

Abstract
Molecular interactions of importance to cell biology are subject to sol-gel transitions: large clusters of weakly interacting multivalent molecules (gel phase) are produced at a critical concentration of monomers. Examples include cell-cell and cell-matrix adhesions, nucleoprotein bodies, and cell signaling platforms. We use the term pleomorphic ensembles (PEs) to describe these clusters, because they have dynamic compositions and sizes and have rapid turnover of their molecular constituents; this plasticity can be highly responsive to cellular signals. The classical polymer physical chemistry theory developed by Flory and Stockmayer provides a brilliant framework for treating multivalent interactions for simple idealized systems. But the complexity and variability of PEs challenges existing modeling approaches. Here we describe and validate a computational algorithm that extends the Flory-Stockmayer formalism to overcome the limitations of analytic theories. We divide the problem by deterministically calculating the fraction of bound sites for each type of binding site, followed by the stochastic assignment of the bonds to a finite number of molecules. The method allows for high valency within many different kinds of interacting molecules and site types, permits simulation of steady-state distributions, as well as assembly kinetics, and can treat cooperative binding within one of the interacting molecules. We then apply our method to the analysis of interactions in the nephrin-Nck-N-Wasp signaling system, demonstrating how multivalent layered scaffolds produce PEs at low monomer concentrations despite weak binding interactions. We show how the experimental data for this system are most consistent with synergistic cooperative interactions between Nck and N-Wasp.
Introduction
Multivalency may lead to the formation of large molecular clusters or polymers even when the individual binding affinities are weak. Several examples serve to illustrate the diversity and ubiquity of such systems in cells: nephrin-Nck-N-Wasp, believed to be essential for maintenance of the filtration barrier in the kidney (1,2); P-granules, characteristic of embryo development (3); mRNA granules, an essential mechanism for delivering messages over large distances in neuronal cells (4); the assembly and dynamics of focal adhesions during cell migration (5); postsynaptic density regions in excitatory neuronal synapses (6); and the aggregation of receptor signaling platforms (7). Such interactions lead to the formation of molecular clusters that increase local concentration of biomolecules, potentially triggering signaling events. Because these complexes are often plastic, with dynamic and variable composition, they have been called pleomorphic ensembles (PEs), to distinguish them from machines, assemblies of strongly and specifically interacting molecules (8).
Analytical and numerical modeling of multimeric complexes have a rich history, originating with the classical theories of Paul Flory and Walter Stockmayer (7,9–16). The Flory-Stockmayer (F-S) theory provides nonspatial analytical expressions to predict the properties of polymers arising from multivalent monomers. Given the valencies and binding-site affinities of the monomers, the F-S theory predicts the concentration dependence of cluster-size distribution and the composition of the clusters (9,17). The existence of a soluble (sol) phase and a macroscopic aggregate, or gel phase is a common feature of these systems, and the F-S theory can predict the critical concentration of monomers for the sol-gel transition. This is possible via the parameter pc, commonly referred to as the percolation threshold, or the critical degree of reaction between individual binding sites that leads to the formation of an infinitely large polymer (gel phase). In these studies, p corresponds to the fraction of reacted sites, or probability of bond formation. Thus, the configuration of the system is characterized by the valency of the monomers and the value p, regardless of the history of the system. Therefore, if the forward and reverse rate constants for site binding are known, the F-S theory can predict the kinetics of cluster formation as readily as the final steady state, as recently reviewed (18). The key equations arising from the F-S theory are provided in the Supporting Material for the convenience of the reader, although full treatments can be found elsewhere (9,10,13,17); we also provide a web-based tool (http://vcell.org/SimGel/) for generating sol-gel transition diagrams based on the classical F-S theory.
Analytical models based on the original F-S theory are restricted to simple cases (e.g., no more than two types of binding sites)—a fundamental limitation (9,10,19,20). In addition, a key simplifying assumption of the F-S theory is that all sites are subject to equal reactivity. This states that the probability of bond formation for any binding site is independent of the state of the molecule it belongs to (whether it is a monomer or part of a PE). It is justified for systems with both noncooperative binding-site interactions and binding rates slower than the diffusion-limited rate (where most collisions do not result in binding). In particular, this does not allow for the possibility of increased rates of formation of internal rings within clusters. As a consequence, the equal-reactivity assumption makes small rings highly unlikely in the sol phase, where the probability of binding with sites outside a given cluster will be dominant. However, a series of spatial numerical simulations confirmed the validity of the F-S theory for 3D systems of rigid monomers (21–23), demonstrating the applicability of the equal-reactivity assumption for this scenario.
