

Background: The cyclin-dependent kinase inhibitor p16INK4a is also a modulator of gene expression through an unknown mechanism.
Results: p16INK4a-CDK4 forms a heterocomplex with Sp1, which induces the expression and UV-dependent up-regulation of miR-141 and miR-146b-5p.
Conclusion: p16INK4a-CDK4 complex has transcriptional activity through interaction with the transcription factor Sp1.
Significance: The microRNAs are novel effectors of the p16INK4a-CDK4 complex.
Keywords: Protein Complexes, Protein-DNA Interaction, shRNA, Signaling, Tumor suppressor gene
Abstract
p16INK4a is a tumor suppressor protein involved in several stress-related cellular responses, including apoptosis. Recent lines of evidence indicate that p16INK4a is also a modulator of gene expression. However, the molecular mechanisms underlying this novel function are still obscure. Here, we present clear evidence that p16INK4a modulates the levels of various microRNAs, with marked positive effect on miR-141 and miR-146b-5p. This effect is mediated through the formation of the p16-CDK4-Sp1 heterocomplex, which binds to Sp1 consensus-binding motifs present in the promoters of miR-141 and miR-146b-5p, and it enables their transcription. In addition, we have shown that p16INK4a interacts with Sp1 through the fourth ankyrin repeat, which is crucial for Sp1 binding to the miR-141 and miR-146b-5p promoters and their transcriptional activation. The physiological importance of this association was revealed by the inability of cancer-related p16INK4a mutants to interact with Sp1. Moreover, we have shown p16-CDK4-Sp1-dependent up-regulation of miR-141 and miR-146b-5p following UV light-induced DNA damage and the role of these two microRNAs in mediating p16-related induction of apoptosis in response to this genotoxic stress. Together, these results indicate that p16INK4a associates with CDK4 not only to inhibit the cell cycle but also to enable the transcription of two important onco-microRNAs, which act as downstream effectors.
Introduction
p16INK4a (hereafter referred to as p16) is a cyclin-dependent kinase (CDK)2 inhibitor encoded by the CDKN2A gene (1–3). In response to various stresses, p16 binds to CDK4/6 and abrogates their binding to D-type cyclins. This inhibits the CDK4/6-mediated phosphorylation of the pRB protein, which prevents the E2F-mediated transcription of genes required for DNA synthesis. This causes the arrest of the cell cycle in G1 phase (4, 5). p16 is also involved in the cellular response to DNA damage, which leads to cell cycle arrest (6), senescence (7, 8), and/or apoptosis (9, 10). CDKN2A is an important tumor suppressor gene, which is frequently inactivated by point mutations, promoter methylation, or deletion in various types of human cancer (11, 12). Additionally, several lines of evidence indicate that p16 is overexpressed at both protein and mRNA levels in certain tumors, and this has been shown to be associated with poor prognosis (13, 14). Furthermore, it became clear that p16 interacts with several proteins (15, 16), and it also regulates the expression of different genes involved in various cellular processes (17, 18). We have recently shown that p16 negatively regulates the expression of AUF1 through activating the turnover of its mRNA (18). However, the molecular mechanism(s) underlying this process were not delineated. The most appealing hypothesis is the possible implication of microRNAs (miRNAs) in this p16-dependent post-transcriptional regulation. As a case in point, Chien et al. (19) have recently shown that p16 overexpression inhibits CDK1 expression through induction of miR-410 and miR-650 in cancer cells.
miRNAs, a new class of endogenous, noncoding, and single-stranded RNAs, provide a novel layer of regulation of gene expression. These small RNAs regulate the expression of protein coding genes through binding to the 3′-untranslated regions of their mRNAs in a sequence-specific manner. Therefore, miRNAs regulate a wide range of fundamental biological processes such as cell proliferation, differentiation, senescence, and apoptosis (20, 21). Alterations in miRNA expressions have been linked to various human diseases, including cancer, where they act as tumor suppressors or oncogenes (21–23).
In this study, we have shown that p16 modulates the expression of several miRNAs, and positively regulates the transcription of miR-141 and miR-146b-5p through physical interaction with CDK4 and the transcription factor Sp1. We have also shown that these two miRNAs are up-regulated in response to UV light-induced DNA damage in a p16/CDK4/Sp1-dependent manner, and act as p16 effectors during UV light-induced apoptosis.
EXPERIMENTAL PROCEDURES
Cell Lines, Cell Culture, and Chemicals
U2OS, EH1, and EH2 cell lines were the generous gift from Dr. G. Peters. Although U2OS cells are p16-deficient, EH1 and EH2 are U2OS derivatives that express ectopic p16 under the control of an IPTG-inducible promoter. However, there is low p16 expression in these cells even in absence of IPTG, with higher levels in EH1 than in EH2 (24). HFSN1 (primary normal human skin fibroblast) was used as described previously (27). These cells were routinely cultured in DMEM supplemented with 10% FCS. IPTG was purchased from Sigma.
RNA Purification, RT-PCR, and Quantitative RT-PCR
Total RNA was purified using TRI Reagent® (Sigma) according to the manufacturer's instructions and was treated with RNase-free DNase before cDNA synthesis using the miScript® II RT kit (Qiagen) for both miRNAs and mRNAs. For RT-PCR, cDNA was amplified using the Platinum® TaqDNA polymerase (Invitrogen). After electrophoresis on ethidium bromide-stained 2% agarose gels, the intensity of the PCR products was determined with the Quantity One program (Bio-Rad) and was normalized against GAPDH. For quantitative RT-PCR (qRT-PCR), the RT2 real time SYBR® Green qPCR master mix (Qiagen) was used, and the amplifications were performed utilizing the Bio-Rad iQ5 multicolor real time PCR detection system. The melting curve data were collected to check PCR specificity, and the amount of PCR products was measured by threshold cycle (Ct) values, and the relative ratio of specific genes to GAPDH for each sample was then calculated. The respective primers are depicted in Table 1.
TABLE 1.
Primers used for quantitative RT-PCR
F indicates forward, and R indicates reverse.
| Primer | Sequence 5′ → 3′ |
|---|---|
| CDKN2A F | CAACGCACCGAATAGTTACG |
| CDKN2A R | CAGCTCCTCAGCCAGGTC |
| miR-141 F | CCTGGGTCCATCTTCCAGTA |
| miR-141 R | ACCCGGGAGCCATCTTTAC |
| miR-146b-5p F | CCTGGCACTGAGAACTGAAT |
| miR-146b-5p R | GCACCAGAACTGAGTCCACA |
| miR-365 F | ATAGGATCCTGAGGTCCCTTTCGTG |
| miR-365 R | GCGAAGCTTAAAAACAGCGGAAGAGTTTGG |
| miR-1243 F | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGCAC |
| miR-1243 R | ACACTCCAGCTGGGAACTGGATCAATTATA |
| miR-27b F | TGCAGAGCTTAGCTGATTGG |
| miR-27b R | CCTTCTCTTCAGGTGCAGAAC |
| miR-1255b F | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGAACCACTT |
| miR-1255b R | ACACTCCAGCTGGGCGGATGAGCAAAGAAA |
| miR-324-5p F | CTGACTATGCCTCCCCGC |
| miR-324-5p R | GACTACAACCCCCAGCAGC |
| miR-1246 F | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGCCT |
| miR-1246 R | ACACTCCAGCTGGGAATGGATTTTTGG |
| miR-298 F | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGTGG |
| miR-298 R | ACACTCCAGCTGGGAGCAGAAGCAGGGAGGTT |
| miR-194 F | ATGGTGTTATCAAGTGTAACAGCA |
| miR-194 R | TTGGTAACCATCAAAAGTAACAGC |
| Sp1 F | TTGAAAAAGGAGTTGGTGGC |
| Sp1 R | TGCTGGTTCTGTAAGTTGGG |
| SLC2A3 F | GTCAACCTGTTGGCTGTCAC |
| SLC2A3 R | GGAAGGATGGTAAAACCCAG |
| CEBPδ F | CTGCGAGAGAAGCTAAACGTG |
| CEBPδ R | CTTAGCTGCATCAACAGGAG |
| GAPDH F | GAGTCCACTGGCGTCTTC |
| GAPDH R | GGGGTGCTAAGCAGTTGGT |
MicroRNA Array
Total RNA was prepared from HFSN1C and HFSN1p16sh using Qiagen RNeasy mini kit (Qiagen). One microgram of total RNA from samples and reference was labeled with Hy3TM and Hy5TM fluorescent label, respectively, using the miRCURYTM LNA array power labeling kit (Exiqon, Denmark) following the manufacturer's instructions. The Hy3TM-labeled samples and a Hy5TM-labeled reference RNA sample were mixed pairwise and hybridized to the miRCURYTM LNA array version 11.0 (Exiqon, Denmark), which contains capture probes targeting all miRNAs for human, mouse, or rat registered in the miRBASE version 13.0 at the Sanger Institute. The miRCURYTM LNA array microarray slides were scanned using the Agilent G2565BA microarray scanner system (Agilent Technologies, Inc.), and the image analysis was carried out using the ImaGene 8.0 software (BioDiscovery, Inc.). The quantified signals were background corrected and normalized using the global Lowess (locally weighted scatterplot smoothing) regression algorithm.
