahhou S., Marionneau C., Haurogne K., Larroque M.-M., Derand R., Szuts V., Escande D., Demolombe S., Barhanin J. (2005) In vitro molecular interactions and distribution of KCNE family with KCNQ1 in the human heart. Cardiovasc. Res. 67, 529–538 [DOI] [PubMed] [Google Scholar]
- 11. Nicolas C. S., Park K.-H., El Harchi A., Camonis J., Kass R. S., Escande D., Mérot J., Loussouarn G., Le Bouffant F., Baró I. (2008) IKs response to protein kinase A-dependent KCNQ1 phosphorylation requires direct interaction with microtubules. Cardiovasc. Res. 79, 427–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ehrlich J. R., Pourrier M., Weerapura M., Ethier N., Marmabachi A. M., Hébert T. E., Nattel S. (2004) KvLQT1 modulates the distribution and biophysical properties of HERG. A novel α-subunit interaction between delayed rectifier currents. J. Biol. Chem. 279, 1233–1241 [DOI] [PubMed] [Google Scholar]
- 13. Melnyk P., Ehrlich J. R., Pourrier M., Villeneuve L., Cha T. J., Nattel S. (2005) Comparison of ion channel distribution and expression in cardiomyocytes of canine pulmonary veins versus left atrium. Cardiovasc. Res. 65, 104–116 [DOI] [PubMed] [Google Scholar]
- 14. Rasmussen H. B., Moller M., Knaus H.-G., Jensen B. S., Olesen S.-P., Jorgensen N. K. (2003) Subcellular localization of the delayed rectifier K+ channels KCNQ1 and ERG1 in the rat heart. Am. J. Physiol. Heart Circ. Physiol. 286, H1300–H1309 [DOI] [PubMed] [Google Scholar]
- 15. Wu D.-M., Jiang M., Zhang M., Liu X.-S., Korolkova Y. V., Tseng G.-N. (2006) KCNE2 is colocalized with KCNQ1 and KCNE1 in cardiac myocytes and may function as a negative modulator of IKs current amplitude in the heart. Heart Rhythm 3, 1469–1480 [DOI] [PubMed] [Google Scholar]
- 16. Furukawa T., Ono Y., Tsuchiya H., Katayama Y., Bang M.-L., Labeit D., Labeit S., Inagaki N., Gregorio C. C. (2001) Specific interaction of the potassium channel β-subunit minK with the sarcomeric protein T-cap suggests a T-tubule-myofibril linking system. J. Mol. Biol. 313, 775–784 [DOI] [PubMed] [Google Scholar]
- 17. Mashanov G. I., Nobles M., Harmer S. C., Molloy J. E., Tinker A. (2010) Direct observation of individual KCNQ1 potassium channels reveals their distinctive diffusive behavior. J. Biol. Chem. 285, 3664–3675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ellingsen O., Davidoff A. J., Prasad S. K., Berger H.-J., Springhorn J. P., Marsh J. D., Kelly R. A., Smith T. W. (1993) Adult rat ventricular myocytes cultured in defined medium. Phenotype and electromechanical function. Am. J. Physiol. 265, H747–H754 [DOI] [PubMed] [Google Scholar]
- 19. Qu J., Barbuti A., Protas L., Santoro B., Cohen I. S., Robinson R. B. (2001) HCN2 overexpression in newborn and adult ventricular myocytes. Distinct effects on gating and excitability. Circ. Res. 89, E8–14 [DOI] [PubMed] [Google Scholar]
- 20. O'Rourke B., Kass D. A., Tomaselli G. F., Kääb S., Tunin R., Marbán E. (1999) Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure, I. Experimental studies. Circ. Res. 84, 562–570 [DOI] [PubMed] [Google Scholar]
- 21. Balse E., Steele D. F., Abriel H., Coulombe A., Fedida D., Hatem S. N. (2012) Dynamic of ion channel expression at the plasma membrane of cardiomyocytes. Physiol. Rev. 92, 1317–1358 [DOI] [PubMed] [Google Scholar]