Biological PE systems (8), such as interactions between modular protein binding domains or between proteins and nucleic acids, involve several different multivalent monomers interacting through many more than just two types of binding sites. Therefore, they cannot be treated with the analytical F-S theory. Because systems of multimeric complexes may lead to the formation of an infinite number of species, it is necessary to solve such problems stochastically with particle-based or network-free simulators, and a number of nonspatial software tools are available (24–26). However, none of these solvers takes advantage of the aforementioned simplifications inherent in the F-S theory.
In this article, we describe an efficient hybrid (deterministic + stochastic) nonspatial algorithm that makes use of the principles used to derive the F-S theory to treat PEs subject to sol-gel transitions. The algorithm extends the theory to account for ring formation and can treat both equal reactivity and cooperative binding. The deterministic part of the algorithm identifies the probabilities of interactions at a given time point, which serves as an input to the stochastic part. The latter consists of the random establishment of bonds, clustering, and statistics. We show that using our approach we reach the same results as fully stochastic solvers. We find that the percolation threshold (the transition to the gel phase) can be robustly identified by the occurrence of rings in the largest cluster, rather than the fraction of molecules in the largest cluster. Because specific sequences of binding sites may act as supramodules (6,27), we address the impact of synergistic interactions on the sol-gel transition diagrams. Finally, we apply our algorithm to the multidomain interactions between nephrin, Nck, and N-Wasp.
Materials and Methods
The deterministic solutions were obtained using Mathematica (Wolfram, Champaign, IL), Matlab (The MathWorks, Natick, MA), or BioNetGen (28). The algorithm for the stochastic part was written in Perl. The average properties and plots were obtained using Mathematica. RuleMonkey 2.0.25 (26) and KaSim 2.01 (25) were used to compare to compare fully stochastic simulations to our hybrid method (see Fig. 2). The corresponding input files are presented in the Supporting Material.
Figure 2.
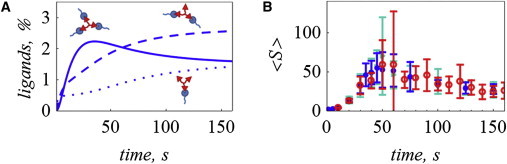
Comparison between simulations using our method and fully stochastic methods. Simulations were performed with 300 receptors, 5000 ligands, k1+ = 1.8 × 10−6 (molecules s)−1, k2+ = 5.6 × 10−4 (molecules s)−1, and k- = 0.01 s−1. (A) The percentages of ligands with one to three bonds are calculated deterministically, as described in Step1. (B) Steps 2 and 3 are repeated 40 times and the average value for <S> (mean PE size) is reported, using the results from A at the time points indicated by the blue dots in B. The results are equivalent to fully stochastic simulations using RuleMonkey (red open circles) and KaSim (green triangles) (means over 40 independent simulations). To see this figure in color, go online.
Results
A multistep deterministic-stochastic algorithm for modeling PEs
We developed a numerical algorithm, substantially extending the F-S theory, that allows for the efficient nonspatial simulation of the kinetics of cluster formation from multivalent monomers. The algorithm can accommodate multiple monomer types and multiple binding-site types. The central concept of the F-S theory is that the reactions between individual binding sites can be treated independently (equal reactivity). Therefore, in our algorithm, the multivalent monomers are first broken into the appropriate concentrations of binding sites. The reactions between sites are evaluated deterministically at a series of time points as a function of binding and unbinding-rate constants and the concentration of sites. This results in a fraction of bound sites for each type of binding site at each time point. We then assign indices to a fixed number of monomers and randomly assign these identifiers to the previously determined fraction of bound sites. The sites are then reconnected into the corresponding multivalent molecules and the clusters are identified. The procedure is repeated multiple times to generate statistics. Molecules formed by sites subject to the equal-reactivity assumption are broken into as many modules as the valency. Molecules with all binding sites subject to cooperativity cannot be broken into smaller independent modules, and the algorithm is adapted for such a scenario. By cooperativity we mean that the binding-rate constant for a given site is a function (activating or inhibiting) of the current site occupancy of the molecule it belongs to (29). With the aid of Fig. 1, we now explain each of the four steps in detail:
Figure 1.