Transfection-CDKN2A
shRNA expressed in pRNAT-U6/Neo vector (GenScript Corp.), Sp1-shRNA (SABiosciences), Sp1-ORF (Origene), CDK4-shRNA (SABiosciences), and their control plasmids were used to carry out transfection using human dermal fibroblast nucleofector kit (Amaxa Biosystems) following the protocol recommended by the manufacturer.
pLKO.1-miRZip146b-5p, pLKO.1-miRZip141, pCDH-pre-miR-141, and pCDH-pre-miR-146b-5p plasmids were purchased from System Biosciences (SBI) and used to prepare the lentiviral supernatant. Media were removed from the target cells and replaced with lentiviral supernatant and incubated for 6 h. Transduced cells were selected after 48 h with puromycin or G418. Human wild-type and mutant p16 (16) were introduced into U2OS cells with Lipofectamine 2000 following the manufacturer's instructions (Invitrogen).
Cloning of Human miR-141 and miR-146b-5p Promoter Regions
miR-141 and miR-200c primary RNA sequences are present 90 bp apart in chromosome XII. Examination of spliced expressed sequence tags matching this region revealed that the two microRNAs are clustered in the only intron of six overlapping expressed sequence tags with two exons, BX094730, BM694730, AA863389, AA863395, BQ370636, and AI969882. One expressed sequence tag, AI695443, has an additional small 5′ exon, and the microRNAs are located in its second intron. The first base of the cDNA, AI695443, was assumed as the putative transcription start site. The 5′ sequence of miR-141, 995-bp DNA fragment spanning from −930 to +65, was amplified from human genomic DNA by PCR. The forward primer (ACTGGTCCCGGGCGTCACAGGCATTCACAGTC) and reverse primer (GATCCTAAGCTTTCTTCCTCCTTCCTTCTCCG) were tagged with SmaI and HindIII sites, respectively, to facilitate directional cloning.
Human miR-146b is processed from a noncoding and single exon transcript of a gene present on chromosome 10 (26). The presence of a transcription start site 700 bp upstream of mature miR-146b was previously reported without further validation (26). miR-146b exon sequence, revealing the location of transcription start site or promoter elements, is not available in the public databases. Therefore, we searched for the presence of promoters in 3000 bp of the 5′-flanking sequences of miR-146b with a neural network promoter prediction tool. This software predicted a promoter with a transcription start site at 832 bp upstream of pre-miR-146b and a poorly conserved TATA box further upstream of the transcription start site (GTGGGGGCCTTATTTAAGGAGCACCGGCTGAGAACATAGTTGCAGATCCA, the TATA box and transcription start site are underlined). The 919-bp promoter fragment encompassing the region from −856 to +63 was amplified by PCR from human genomic DNA using the forward primer (TATGCACCCGGGCCCTATCCCCTGTTTGCTGG) and reverse primer (CGTACTAAGCTTGAGAGAGGGACAGCCACTTAC), both tagged with restriction sites SmaI and HindIII, respectively.
Luciferase Assay
To evaluate the function of the 5′-flanking sequences of miR-141 and miR-146b, the candidate sequences of these promoters were amplified from genomic DNA isolated from human peripheral blood cells and cloned into the pGL3 vector (Promega). 80% confluent EH1 and U20S cells were subcultured in 24-well plates. Transfection of pGL3 constructs (800 ng) together with Renilla luciferase phRLTK vector (200 ng) was performed with Lipofectamine 2000. After 24 h, cell extracts were assayed for firefly and Renilla luciferases by Dual-Luciferase assay kit (Promega). Luciferase activity was measured using bio-imaging apparatus and software (BD Biosciences). For each construct, assays were done in triplicate in three independent experiments. To normalize difference in the transfection efficiencies between wells, firefly luciferase activity of each well was divided with Renilla luciferase activity. The mean ± S.E. were calculated from three wells for each promoter activity.
Northern Blot
Northern blot analysis was performed using the highly sensitive miRNA Northern blot assay kit as recommended by the manufacturer (Signosis, Inc.). Briefly, total RNA was prepared using the RNeasy mini kit (Qiagen), and 5 μg of RNA were separated using precast 15% TBE/urea gel (Signosis). After transfer, nylon membranes (Signosis) were UV-cross-linked and hybridized with DNA oligonucleotides complementary to miRNA or U6 that had been end-labeled with biotin (Signosis). Images were acquired with a CCD camera (LAS 4000, GE Healthcare).
Analysis of mRNA Stability
Cells were challenged with 5 μg/ml actinomycin D for various periods of time (0–6 h), and then total RNA was purified and assessed using RT-PCR.
Chromatin Immunoprecipitation (ChIP) Assay
Cells (106) were treated with 2% formaldehyde for 10 min at room temperature to cross-link the transcription factor to DNA. The cross-linking was terminated by addition of glycine (0.125 m). After washing with PBS, cells were collected and resuspended in SDS lysis buffer (1% SDS, 10 mm EDTA, 50 mm Tris, pH 8.1) with protease inhibitors. The sonicated lysate was processed using the ChIP assay kit following the manufacturer's instructions (USB Bio-system). ChIP experiments were performed using antibodies against Sp1 (Cell Signaling Technology), p16 (BD Biosciences), and CDK4 (C-22). Anti-IgG antibody was used as control. In addition, sonicated chromatin with no antibody (mock) served as control for nonspecific interactions between chromatin and the agarose beads. Input is the supernatant of the negative control (mock) and served as a positive PCR control, which represents the starting genomic DNA that was used for normalization. Immunoprecipitated chromatin was analyzed by qPCR using GAPDH as unlinked locus control. The sequences of the primers used were as follows: miR-141, 5′-GGCGTCACAGGCATTCACAGTC-3′ (forward) and 5′-TCTTCCTCCTTCCTTCTCCG-3′ (reverse); miR-146b-5p, 5′-CCCCTATCCCCTGTTTGCTGG-3′ (forward) and 5′-AGAGAGAGGGACAGCCACTTA-3′ (reverse); GAPDH, 5′-TACTAGCGGTTTTACGGGCG-3′ (forward) and 5′-TACTAGCGGTTTTACGGGCG-3′ (reverse); FGF21, 5′-CTGTAGCTCCTGCCAAATGG-3′ (forward) and 5′-GTGGTTTAGAATTGGTGCCAG-3′ (reverse); and IHPK2, 5′- GTTCGAAGTAGCGTGGGAAG-3′ (forward) and 5′-CCTGCCTGTCGGCATCTAAC-3′ (reverse).
Electrophoretic Mobility Shift Assay (EMSA)
Complementary oligonucleotides representing the wild-type Sp1-binding site on the miR-141 promoter (5′-GGCCCCTGGGAGAGGGTGGGAGGCCTAGAGGAG-3′), the mutated Sp1-binding site (5′-GGCCCCTCTATCGAATCCTTACCATTCGAGGAG-3′),the wild-type Sp1-binding site on the miR-146b-5p promoter (5′-GAGGCGGCTGGGGCGGGGGTGGGGGCCT-3′), and the mutated Sp1-binding site (5′-GATTAAACTTACAGTACGGACTCTACCT-3′) as well as Sp1-binding site 1 on the miR-365 promoter (5′-GCAAGAAAAATATTTTTCACCTCCTGTATTCCCC-3′), Sp1-binding site 2 (5′-GGGGAGGACCAGGGCTCTGAATACTTGC-3′), and Sp1-binding site 3 (5′-CATCCCTAACGGGCGTGTCTTAGCTGAAG-3′) were designed and purified by HPLC. Nuclear extracts were prepared from ∼8 × 107 cells. For EMSA, nuclear proteins (3 μg) or pure proteins were preincubated for 10 min on ice with 1 μg of poly(dI-dC) in a binding buffer (4% Ficoll, 20 mm HEPES, pH 7.9, 1 mm EDTA, 1 mm DTT, 50 mm KCl). Nuclear proteins or pure proteins were then incubated with biotin-labeled oligonucleotide probes (20 fmol) for 20 min at room temperature and then analyzed on 6% polyacrylamide gels and electroblotted onto a positively charged nylon membrane (Pierce). The membrane was UV-cross-linked, and the biotin end-labeled probe was detected with streptavidin-horseradish peroxidase using a luminal enhancer substrate (Lightshift Chemiluminescent EMSA kit; Pierce). Fifty-fold excess of unlabeled oligonucleotides were used as cold competitor to test for the specificity of the DNA protein binding. Light emission was captured using a CCD camera (LAS 4000, GE Healthcare). In supershift assays, the protein was preincubated with 1 μg of the antibody to p16 (BD Biosciences), Sp1 (Cell Signaling Technology), or CDK4 (C-22) before addition of the probe.
Cellular Lysate Preparation and Immunoblotting
This has been performed as described previously (27). Antibodies directed against GAPDH (FL-335), CDK4 (C-22), proliferating cell nuclear antigen (PC-10), pRB (F-8), and normal mouse IgG were purchased from Santa Cruz Biotechnology (Santa Cruz, CA); Sp1, cleaved PARP (Asp-214), P-pRB (Ser-780), and cleaved caspase-3 (Asp-175) were purchased from Cell Signaling (Danvers, MA); p16 was from BD Biosciences; P-pRB (Thr-356) was from Abnova (Taipei, Taiwan).