- 22. Simpson P., Savion S. (1982) Differentiation of rat myocytes in single cell cultures with and without proliferating nonmyocardial cells. Cross-striations, ultrastructure, and chronotropic response to isoproterenol. Circ. Res. 50, 101–116 [DOI] [PubMed] [Google Scholar]
- 23. Kenworthy A. K. (2001) Imaging protein-protein interactions using fluorescence resonance energy transfer microscopy. Methods 24, 289–296 [DOI] [PubMed] [Google Scholar]
- 24. Haitin Y., Wiener R., Shaham D., Peretz A., Cohen E. B., Shamgar L., Pongs O., Hirsch J. A., Attali B. (2009) Intracellular domains interactions and gating motions of IKs potassium channel subunits. EMBO J. 28, 1994–2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Subramanian K., Meyer T. (1997) Calcium-induced restructuring of nuclear envelope and endoplasmic reticulum calcium stores. Cell 89, 963–971 [DOI] [PubMed] [Google Scholar]
- 26. Hunyady L., Catt K. J. (2006) Pleiotropic AT1 receptor signaling pathways mediating physiological and pathological actions of angiotensin II. Mol. Endocrinol. 20, 953–970 [DOI] [PubMed] [Google Scholar]
- 27. Friedman J. R., Voeltz G. K. (2011) The ER in 3D. A multifunctional dynamic membrane network. Trends Cell Biol. 21, 709–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Michalak M., Opas M. (2009) Endoplasmic and sarcoplasmic reticulum in the heart. Trends Cell Biol. 19, 253–259 [DOI] [PubMed] [Google Scholar]
- 29. Seebohm G., Strutz-Seebohm N., Birkin R., Dell G., Bucci C., Spinosa M. R., Baltaev R., Mack A. F., Korniychuk G., Choudhury A., Marks D., Pagano R. E., Attali B., Pfeufer A., Kass R. S., Sanguinetti M. C., Tavare J. M., Lang F. (2007) Regulation of endocytic recycling of KCNQ1/KCNE1 potassium channels. Circ. Res. 100, 686–692 [DOI] [PubMed] [Google Scholar]
- 30. Tohse N. (1990) Calcium-sensitive delayed rectifier potassium current in guinea pig ventricular cells. Am. J. Physiol. 258, H1200–H1207 [DOI] [PubMed] [Google Scholar]
- 31. Shamgar L., Ma L., Schmitt N., Haitin Y., Peretz A., Wiener R., Hirsch J., Pongs O., Attali B. (2006) Calmodulin is essential for cardiac IKs channel gating and assembly. Impaired function in long-QT mutations. Circ. Res. 98, 1055–1063 [DOI] [PubMed] [Google Scholar]
- 32. Nguyen N., Kozer-Gorevich N., Gilddon B. L., Smolka A. J., Clayton A. H., Gleeson P. A., van Driel I. R. (2013) Independent trafficking of the KCNQ1 K+ channel and H+-K+-ATPase in gastric parietal cells from mice. Am. J. Physiol. Gastrointest. Liver Physiol. 304, G157–G166 [DOI] [PubMed] [Google Scholar]
- 33. McDonald T. V., Yu Z., Ming Z., Palma E., Meyers M. B., Wang K. W., Goldstein S. A., Fishman G. I. (1997) A minK-HERG complex regulates the cardiac potassium current I(Kr). Nature 388, 289–292 [DOI] [PubMed] [Google Scholar]
- 34. Organ-Darling L. E., Vernon A. N., Giovanniello J. R., Lu Y., Moshal K., Roder K., Li W., Koren G. (2013) Interactions between hERG and KCNQ1 α-subunits are mediated by their COOH termini and modulated by cAMP. Am. J. Physiol. Heart Circ. Physiol. 304, H589–H599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Abraham R. L., Yang T., Blair M., Roden D. M., Darbar D. (2010) Augmented potassium current is a shared phenotype for two genetic defects associated with familial atrial fibrillation. J. Mol. Cell Cardiol. 48, 181–190 [DOI] [PMC free article] [PubMed] [Google Scholar]