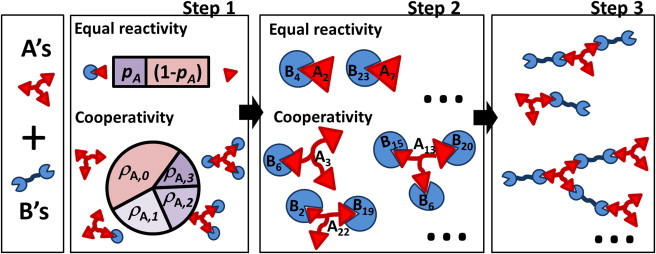
Method workflow. This illustrative example is for a system with trivalent molecules A (m = 3) and bivalent molecules B (n = 2). In Step1, all molecules subject to equal reactivity are split into sites. If both molecule types, A and B, are subject to equal reactivity (Step1, upper), the probability of bond formation, pA, is calculated. If A is subject to cooperativity (Step 1, lower), the fractions ρ0,…,ρm of molecules with 0 to m occupied sites are determined. In Step 2, sites are randomly chosen to establish bonds between a fixed number of molecules, Ai and Bj. In the equal-reactivity case, the bonds are established between pairs removed from the list of type A sites and the list of type B sites. In the cooperativity case, the vector of type A molecules (with a single copy per molecule), is used instead. Bonds are organized into clusters at Step 3; the clustering algorithm is fully described in Fig. S1. To see this figure in color, go online.
Step 1
The objective of this step is to reduce the potentially infinite system of equations (with one equation describing each potential PE composition and connectivity) to a finite system. For molecules subject to equal reactivity, this involves calculating the fractions of occupied sites at each time point, pi(t), where index i corresponds to one of the binding-site types. For steady-state calculations (t = ∞), this simply requires solution of algebraic equations using the given binding-site affinities. To model a kinetic time course, a system of ordinary differential equations (ODEs) is integrated, with one rate equation per site type. Conventional units, such as concentrations in micromolar, can be used. Conservation of mass provides the algebraic equations relating number of occupied and free sites. This step of the process is completely equivalent to treating all the binding sites as monovalent molecules and is therefore computationally trivial. The key output, pi(t), corresponds to the probability of a site of type i being occupied; it is simply calculated as the concentration of occupied i sites divided by the total concentration of i sites. This step represents a reduction of the system from an infinite number of species (monomers, dimers, trimers …) to a finite number of binding-site types.
A finite system is also possible for binding sites subject to cooperativity within a molecule, as long as their respective binding partners are still subject to equal reactivity. The number of required ODEs corresponds to the number of states the cooperative molecules have plus the number of types of equal-reactivity sites in the system. The output for this case is ρA,l(t), computed as the ratio between the concentration of cooperative molecules A in state l divided by the total concentration of A molecules.
Step 2
The stochastic part of the algorithm consists of translating the results from the deterministic step, which characterize the states of sites and molecules, into a discrete list of bonds for each time point. In this step, we choose a specified number of molecules, proportional to the respective concentration of each type of monomer, with each molecule assigned an individual index number; 1000 molecules usually suffices to produce robust results (see below). Sites displaying equal-reactivity interactions are stored in lists that carry the name and index number of the molecule. There is a list for each site type, and the valencies determine the number of identical objects in the list. Bonds between two sites that are subject to equal reactivity are randomly assigned to generate a list of paired site indices until the number of bonds relative to total sites satisfies the probability pi(t). The remaining unbound sites are left in their own lists, with a number corresponding to 1 − pi(t).
Cooperative molecules are stored in lists as complete objects, containing molecule name and index number. The bond assignments corresponding to each state l are performed sequentially. For a given state l, one cooperative molecule is randomly removed from its list and paired with as many sites as determined by its state. For example, let ρA,2B represent the fraction of molecules A that have two sites bound to sites of type B. For every object removed from the list of molecules A, two objects are randomly removed from the list of type B sites, and two bonds are stored in the list of bonds. If ρA,2B = 0.3 in a system with 1000 molecules A, this procedure is repeated 300 times.