Immunoprecipitation
Cell lysates were prepared using RIPA buffer containing protease inhibitors and then centrifuged at 14,000 rpm at 4 °C. Three hundred micrograms of protein extracts were pre-cleaned with 20 μl of protein A/G-agarose for 2 h at 4 °C and then incubated for 2 h with 2 μg of specific antibody at 4 °C. Subsequently, 50 μl of A/G-agarose was added for 2 h at 4 °C. After centrifugation, the pellet was washed with the RIPA buffer, and the proteins were recovered by boiling in Laemmli buffer.
Ki-67 Immunostaining
Ki-67 level was assessed by immunostaining as described previously (28). Cells were fixed in 1:1 acetone/methanol, and a standard indirect immunoperoxidase procedure was applied using Ki-67 antibody (Abcam), followed by peroxidase-conjugated anti-rabbit antibody (Dako). Sites of antibody binding were visualized by the deposition of brown polymer of 3,3′-diaminobenzidine chromogen (Novocastra Laboratories Ltd.). The percentage of Ki-67-labeled cells (labeling index) was determined for at least 500 cells per data point and expressed as mean ± S.E. of triplicate determinations. All labeled cells were counted positive.
Cell Proliferation Assay
Exponentially growing HFSN1 cells (2 × 104) expressing either control shRNA (HFSN1C) or CDKN2A-shRNA (HFSN1p16sh) were seeded in E-16 plates and reincubated for 48 h. The proliferation rate was measured using the RTCA-DP xCELLigence system (Roche Applied Science). Cell index represents cell status based on the measured electrical impendence change divided by a background value.
Flow Cytometry
Cells were harvested and resuspended in 1 ml of PBS before being fixed by dropwise addition of 3 ml of 100% methanol. Fixed cells were centrifuged, resuspended in 50 μl of RNase (1 mg/ml), and incubated for 30 min at room temperature, followed by addition of 1 ml of 0.1 mg/ml of propidium iodide. Cells were analyzed for DNA content by flow cytometry (BD Biosciences). The percentage of cells in various cell cycle phases was determined by using CellQuest software (BD Biosciences).
UV Irradiation
The medium was removed, and the monolayers in dishes were covered with PBS and exposed to a germicidal UV lamp (254 nm) at a fixed distance. The UV dosimetry was performed using an ultraviolet meter (Spectronics Corp., New York).
Apoptosis Analysis by Annexin V/Flow Cytometry
Cells were either not treated or challenged with UV light (10 Jm−2), then harvested after 72 h, centrifuged, and stained by propidium iodide and Alexa Fluor 488 annexin V (Molecular Probes, Eugene, OR) as described previously (18).
Quantification of Protein Expression Level
The expression levels of the immunoblotted proteins were measured using the densitometer (GS-800 calibrated densitometer, Bio-Rad) as described previously (27).
Statistical Analysis
Statistical analysis was performed by Student's t test, and p values of 0.05 and less were considered as statistically significant.
RESULTS
p16 Modulates the Expression of Several MicroRNAs
In an attempt to elucidate the molecular mechanisms that underlie p16-dependent post-transcriptional modulation of gene expression, we have first investigated the possible implication of p16 in regulating the expression of microRNAs. Therefore, p16 was down-regulated by shRNA in human HFSN1 skin primary fibroblast cells, and scrambled shRNA sequence was used as control. The utilized shRNA targeted specifically CDKN2A and not the ARF gene as has been previously shown (29). The generated cells (HFSN1p16sh and HFSN1C, respectively) proliferated similarly with slightly higher growth rate of HFSNp16sh than HFSN1C cells (Fig. 1A), and they both exhibited similar cell cycle distribution and DNA content (Fig. 1B). To further evaluate the effect on cell proliferation, we assessed the levels of the two main proliferation markers, Ki-67 and proliferating cell nuclear antigen, and have shown no effect on the level of these two proteins (Fig. 1, C and D). Furthermore, we tested the effect of p16 down-regulation on pRB expression and activation. To this end, whole cell lysates were prepared from HFSN1p16sh and HFSN1C and used for immunoblotting analysis using specific antibodies and anti-GAPDH as internal control. Fig. 1D shows that although the level of p16 decreased 3.6-fold in HFSN1p16sh cells, the levels of pRB and phospho-pRB (Ser-780 and Thr-365) were not affected. These results show that p16 down-regulation in these cells did not affect their proliferation nor activate pRB.
FIGURE 1.

p16 down-regulation does not affect cell proliferation in primary skin fibroblasts. A, HFSN1 cells expressing either control-shRNA (HFSN1C) or CDKN2A-shRNA (HFSN1p16sh) were seeded in E-16 plates and reincubated for 48 h. The proliferation rate was measured using the RTCA-DP xCelligence system. B, cell cycle analysis using flow cytometry. C, immunostaining using Ki-67 antibody. Scale bars, 50 μm. Labeling index for Ki-67 staining was determined for at least 500 cells per data point and expressed as mean ± S.D. of triplicate determinations. D, whole cell lysates were prepared from the indicated cells and used for immunoblotting using the indicated antibodies. The numbers below the bands indicate protein expression relative to total pRB and GAPDH.
Next, total RNA was prepared from HFSN1C and HFSN1p16sh cells and used to perform transcriptome-wide analysis of microRNAs using miRCURY LNA arrays (Fig. 2A). The difference in miRNA expression was considered significant only when the variation was ≥1.5-fold. Interestingly, out of 1266 miRNAs contained on the expression matrix, down-regulation of p16 modulated the expression of 23 miRNAs, with four miRNAs (17.4%) up-regulated and 19 miRNAs (82.6%) down-regulated (Fig. 2A). This implicates p16 in the regulation of several miRNAs. To validate these results, we confirmed the differential expression of nine different miRNAs by qRT-PCR. Fig. 2, B and C, shows that p16 down-regulation increased more than 1.5-fold the expression of three miRNAs (miR-27b, miR-1255b, and miR-324–5p) but decreased the expression of six other miRNAs (miR-1243, miR-1246, miR-298, miR-194, miR-141, and miR-146b-5p), which mirrors the microarray results.
FIGURE 2.

p16-dependent differential expression of miRNAs. A, total RNA samples were prepared from the indicated cells and were used to perform microRNA array. B and C, qRT-PCR using primers corresponding to the indicated genes. The relative transcript values were normalized to GAPDH transcript levels. Error bars represent means ± S.D. D, total RNA was extracted from the indicated cells utilizing the RNeasy mini kit (Qiagen), and then was used for Northern blot analysis.
p16 Modulates the Expression of miR-141 and miR-146b-5p
Among the 23 differentially expressed miRNAs, the levels of miR-141 and miR-146b-5p were significantly reduced in the HFSN1p16sh as compared with their levels in the control HFSN1C counterparts (Fig. 2A). Indeed, qRT-PCR showed a 3- and 5-fold decrease in their levels in p16-deficient cells as compared with control cells, respectively (Fig. 2C). To confirm p16 implication in modulating the expression of miR-141 and miR-146b-5p, we made use of two other experimental systems as follows: p16-defective osteosarcoma U2OS cell line in which we expressed either CDKN2A-ORF (U2OSp16ORF) or empty vector (U2OS-C) (EH1 and EH2 cells were used as control), and IPTG-dependent up-regulation of p16 in EH2 cells. Fig. 2C shows clear p16-dependent expression of both miR-141 and miR-146b-5p. Indeed, ectopic expression of p16 in U2OS cells strongly increased miR-141 and miR-146b-5p levels (Fig. 2C). A similar increase was obtained in EH1 cells and to a lesser extent in EH2 cells as compared with U2OS (Fig. 2C). Interestingly, treatment of EH2 with IPTG (1 mm) further increased the expression of miR-141 and miR-146b-5p to levels similar to those observed in U2OSp16ORF and EH1 cells (Fig. 2C). These results show a direct correlation between the level of p16 and the expression of miR-141 and miR-146b-5p.
To show that the levels of the mature forms of these miRNAs were also affected, total RNA was prepared, and a highly sensitive miRNA Northern blot assay was performed. Fig. 2D shows that the levels of the mature forms of both miR-141 and miR-146b-5p decreased in p16-defective cells (HFSN1p16sh and U2OS-C) as compared with their respective controls (HFSN1C and U2OSp16ORF).
To show the lack of cell cycle effect on p16-dependent modulation of miR-141 and miR-146b-5p expression, the levels of these two miRNAs were assessed in HFSN1C and HFSN1p16sh quiescent cells (Fig. 3, A and B). Fig. 3B shows a 4-fold decrease in the expression level of both miR-141 and miR-146b-5p in p16-deficient cells as compared with control cells. This indicates that p16 modulates the expression of these two miRNAs in a cell cycle-independent manner.
FIGURE 3.
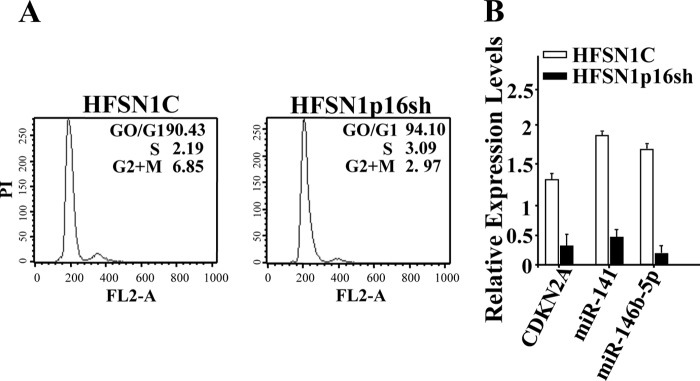
p16 modulates the expression of miR-141 and miR-146b-5p in a cell cycle-independent manner. A, confluent cells were starved for 24 h in serum-free media, and then the cell cycle was analyzed by flow cytometry. The numbers in the boxes indicate the proportion of cells in each phase of the cell cycle. PI, propidium iodide. B, total RNA was purified from the indicated cells and was amplified by qRT-PCR using specific primers for the indicated genes. Error bars represent means ± S.D.
p16 Positively Controls miR-141 and miR-146b-5p at the Transcriptional Level
To determine the mechanism whereby p16 positively regulates the expression of miR-141 and miR-146b-5p, we first assessed the half-lives of these miRNAs in HFSN1p16sh cells and their control counterparts using actinomycin D. The pre-miR-141 and pre-miR-146b-5p turnovers were not effected by the status of p16 (Fig. 4A), suggesting that p16 may control their expression at the transcriptional level. To investigate this possibility, we first determined the genomic regions encompassing the promoters of miR-141 and miR-146b-5p and amplified them from human genomic DNA by PCR. The putative promoter of miR-141 is a 995-bp DNA fragment spanning from −930 to +65, and the putative promoter of miR-146b-5p is a 919-bp DNA fragment encompassing the region from −856 to +63 (Fig. 4B). miR-141 and miR-146b-5p promoters were directionally cloned by insertion into SmaI and HindIII sites of the luciferase reporter pGL3 vector. p16-deficient (U2OS) and p16-proficient (EH1) cells were transfected with these vectors together with the Renilla luciferase phRLTK vector to normalize for variation in transfection efficiency, and the firefly and Renilla luciferase activities were measured 24 h post-transfection. We first examined the activity of these promoters in U2OS cells. The 5′-flanking sequence of miR-141 in the pGL3 vector (pGL3-pMR141) showed a 3-fold increase in luciferase activity compared with the activity of the promoter-less pGL3 vector (Fig. 4C). However, the promoter of the miR-146b-5p in the pGL3 vector (pGL3-pMR146b-5p) showed a 100-fold increase in luciferase activity, compared with the control vector (Fig. 4C). Interestingly, the miR-141 and miR-146b-5p promoters showed 8- and 3-fold greater luciferase activity in the p16-expressing EH1 cells as compared with the p16-defective U2OS cells, respectively (Fig. 4C). This suggests that p16 controls the expression of miR-141 and miR-146b-5p at the transcriptional level.
FIGURE 4.

p16 positively regulates miR-141 and miR-146b-5p at the transcriptional level. A, Cells were treated with actinomycin D for the indicated periods of time. Total RNA was extracted, and the remaining levels of miR-141 and miR-146b-5p were assessed by RT-PCR following electrophoresis on 2% ethidium bromide-stained agarose gels. B, schematic diagrams of miR-141 and miR-146b-5p genomic loci and their putative promoters. Chr, chromosome; EST, expressed sequence tag; TSS, transcription start site. C, miR-141 and miR-146b-5p promoters were inserted into SmaI/HindIII sites in the pGL3 luciferase reporter plasmid. The pGL3 vector was used to transfect U2OS and EH1 cells together with Renilla luciferase phRLTK vector to normalize for variation in transfection efficiency, and the firefly (FL) and Renilla (RL) luciferase activities were measured 24 h post-transfection using bio-imaging apparatus and software (BD Biosciences). Histogram shows the activity of luciferase under miR-141 and miR-146b-5p promoters. Data are presented as means ± S.E. of three independent experiments and each is measured in triplicate (*, p < 0.05).
Sp1 Binds miR-141 and miR-146b-5p Promoters in a p16-dependent Manner
Because p16 is not a DNA-binding protein, we hypothesized that it may modulate the transcription of miR-141 and miR-146b-5p indirectly through a transcription factor. Analysis of the miR-141 and miR-146b-5p promoters revealed the presence of several consensus binding sites for the Sp1 transcription factor (Fig. 5A). To test the possible p16-dependent regulation of miR-141 and miR-146b-5p through Sp1, we first down-regulated Sp1 by specific shRNA in HFSN1 cells (Sp1-shRNA); a scrambled sequence was used as control (HFSN1C) (Fig. 5B). Next, EMSA was performed to investigate the binding of Sp1 to its cognate sites on the miR-141 and miR-146b-5p promoters. To this end, biotin-labeled oligonucleotides were incubated with nuclear extracts prepared from p16- and Sp1-deficient cells as well as their control cells. Fig. 5C shows that upon electrophoresis the nuclear extracts from control cells enabled the shift of a single DNA-protein complex band. This band disappeared following preincubation with a 50-fold excess of cold probe used as competitor, which indicates specific binding to this sequence (Fig. 5C, lane 2). To further show this specificity, a mutated sequence of the Sp1-binding site was used, and no band shift was obtained with the same nuclear extract (Fig. 5C, lane 3). Furthermore, when extract from Sp1-deficient cells (HFSN1Sp1sh) was used, the intensity of the shifted band strongly decreased (Fig. 5C, lane 5). A similar result was obtained with nuclear extract from the p16-defective (HFSN1p16sh) cells (Fig. 5C, lane 4), despite the fact that the Sp1 level changed only marginally in p16-defective cells (Fig. 5D). Interestingly, it was possible to normalize the binding efficiency to the miR-146b-5p promoter by complementing HFSN1Sp1sh nuclear extract with 10 ng of pure Sp1 protein (Fig. 5E). Similar results were obtained for miR-141, indicating that Sp1 binds to its consensus binding sites present in the miR-141 and miR-146b-5p promoters and that this binding is affected by the status of p16.
FIGURE 5.

p16 positively regulates the expression of miR-141 and miR-146b-5p through the Sp1 transcription factor. A, nucleotide sequences of the miR-141 and miR-146b-5p promoters, −39 to +9 nucleotides and −93 to −26 nucleotides, respectively, relative to the putative transcription start site. Highlighted are the various overlapped Sp1-binding sites in the miR-141 and miR-146b-5p promoters. B and D, whole cell lysates were prepared from the indicated cells and used for immunoblotting using the indicated antibodies. C and E, EMSAs were performed using nuclear extracts from the indicated cells and biotin-labeled Sp1 consensus binding site of the miR-146b-5p promoter. Oligos, oligonucleotides.
To further confirm these important results, we performed ChIP assay to study the binding of Sp1 to the miR-141 and miR-146b-5p promoters and the effect of p16 on this binding in vivo. Chromatin was prepared from HFSN1p16sh and HFSN1C cells, and the Sp1-DNA complex was pulled down using anti-Sp1 antibody, and IgG was used as control. Subsequently, the miR-141 and miR-146b-5p promoters as well as the GAPDH gene (used as unlinked locus negative control) were amplified by PCR and qPCR. Fig. 6A (upper panel) shows a strong amplification of both promoters in control cells following immunoprecipitation with anti-Sp1 antibody but not with the IgG, showing the binding of Sp1 to these promoters in vivo. However, in p16-deficient cells, the level of the immunoprecipitated miR-141 and miR-146b-5p promoters decreased 3- and 4-fold, respectively (Fig. 6A, lower panel). This shows that the Sp1 binding to the miR-141 and miR-146b-5p promoters is p16-dependent in vivo as well.
FIGURE 6.

Sp1 binds the miR-141 and miR-146b-5p promoters and activates their transcription in a p16-dependent manner. A, ChIP assay. Chromatin was purified from HFSN1C and HFSN1p16sh, and then immunoprecipitated (IP) using anti-Sp1 antibody. miR-141 and miR-146b-5p promoters were amplified by PCR (upper panel) and qPCR (lower panel) using specific primers, and their abundances were plotted relative to the input, and GAPDH (unlinked locus) was used as negative control. These experiments were performed in triplicate (*, p < 0.05). B, ChIP assay. The promoters of the indicated genes were amplified using qPCR. C, total RNA was extracted from HFSN1 cells expressing either Sp1-shRNA (scrambled sequence was used as control) or Sp1-ORF (empty vector was used as control) and was utilized to assess the expression level of the indicated genes by qRT-PCR. Error bars represent means ± S.D.
To determine whether p16 influence is global or specific for some Sp1 target genes, the effect of p16 on the binding of Sp1 to the promoter of the FGF21 and IHPK2 genes, which are positively regulated by Sp1 (30) but are not p16 targets (18), was investigated by the ChIP assay. Fig. 6B shows that p16 down-regulation did not affect the binding of Sp1 to the promoters of these two genes (Fig. 6B). This indicates that not all Sp1 target genes are also under the control of p16.