To summarize, the output of this step is a list of randomly selected bonds between a population of molecules, representing one instantiation of the fractional binding-site occupancies calculated in step 1 for time t.
Step 3
This step identifies the molecular clusters by searching through the list of bonds for molecules linked by more than one bond (i.e., a molecule’s index appears in several bonds). The first element in the list is compared to the last element of the list to see if they have any molecules in common. If they do, the first element is removed and the last element now has the two bonds connecting the three molecules and represents a cluster. We move on to the new first element and compare it to the last element of the list. If false, we compare the first element to the second element from the end and so on until the test is true (and the first element is again removed and added to the cluster containing at least one of its molecules); if the first element is reached without finding any molecules in common with any of the other elements in the list, this element corresponds to a simple dimer and is kept in the list. The process then continues by moving to the second element, and the iterations are repeated until the last two elements of the list are compared. This step is represented in Fig. S1 in the Supporting Material. The output of this step is thus a list of all the clusters, with explicit account of their sizes, composition and bond connectivity.
Step 4
The average properties of the system are obtained by repeating Steps 2 and 3. Among the average properties that we compute are the size of the largest cluster, molecular composition of clusters, number of bonds of each type, number of rings (one plus the difference between the total number of bonds and the total number of molecules in the cluster (10)), etc.
The following sections illustrate the multiple types of data we can produce. The algorithm is validated against both analytical theory and existing fully stochastic numerical algorithms. Although it is easier to conceptualize in terms of two monomer species, each with identical binding sites, the method is fully general for multiple monomeric species, each with multiple binding domain types and multiple valencies. Indeed, below, we treat the interaction between trivalent nephrin and a single binding site on Nck (SH2 domain), together with the interaction of three SH3 domains on Nck with pentavalent N-Wasp proline-rich motifs. Furthermore, we show how outputs of our numerical approach can be used to extend the predictive power of the F-S theory to more complex systems that cannot be treated analytically.
Demonstration and validation: kinetics of PE formation in a cooperative system
We first illustrate our method with the trivalent-ligand-bivalent-receptor system, where a trivalent soluble ligand L binds to one of two sites on a membrane-associated receptor R and can then cross-link on the membrane by binding to other receptors. This is a convenient numerical example, since its rings-forbidden version has been extensively studied both analytically (7) and numerically (24,26,30,31), and its properties are discussed in the literature in great detail. Here we focus on validating our method. Of course, the ability to form internal rings will be dependent on the flexibility of the monomers within the cluster (32). To show that our algorithm can reproduce time-dependent responses, we treat this system under the condition that ring formation is allowed.
The trivalent-ligand-bivalent-receptor system is solved using two independent binding-rate constants. The first, k1+, concerns the binding of a free ligand and a free site on a membrane-bound receptor. The second, k2+, concerns the binding of a ligand that is already linked to the membrane via any receptor site. All bonds dissociate at the fixed rate constant k−. Although there may not be cooperativity at the molecular level, the biological context forces cooperative behavior: the targeting of the ligand to the membrane results in a modified binding rate for the binding to the next two receptors. Specifically, this behavior results in different affinities for the first versus the second and third L binding sites, thus defining L as a cooperative molecule.
In Step1, the receptor sites sR can be treated as subject to the equal-reactivity assumption. The ligand L must be treated as a cooperative molecule: the binding rate depends on the state of the ligand (whether it is free or with at least one bond). The ODEs for this system are presented in the Supporting Material. The deterministic solution for this small system of equations as a function of time (Fig. 2 A) is used as an input for the stochastic part of the algorithm. We generate clusters using the values of ρL,l for specific time points and compare the results with the numerical solutions from two fully stochastic solvers (RuleMonkey and KaSim (25,26)) that compute binding and unbinding reactions for all necessary time steps up to the time point of interest. The mean PE size, <S>, is a common output of interest for this system. It is defined as the average number of receptors in clusters with at least two receptors (30). Fig. 2 B shows that the three solvers result in equivalent numerical solutions.