Sp1 Positively Regulates the Expression of miR-141 and miR-146b-5p
After showing the binding of Sp1 to the miR-141 and miR-146b-5p promoters, we sought to investigate the effect of Sp1 on the transcription of these microRNAs. To this end, Sp1 was either down-regulated (specific shRNA, a scrambled sequence was used as control) or ectopically expressed in HFSN1 cells, and total RNA was prepared from these cells and used to assess the levels of both miRNAs by qRT-PCR. Fig. 6C shows that down-regulation of Sp1 decreased the levels of miR-141 and miR-146b-5p 14- and 10-fold, respectively. However, Sp1 up-regulation increased the expression of miR-141 and miR-146b-5p (Fig. 6C). This provides clear evidence that Sp1 is involved in the transcription of miR-141 and miR-146b-5p.
p16, Sp1, and CDK4 Form a Heterocomplex That Binds the miR-141 and miR-146b-5p Promoters Both in Vitro and in Vivo
After showing the p16-dependent binding of Sp1 to the miR-141 and miR-146b-5p promoters, and the role of Sp1 in the expression of these two miRNAs, we sought to investigate the possible physical interaction between the p16 and Sp1 proteins. To this end, reciprocal immunoprecipitation using anti-p16 or anti-Sp1 antibodies was carried out on whole cell extracts prepared from HFSN1 cells. The co-immunoprecipitated proteins were identified by immunoblotting using specific antibodies. Fig. 7A shows that Sp1 was pulled down with anti-p16 antibody. CDK4, a major target and partner of p16, was also among the p16-immunoprecipitated proteins (Fig. 7A). Likewise, p16 was also pulled down with anti-Sp1 antibody from HFSN1 protein extract (Fig. 7A).
FIGURE 7.

p16, Sp1, and CDK4 proteins form a heterocomplex that binds the miR-141 and miR-146b-5p promoters. A, whole cell extracts were prepared from HFSN1 cells and immunoprecipitated (IP) with anti-p16, anti-Sp1, or anti-CDK4 antibodies (mouse IgG was used as control), and immunoblotting was performed using the indicated antibodies. B, EMSA. Supershift assay was performed using nuclear extracts from the indicated cells that were preincubated with the indicated antibodies (1 μg) for an hour before adding the biotin-labeled oligonucleotides of the Sp1 consensus-binding site of the miR-146b-5p promoter. C, EMSA. Pure proteins were used in the indicated combinations and biotin-labeled oligonucleotides of the Sp1 consensus binding sites of the miR-141 and miR-146b-5p promoters. The numbers indicate the amount of proteins in nanograms. D, ChIP assay. Chromatin was purified from the indicated cells and then immunoprecipitated using anti-p16 or anti-CDK4 antibodies. miR-141 and miR-146b-5p promoters were amplified by PCR (upper panel) and qPCR (lower panel), and their abundances were plotted relative to the input. E, total proteins and RNA were prepared from HFSN1 cells expressing either control-shRNA or CDK4-shRNA and were used to assess the level of CDK4 by immunoblotting and the level of miR-141 and miR-146b-5p by qRT-PCR. Error bars represent means ± S.D.
It has recently been shown that Sp1 protein interacts with the CDK4 protein kinase (31). Therefore, we investigated the presence of CDK4 in the Sp1-dependent pulled down proteins. Fig. 7A shows that as with anti-p16 antibody, CDK4 was also pulled down with anti-Sp1 antibody. Furthermore, immunoprecipitation with anti-CDK4 antibody pulled down both p16 and Sp1 (Fig. 7A), suggesting the presence of a p16-Sp1-CDK4 heterocomplex.
To confirm the presence of this heterocomplex and show its binding to the miR-141 and miR-146b-5p promoters, the EMSA was performed using nuclear extracts from HFSN1 cells and biotin-labeled oligonucleotides for the Sp1-binding sites present in the miR-146b-5p promoter. In addition, specific antibodies against Sp1, p16, or CDK4 were separately added to the extracts to endorse the presence of their respective proteins in the heterocomplex. IgG was used as negative control and, as expected, it did not change the band shift corresponding to the binding of the Sp1 heterocomplex to the oligomer (Fig. 7B, lanes 1 and 2). By contrast, adding anti-Sp1, anti-p16, or anti-CDK4 antibodies resulted in a supershift of the band, indicating the presence of the three proteins in the binding complex (Fig. 7B, lanes 3–5). Importantly, when nuclear proteins were prepared from Sp1-shRNA-, p16-shRNA-, or CDK4-shRNA-expressing cells, the intensities of the supershifted bands were reduced (Fig. 7B, lanes 6–8). These results further confirm the binding of the Sp1-p16-CDK4 heterocomplex to the miR-141 and miR-146b-5p promoters.
To demonstrate the formation of the Sp1-p16-CDK4 complex and its binding to the miR-141 and miR-146b-5p promoters, we made use of pure proteins and the EMSA as described above. Adding Sp1 (1 ng) alone resulted in a weak binding and shift of the miR-146b-5p oligomer (Fig. 7C, lane 2). Increasing the amount of Sp1 to 2 ng increased the binding but only slightly (Fig. 7C, lane 3). Adding 1 ng of p16 or CDK4 did not improve the binding of Sp1 to the oligomer (Fig. 7C, lanes 4 and 5), and when p16 or CDK4 was mixed alone with the labeled oligomer, no band shift was observed (Fig. 7C, lane 6). This indicates that p16 and CDK4 do not bind to the miR-146b-5p promoter in absence of Sp1. Interestingly, the presence of the three proteins (1 ng each) yielded great binding and supershift similar to that obtained with the nuclear extract (Fig. 7C, lanes 7–10). This demonstrates the formation and efficient binding of the complex Sp1-p16-CDK4 to the Sp1-binding sites present in the miR-141 and miR-146b-5p promoters.
Next, we sought to show the binding of p16 and CDK4 at the miR-141 and miR-146b-5p promoters in vivo. To this end, chromatin was prepared from HFSN1C and HFSN1Sp1sh cells, and then p16-DNA and CDK4-DNA complexes were pulled down using anti-p16 and anti-CDK4 antibodies, respectively. IgG was used as negative control. Subsequently, the miR-141 and miR-146b-5p promoters were amplified by PCR and qPCR. Although no amplification was obtained following the immunoprecipitation with the IgG antibody, a strong amplification of both promoters was obtained when anti-p16 and anti-CDK4 antibodies and Sp1-proficient cells were used (Fig. 7D). However, when chromatin was immunoprecipitated from Sp1-deficient cells, the level of p16 and CDK4 binding to the miR-141 and miR-146b-5p promoters was decreased (Fig. 7D). This shows Sp1-dependent binding of p16 and CDK4 to the miR-141 and miR-146b-5p promoters in vivo.
These results prompted us to suggest that CDK4 may also be involved in the expression of miR-141 and miR-146b-5p. To show this, we assessed the levels of these miRNAs by qRT-PCR in HFSN1 cells expressing control-shRNA or CDK4-shRNA. CDK4-shRNA significantly reduced the expression of the CDK4 protein (Fig. 7E, upper panel), and concomitantly reduced the level of both miR-141 and miR-146b-5p (Fig. 7E, lower panel), proving the importance of CDK4 in the transcription of miR-141 and miR-146b-5p.
p16 Interacts with Sp1 through the Fourth Ankyrin Repeat
To determine the p16 domain(s) required for the interaction with Sp1, we made use of mutants with deletions in each of the four ankyrin repeats and the C-terminal 21 amino acids of the p16 protein (16), and we investigated their binding to Sp1 in the p16-defective U2OS cells using immunoprecipitation. Like the wild-type, the mutant lacking the first ankyrin repeat bound to Sp1. Deletion of ankyrin repeats 2 and 3 reduced but allowed Sp1 binding. However, the loss of ankyrin repeat 4 completely abolished p16/Sp1 interaction (Fig. 8A). The mutated p16 proteins were expressed at levels similar to that of the wild-type protein (Fig. 8A). Similarly, the level of the Sp1 protein did not change in the presence of mutated p16 proteins (Fig. 8B). This indicates that the sharp decrease in the Sp1 binding to the mutated p16 protein (p16ΔAR4) is not due to a decrease in their levels.
FIGURE 8.

p16 interacts with Sp1 through the fourth ankyrin repeat. A, p16-negative U2OS cells were transfected with the indicated constructs, and then whole cell extracts were prepared and used for immunoprecipitation (IP) with anti-p16 antibody (mouse IgG was used as control), and immunoblotting was performed utilizing the indicated antibodies. B, whole cell extracts were prepared from U2OS expressing the indicated constructs, and immunoblotting was performed for the indicated antibodies. C, ChIP assay. Chromatin was purified from the indicated cells and then immunoprecipitated using anti-Sp1 antibody. miR-141 and miR-146b-5p promoters were amplified by qPCR using specific primers. D, total RNA was prepared from U2OS expressing the indicated constructs and used to assess the level of the indicated genes by qRT-PCR using GAPDH as internal control. Error bars represent means ± S.D. E, U2OS cells were transfected with constructs bearing the indicated tumor-associated point mutations in the fourth ankyrin repeat of human p16. Whole cell extracts were prepared and used for immunoprecipitation with anti-p16 antibody (mouse IgG was used as control), and immunoblotting was performed utilizing the indicated antibodies.
To investigate the role of the p16/Sp1 interaction in the binding of Sp1 to the two miRNA promoters, ChIP assay was performed on chromatin prepared from U2OS cells containing either the control plasmid, plasmids expressing wild-type p16, or the p16ΔΑR4 mutant, and the Sp1-DNA complex was pulled down using anti-Sp1 antibody. Subsequently, the miR-141 and miR-146b-5p promoter sequences were amplified by qPCR. Fig. 8C shows that although both promoters were strongly amplified in the presence of normal p16 as compared with their levels in the control U2OS cells, their amplification was significantly reduced in the presence of p16ΔAR4 mutant. This suggests that the binding of Sp1 to the miR-141 and miR-146b-5p promoters was strongly affected by the presence of the mutated p16ΔAR4 protein.
To further show the importance of this Sp1/p16 interaction, we investigated the effect of the deletion of the p16 fourth ankyrin repeat on the expression of miR-141 and miR-146b-5p. Therefore, the levels of these miRNAs were assessed by qRT-PCR in U2OS cells containing either the control plasmid or plasmids expressing wild-type p16 or the p16ΔΑR4 mutant. Fig. 8D shows that although expression of normal p16 increased the level of miR-141 and miR-146b-5p, the presence of p16ΔAR4 mutant significantly decreased their levels reaching a level similar to that of U2OS cells. This further supports our hypothesis that p16/Sp1 is critical for the expression of miR-141 and miR-146b-5p.
Several p16 mutations have been reported to be located in the fourth ankyrin repeat of the protein in different human tumors (11). Therefore, we sought to investigate whether such mutations affect p16 interaction with Sp1. These mutations were specifically generated in vitro in AR4 of human p16, as described previously (16), and the plasmids bearing them were introduced into U2OS cells. Cell lysates were immunoprecipitated with anti-p16 antibody, and the presence of Sp1 was assessed by immunoblotting using specific antibody. Unlike the wild-type p16 protein, the p16-Sp1 interaction was reduced in all the AR4 mutants; however, the R128W and A147G mutants were essential for this interaction, with an effect similar to that observed with the deletion of the whole ankyrin 4 repeat (Fig. 8E).
p16 Modulates the Expression of Different Sp1 Targets
It has been previously shown that the Sp1 protein has three different binding sites in the miR-365 promoter and that Sp1 regulates the transcription of this miRNA (32). Because p16 interacts with Sp1, we sought to investigate the possible implication of p16 in the binding of Sp1 to its cognate binding sites and the expression of the corresponding miR-365. Fig. 9A shows that the expression of miR-365 decreased in p16-deficient cells as compared with control cells. Furthermore, using EMSA we have shown that p16 depletion affected the binding of Sp1 to one of its binding sites (site 1), although it did not affect the binding to the two other sites (Fig. 9B). This indicates that p16 is also important for the binding of Sp1 to other miRNAs. In addition, we investigated the possible role of p16 in regulating the expression of different Sp1 target genes, which also belong to p16 regulon (18, 30). Fig. 9C shows that p16 down-regulation affected the expression of SLC2A3 and CEBPδ. This indicates that p16 modulates the expression of various Sp1 target genes.
FIGURE 9.
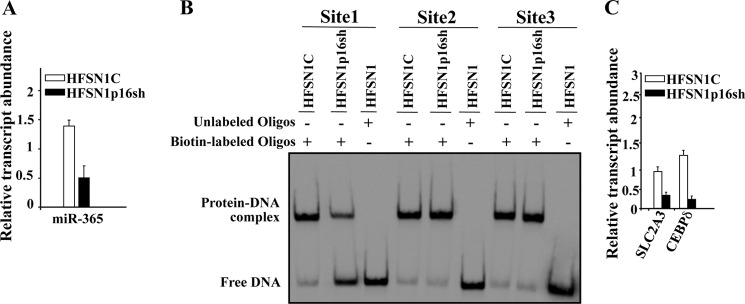
p16 modulates the expression of different Sp1 targets. A and C, total RNA was prepared from the indicated cells, and quantitative RT-PCR was performed using specific primers for the indicated genes. B, EMSA was performed using nuclear extracts from the indicated cells and biotin-labeled oligonucleotides corresponding to three different Sp1 consensus-binding sites of the miR-365 promoter. Error bars represent means ± S.D. Oligos, oligonucleotides.
UV Light Up-regulates miR-141 and miR-146b-5p in a p16-CDK4-Sp1-dependent Manner
p16 protein is up-regulated in response to UVC (34). Therefore, we investigated the possible UV light-dependent up-regulation of miR-141 and miR-146b-5p in a p16-dependent manner. To this end, HFSN1p16sh and HFSN1C cells were either mock-treated or challenged with UVC (10 Jm−2) and incubated for different periods of time. Total RNA was prepared, and RT-PCR was performed using specific primers for miR-141 and miR-146b-5p. Fig. 10A shows that UV treatment of control cells increased the expression of miR-141 and miR-146b-5p reaching levels 4.3- and 3.8-fold higher as compared with nontreated cells, respectively. However, in the HFSN1p16sh cells, their levels increased only marginally (Fig. 10A). This result was confirmed by qRT-PCR using mRNAs from time 0 and 2 h post-irradiation (Fig. 10B). This further shows a molecular link between p16, miR-141, and miR-146b-5p and provides the first indication that these two microRNAs are UV damage-inducible in a p16-dependent manner.
FIGURE 10.

p16-CDK4-Sp1-dependent up-regulation of miR-141 and miR-146b-5p and their role in apoptosis following UV damage. A, cells were either mock-treated or challenged with UV light (10 Jm−2) and then re-incubated for the indicated periods of time. Total RNA was then isolated and used for RT-PCR using specific primers for the indicated genes. The numbers below the bands indicate the corresponding expression levels following loading correction against GAPDH. B, qRT-PCR on mRNAs corresponding to 2 h of post-irradiation relative to time 0. These experiments were performed in triplicate (*, p < 0.05). C, cells were either mock-treated or challenged with UV light (10 Jm−2) and incubated for 72 h. Apoptosis was analyzed by annexin V/propidium iodide flow cytometry. D, the histogram shows the proportions of spontaneous and induced apoptosis (early + late). The error bars represent standard deviations of at least three different experiments. E, HFSN1C and HFSN1p16sh cells expressing the control plasmid, pre-miR-141 or pre-miR-146b-5p, were treated as above and collected after 48 and 72 h. Whole cell lysates were prepared and used for immunoblotting analysis. Error bars represent means ± S.D.
Because CDK4 and Sp1 control also the expression of miR-141 and miR-146b-5p, we investigated the effect of Sp1 and CDK4 down-regulation using specific shRNA on the induction of miR-141 and miR-146b-5p in response to UV light. CDK4-shRNA- and Sp1-shRNA-expressing HFSN1 cells were treated as above; RNA was prepared, and RT-PCR was performed to assess the levels of miR-141 and miR-146b-5p. Fig. 10, A and B shows that, as in p16-defective cells, UV treatment of Sp1- and CDK4-defective cells led to only a marginal increase in the level of miR-141 and miR-146b-5p. This indicates that miR-141 and miR-146b-5p are UV damage-inducible in a p16-CDK4-Sp1-dependent manner.
miR-141 and miR-146b-5p Contribute to p16-mediated UV Light-induced Apoptosis
Next, we investigated the possible role of p16, miR-141 and miR-146b-5p in UV light-induced apoptosis in human skin fibroblast cells. To this end, p16, miR-141 and miR-146b-5p were down-regulated in HFSN1 cells, and then these cells as well as HFSN1C were challenged with a UV fluence of 10 Jm−2 and incubated for 72 h. Apoptosis was assessed by propidium iodide/annexinV associated with flow cytometry. Fig. 10, C and D, shows that although about 35% of normal cells underwent apoptosis in response to UV light, only about 10% of p16-defective cells (HFSN1p16sh) were apoptotic. Similar results were obtained in cells wherein miR-141 and miR-146b-5p were inhibited (Fig. 10, C and D). This reveals a role of p16, miR-141, and miR-146b-5p in UV light-induced apoptosis. To confirm this, pre-miR-141 and pre-miR-146b-5p were expressed in HFSN1p16sh and then cells were treated as above. Fig. 10, C and D, shows that the increase in the expression of miR-141 and miR-146b-5p in p16-defective cells increased the proportion of apoptotic cells to 85 and 65%, respectively.
To support these observations, we assessed the effect of these miRNAs on the activation of the apoptotic proteins caspase-3 and PARP in response to UV damage. Therefore, HFSN1C as well as HFSN1p16sh cells containing either the control plasmid or a plasmid expressing pre-miR-141 or pre-miR-146b-5p were treated as above and harvested at 48 and 72 h post-irradiation. Fig. 10E shows that the level of the active form of both proteins increased upon UV damage in HFSN1C cells. However, no increase in the cleaved caspase-3 was observed in the p16-defective cells, although active PARP increased only slightly (Fig. 10E). Interestingly, the expression of miR-141 or miR-146b-5p in these cells activated both apoptotic markers in response to UV light (Fig. 10E). Together, these results show that miR-141 and miR-146b-5p play a critical role in UV damage-induced apoptosis.
DISCUSSION
We have shown here that p16 modulates the expression of 23 miRNAs in human primary skin fibroblast cells. miR-141 and miR-146b-5p, which are recognized to play important roles in cancer development and spread (35–38), were strongly affected by p16 depletion. Therefore, we further studied the effect of p16 on these two miRNAs using different technical approaches, and we have demonstrated the implication of p16 in their transactivation. This effect is mediated through physical interaction with the transcription factor Sp1, which has consensus binding sites on the promoters of both miR-141 and miR-146b-5p (Fig. 5A). Importantly, efficient binding of Sp1 to both promoters requires association with p16 and CDK4. Consequently, the three proteins are required for normal transcription of miR-141 and miR-146b-5p. The fact that Sp1 is a transcription factor that controls the expression of a plethora of genes suggests that the expression of several of these genes is also under the control of p16 and CDK4. Indeed, we have recently shown that p16 down-regulation with specific shRNA modulates the expression of 2170 genes. Most of these genes (1501) are involved in cell cycle regulation, and the others are implicated in various cancer-related mechanisms such as apoptosis, DNA repair, senescence, angiogenesis, and aging (18). Importantly, 57 of these genes belong also to the Sp1 regulon (supplemental Table S1) (30, 39). This has been confirmed by showing that p16 down-regulation affects the expression of SLC2A3 and CEBPδ, which are Sp1 target genes. Moreover, we have shown that p16 also modulates the expression of miR-365, which has been recently shown to be under the control of Sp1 (32). This indicates that, in addition to miR-141 and miR-146b-5p, Sp1 and p16 also co-regulate miR-365, and there may be others that need to be identified. However, it is also clear that not all the Sp1 targets are also p16 targets. This could be due to the fact that each of these two proteins interacts with several other proteins to regulate specific set of genes. For example, Sp1 interacts with C/EBPβ and HMGI-Y to control the expression of the human insulin receptor gene (40), whereas p16 associates with GRIM-19 to regulate the expression of E2F1 (16).
p16, Sp1, and CDK4 are known to play important roles in various physiological processes and also in carcinogenesis. Indeed, CDKN2A is a master tumor suppressor gene that has been found mutated or silenced in various types of neoplasms, and the inactivation of this gene is among the most frequent and earliest cytogenetic events in human cancer (11, 41). Furthermore, p16 overexpression has also been found in some benign tumors and in some cancer types, such as cervical cancer, breast cancer, and head and neck cancer (13, 14).
Sp1, which is the major transcription factor in the complex, is involved in the transcription of many genes implicated in most cellular processes. Like p16, high expression of the Sp1 protein has been shown in many cancers and is highly correlated with the stage and poor prognosis (13, 42). Furthermore, it has been recently shown that Sp1 level regulates lung tumor progression, and it strongly increases in the early stage and then decreases in the late stage, with high invasiveness of lung adenocarcinoma cells (43).
Likewise, CDK4 has also been found up-regulated in several human cancers and is considered as oncogene. However, it has been recently shown that CDK4 inhibition induces EMT and enhances invasiveness and pancreatic cancer cells (44). Furthermore, Sabir et al. (45) have shown the presence of novel CDK4 mutations in squamous cell carcinoma of the head and neck, suggesting their potential role in the development of these neoplasms. These results suggest that CDK4 may also have a tumor suppressor function. Therefore, the heterocomplex p16-Sp1-CDK4 may have a tumor suppressor function through positive regulation of miR-141 and miR-146b-5p.
Indeed, several sources of evidence indicate that microRNAs play important roles during cancer development and spread (23, 46). They can act as tumor suppressors and oncogenes (21–23). miR-146b-5p was shown to suppress EGF receptor expression and to reduce the migration and invasion of glioma and breast cancer cells in vitro (36, 37, 47) and also breast cancer metastasis (38). Similarly, five members of the miR-200 family, including miR-141, were found to be down-regulated during the EMT, an important invasion and metastasis process (35, 48), suggesting an association between EMT and the loss of miR-141. These miRNAs are almost undetectable in the highly invasive breast cancer MDA-MB-231 cells, which do not express p16 (35).
In addition, we have shown that p16-Sp1 association is mediated through the p16 fourth ankyrin repeat. In the presence of mutated p16 lacking this ankyrin repeat (p16ΔAR4), the level of Sp1 was not affected, although the binding to its cognate site and the trans-activation of miR-141 and miR-146b-5p were severely reduced (Fig. 8). This further shows that the physical interaction between p16 and Sp1 is crucial for the recruitment of Sp1 to its binding sites on the miR-141 and miR-146b-5p promoters. It is noteworthy that the p16 fourth ankyrin repeat is critical for association with GRIM-19 as well (16). Importantly, several cancer-related p16 mutations localized in the fourth ankyrin repeat affected the binding of p16 to Sp1, suggesting an important role of this association during carcinogenesis.
p16 is also involved in the cellular response to UV light-induced DNA damage (18, 27). Interestingly, we have shown that the levels of miR-141 and miR-146b-5p increase upon UV damage in a p16-CDK4-Sp1-dependent manner. This is the first indication that these two microRNAs are DNA damage-inducible and that CDK4 and Sp1 are also implicated in the UV light-related cellular response to UV damage. UV light-dependent up-regulation of miRNAs has been recently reported in various human cells (49–52). Furthermore, miRNA profiling showed modulation in the expression of various miRNAs upon treatment with different genotoxic agents (33). However, none of these studies reported the up-regulation of miR-141 and miR-146b-5p upon UV damage. This indicates that these miRNAs are part of the DNA damage response of human skin fibroblasts, and hence they may play important roles in restoring genomic stability. Indeed, down-regulation of miR-141 and miR-146b-5p reduced UV light-induced apoptosis, but increasing the expression of these two miRNAs activated the caspase-3 and PARP proteins and promoted apoptosis in response to UV damage (Fig. 10). This provides the first indication that these two miRNAs are positive regulators of UV-related apoptosis in human skin fibroblasts. It has been previously shown that miR-22 is also a modulator of UVC-induced apoptosis (50). Importantly, ectopic expression of miR-141 or miR-146b-5p in p16-deficient cells restored their apoptotic response to UV damage, indicating that these two miRNAs are downstream effectors of p16 in UV light-induced apoptosis.
In summary, these results present clear evidence that the tumor suppressor p16 protein is a master regulator of gene expression through association with the transcription factor Sp1 and the control of microRNAs.
Acknowledgments
We are grateful to P. S. Manogaran for help with the FACScan analysis and the Research Center administration for their continuous help and support.
This work was supported, in whole or in part, by National Institutes of Health Grant CA105005 (to D. V. K.). This work was also supported by Research Advisory Council Proposal 2090027.

This article contains Table S1.
- CDK
- cyclin-dependent kinase
- PARP
- poly(ADP-ribose) polymerase
- qRT-PCR
- quantitative RT-PCR
- miRNA
- microRNA
- IPTG
- isopropyl 1-thio-β-d-galactopyranoside
- EMT
- epithelial to mesenchymal transition.
REFERENCES
- 1. Koh J., Enders G. H., Dynlacht B. D., Harlow E. (1995) Tumour-derived p16 alleles encoding proteins defective in cell-cycle inhibition. Nature 375, 506–510 [DOI] [PubMed] [Google Scholar]
- 2. Lukas J., Parry D., Aagaard L., Mann D. J., Bartkova J., Strauss M., Peters G., Bartek J. (1995) Retinoblastoma-protein-dependent cell-cycle inhibition by the tumour suppressor p16. Nature 375, 503–506 [DOI] [PubMed] [Google Scholar]
- 3. Sherr C. J. (2001) The INK4a/ARF network in tumour suppression. Nat. Rev. Mol. Cell Biol. 2, 731–737 [DOI] [PubMed] [Google Scholar]
- 4. Serrano M., Hannon G. J., Beach D. (1993) A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 366, 704–707 [DOI] [PubMed] [Google Scholar]
- 5. Giacinti C., Giordano A. (2006) RB and cell cycle progression. Oncogene 25, 5220–5227 [DOI] [PubMed] [Google Scholar]
- 6. Shapiro G. I., Edwards C. D., Ewen M. E., Rollins B. J. (1998) p16ink4a participates in a G1 arrest checkpoint in response to DNA damage. Mol. Cell. Biol. 18, 378–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Alcorta D. A., Xiong Y., Phelps D., Hannon G., Beach D., Barrett J. C. (1996) Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc. Natl. Acad. Sci. U.S.A. 93, 13742–13747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Di Micco R., Fumagalli M., Cicalese A., Piccinin S., Gasparini P., Luise C., Schurra C., Garre' M., Nuciforo P. G., Bensimon A., Maestro R., Pelicci P. G., d'Adda di Fagagna F. (2006) Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 444, 638–642 [DOI] [PubMed] [Google Scholar]
- 9. Chavey C., Bibeau F., Gourgou-Bourgade S., Burlinchon S., Boissière F., Laune D., Roques S., Lazennec G. (2007) Oestrogen receptor negative breast cancers exhibit high cytokine content. Breast Cancer Res. 9, R15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Al-Mohanna M. A., Manogaran P. S., Al-Mukhalafi Z., A Al-Hussein K., Aboussekhra A. (2004) The tumor suppressor p16(INK4a) gene is a regulator of apoptosis induced by ultraviolet light and cisplatin. Oncogene 23, 201–212 [DOI] [PubMed] [Google Scholar]
- 11. Ruas M., Peters G. (1998) The p16INK4a/CDKN2A tumor suppressor and its relatives. Biochim. Biophys. Acta 1378, F115–F177 [DOI] [PubMed] [Google Scholar]
- 12. Rocco J. W., Sidransky D. (2001) p16(MTS-1/CDKN2/INK4a) in cancer progression. Exp. Cell Res. 264, 42–55 [DOI] [PubMed] [Google Scholar]
- 13. Romagosa C., Simonetti S., López-Vicente L., Mazo A., Lleonart M. E., Castellvi J., Ramon y Cajal S. (2011) p16(Ink4a) overexpression in cancer: a tumor suppressor gene associated with senescence and high-grade tumors. Oncogene 30, 2087–2097 [DOI] [PubMed] [Google Scholar]
- 14. Witkiewicz A. K., Knudsen K. E., Dicker A. P., Knudsen E. S. (2011) The meaning of p16(ink4a) expression in tumors: functional significance, clinical associations and future developments. Cell Cycle 10, 2497–2503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Souza-Rodrígues E., Estanyol J. M., Friedrich-Heineken E., Olmedo E., Vera J., Canela N., Brun S., Agell N., Hübscher U., Bachs O., Jaumot M. (2007) Proteomic analysis of p16ink4a-binding proteins. Proteomics 7, 4102–4111 [DOI] [PubMed] [Google Scholar]
- 16. Sun P., Nallar S. C., Raha A., Kalakonda S., Velalar C. N., Reddy S. P., Kalvakolanu D. V. (2010) GRIM-19 and p16(INK4a) synergistically regulate cell cycle progression and E2F1-responsive gene expression. J. Biol. Chem. 285, 27545–27552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vernell R., Helin K., Müller H. (2003) Identification of target genes of the p16INK4A-pRB-E2F pathway. J. Biol. Chem. 278, 46124–46137 [DOI] [PubMed] [Google Scholar]
- 18. Al-Khalaf H. H., Colak D., Al-Saif M., Al-Bakheet A., Hendrayani S. F., Al-Yousef N., Kaya N., Khabar K. S., Aboussekhra A. (2011) p16(INK4a) positively regulates cyclin D1 and E2F1 through negative control of AUF1. PLoS One 6, e21111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chien W. W., Domenech C., Catallo R., Kaddar T., Magaud J. P., Salles G., Ffrench M. (2011) Cyclin-dependent kinase 1 expression is inhibited by p16(INK4a) at the post-transcriptional level through the microRNA pathway. Oncogene 30, 1880–1891 [DOI] [PubMed] [Google Scholar]
- 20. Bartel D. P. (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297 [DOI] [PubMed] [Google Scholar]
- 21. Koturbash I., Zemp F. J., Pogribny I., Kovalchuk O. (2011) Small molecules with big effects: the role of the microRNAome in cancer and carcinogenesis. Mutat. Res. 722, 94–105 [DOI] [PubMed] [Google Scholar]
- 22. Farazi T. A., Spitzer J. I., Morozov P., Tuschl T. (2011) miRNAs in human cancer. J. Pathol. 223, 102–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lovat F., Valeri N., Croce C. M. (2011) MicroRNAs in the pathogenesis of cancer. Semin. Oncol. 38, 724–733 [DOI] [PubMed] [Google Scholar]
- 24. McConnell B. B., Gregory F. J., Stott F. J., Hara E., Peters G. (1999) Induced expression of p16(INK4a) inhibits both CDK4- and CDK2-associated kinase activity by reassortment of cyclin-CDK-inhibitor complexes. Mol. Cell. Biol. 19, 1981–1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Deleted in proof.
- 26. Taganov K. D., Boldin M. P., Chang K. J., Baltimore D. (2006) NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. U.S.A. 103, 12481–12486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Al-Mohanna M. A., Al-Khalaf H. H., Al-Yousef N., Aboussekhra A. (2007) The p16INK4a tumor suppressor controls p21WAF1 induction in response to ultraviolet light. Nucleic Acids Res. 35, 223–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bond J. A., Haughton M. F., Rowson J. M., Smith P. J., Gire V., Wynford-Thomas D., Wyllie F. S. (1999) Control of replicative life span in human cells: barriers to clonal expansion intermediate between M1 senescence and M2 crisis. Mol. Cell. Biol. 19, 3103–3114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Al-Khalaf H. H., Aboussekhra A. (2013) p16(INK4A) Positively regulates p21(WAF1) expression by suppressing AUF1-dependent mRNA decay. PLoS One 8, e70133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Oleaga C., Welten S., Belloc A., Solé A., Rodriguez L., Mencia N., Selga E., Tapias A., Noé V., Ciudad C. J. (2012) Identification of novel Sp1 targets involved in proliferation and cancer by functional genomics. Biochem. Pharmacol. 84, 1581–1591 [DOI] [PubMed] [Google Scholar]
- 31. Tapias A., Ciudad C. J., Roninson I. B., Noé V. (2008) Regulation of Sp1 by cell cycle related proteins. Cell Cycle 7, 2856–2867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xu Z., Xiao S. B., Xu P., Xie Q., Cao L., Wang D., Luo R., Zhong Y., Chen H. C., Fang L. R. (2011) miR-365, a novel negative regulator of interleukin-6 gene expression, is cooperatively regulated by Sp1 and NF-κB. J. Biol. Chem. 286, 21401–21412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wouters M. D., van Gent D. C., Hoeijmakers J. H., Pothof J. (2011) MicroRNAs, the DNA damage response and cancer. Mutat. Res. 717, 54–66 [DOI] [PubMed] [Google Scholar]
- 34. Al-Khalaf H. H., Hendrayani S. F., Aboussekhra A. (2011) The Atr protein kinase controls UV-dependent up-regulation of p16INK4A through inhibition of Skp2-related polyubiquitination/degradation. Mol. Cancer Res. 9, 311–319 [DOI] [PubMed] [Google Scholar]
- 35. Burk U., Schubert J., Wellner U., Schmalhofer O., Vincan E., Spaderna S., Brabletz T. (2008) A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 9, 582–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Katakowski M., Zheng X., Jiang F., Rogers T., Szalad A., Chopp M. (2010) MiR-146b-5p suppresses EGFR expression and reduces in vitro migration and invasion of glioma. Cancer Invest. 28, 1024–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhao J. L., Rao D. S., Boldin M. P., Taganov K. D., O'Connell R. M., Baltimore D. (2011) NF-κB dysregulation in microRNA-146a-deficient mice drives the development of myeloid malignancies. Proc. Natl. Acad. Sci. U.S.A. 108, 9184–9189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hurst D. R., Edmonds M. D., Scott G. K., Benz C. C., Vaidya K. S., Welch D. R. (2009) Breast cancer metastasis suppressor 1 up-regulates miR-146, which suppresses breast cancer metastasis. Cancer Res. 69, 1279–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mansilla S., Portugal J. (2008) Sp1 transcription factor as a target for anthracyclines: effects on gene transcription. Biochimie 90, 976–987 [DOI] [PubMed] [Google Scholar]
- 40. Foti D., Iuliano R., Chiefari E., Brunetti A. (2003) A nucleoprotein complex containing Sp1, C/EBPβ, and HMGI-Y controls human insulin receptor gene transcription. Mol. Cell. Biol. 23, 2720–2732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ortega S., Malumbres M., Barbacid M. (2002) Cyclin D-dependent kinases, INK4 inhibitors and cancer. Biochim. Biophys. Acta 1602, 73–87 [DOI] [PubMed] [Google Scholar]
- 42. Li L., Davie J. R. (2010) The role of Sp1 and Sp3 in normal and cancer cell biology. Ann. Anat. 192, 275–283 [DOI] [PubMed] [Google Scholar]
- 43. Hsu T. I., Wang M. C., Chen S. Y., Yeh Y. M., Su W. C., Chang W. C., Hung J. J. (2012) Sp1 expression regulates lung tumor progression. Oncogene 31, 3973–3988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Arslan A. A., Silvera D., Arju R., Giashuddin S., Belitskaya-Levy I., Formenti S. C., Schneider R. J. (2012) Atypical ezrin localization as a marker of locally advanced breast cancer. Breast Cancer Res. Treat. 134, 981–988 [DOI] [PubMed] [Google Scholar]
- 45. Sabir M., Baig R. M., Mahjabeen I., Kayani M. A. (2012) Novel germline CDK4 mutations in patients with head and neck cancer. Hered. Cancer Clin. Pract. 10, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Negrini M., Nicoloso M. S., Calin G. A. (2009) MicroRNAs and cancer–new paradigms in molecular oncology. Curr. Opin. Cell Biol. 21, 470–479 [DOI] [PubMed] [Google Scholar]
- 47. Bhaumik D., Scott G. K., Schokrpur S., Patil C. K., Campisi J., Benz C. C. (2008) Expression of microRNA-146 suppresses NF-κB activity with reduction of metastatic potential in breast cancer cells. Oncogene 27, 5643–5647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gregory P. A., Bert A. G., Paterson E. L., Barry S. C., Tsykin A., Farshid G., Vadas M. A., Khew-Goodall Y., Goodall G. J. (2008) The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 10, 593–601 [DOI] [PubMed] [Google Scholar]
- 49. Pothof J., Verkaik N. S., van IJcken W., Wiemer E. A., Ta V. T., van der Horst G. T., Jaspers N. G., van Gent D. C., Hoeijmakers J. H., Persengiev S. P. (2009) MicroRNA-mediated gene silencing modulates the UV light-induced DNA-damage response. EMBO J. 28, 2090–2099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tan G., Shi Y., Wu Z. H. (2012) MicroRNA-22 promotes cell survival upon UV radiation by repressing PTEN. Biochem. Biophys. Res. Commun. 417, 546–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhou X., Wang G., Zhang W. (2007) UV-B responsive microRNA genes in Arabidopsis thaliana. Mol. Syst. Biol. 3, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tan G., Niu J., Shi Y., Ouyang H., Wu Z. H. (2012) NF-κB-dependent microRNA-125b up-regulation promotes cell survival by targeting p38α upon ultraviolet radiation. J. Biol. Chem. 287, 33036–33047 [DOI] [PMC free article] [PubMed] [Google Scholar]