This result shows that the method described can be used to properly model cluster formation for systems with cooperativity in a time-dependent fashion. The impact is that numerical computation of systems with binding-rate constants slower than the diffusion limit can benefit from 1) reducing the number of equations needed (a potentially infinite number of ODEs is replaced by a small system of ODEs); and 2) avoiding computation of individual stochastic reaction events.
The occurrence of rings in the largest cluster robustly identifies the sol-gel transition
A great contribution from analytical F-S theory is the prediction of the percolation threshold, or critical degree of reaction, pc, for the sol-gel transition. In particular, the F-S theory allows calculation of the fraction of monomers that belong to the gel phase, Wg, a parameter that is null for p ≤ pc and increases steeply at p > pc (9) (Fig. 3). However, the analytical expressions are limited to simple systems with up to only two site types. The identification of numerical outputs that are the most robust correlates of the theory would enable the prediction of the sol-gel transition when modeling systems are too complex to be handled analytically. Here, to establish such numerical outputs, we compare the fraction of molecules in the largest cluster, WLC, and the occurrence of rings in the largest cluster with the analytical F-S theory predictions.
Figure 3.
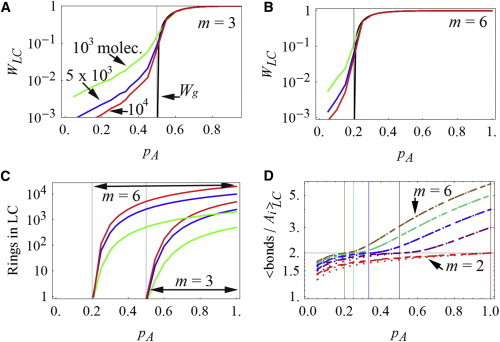
Properties of the largest cluster (LC) in a symmetric system consisting of an equal number of A and B molecules, each with valency m. (A and B) The weight fraction of the largest cluster, WLC, where colored plots correspond to different numbers of molecules (green, 103 molecules; blue, 5 × 103 molecules; and red, 104 molecules), and the weight fraction of the gel phase, Wg, from the analytical theory (9) (black lines). Molecules A and B have identical valency m = 3 (A) or m = 6 (B).The results of the simulations of WLC converge toward the theory (black line) for pA > pA,c. (C) Appearance of rings in the largest cluster is abrupt at pA,c, indicated by the lines at pA = 0.2 for valency m = 6 and at pA = 0.5 for valency m = 3; the color code is as in A and B. (D) Average number of bonds in each molecule A in the largest cluster. When this quantity is >2, there must be rings, and as in C, this condition is robustly met when pA > pA,c. Each color corresponds to a different valency, m = 6 to m = 2 from top to bottom (and left to right for pA,c). For each valency, simulations for 103, 5 × 103, and 104 molecules are plotted as dotted, dot-dashed, and dashed lines, respectively, and it is clear that the number of molecules does not significantly affect the results, especially past the pA,c. All results in A–D are the means of 50 simulations for systems with 103 and 5 × 103 molecules, and 10 simulations for the system with 104 molecules. To see this figure in color, go online.
Consider a system with two molecule types, A and B, with concentrations cA = cB. Each molecule has m binding sites. In such symmetric systems, pA = pB and the F-S theory predicts the existence of a gel phase for values of pA larger than pA,c = 1/(m − 1). As seen in Fig. 3, Wg increases abruptly as soon as pA > pA,c, offering an experimentally accessible observable for the analysis of sol-gel systems. However, Wg can only be calculated from the analytical expressions derived for such simple systems. The fraction of molecules belonging to the largest cluster, WLC, is the numerical correlate of Wg. WLC has been used previously to identify the presence of a gel phase in systems where intramolecular interactions are forbidden, with the gel phase being characterized somewhat arbitrarily by WLC > 0.05 (30). In contrast to Wg, this output has nonzero values for . We wanted to compare the WLC with the analytical expression for Wg in the region . Fig. 3, A and B, shows that the values of WLC at pA < pA,c are sensitive to the number of molecules chosen for the simulations. For m = 3, pA,c = 0.5, and the results for simulations using 1 × 103 (green), 5 × 103 (blue), and 1 × 104 (red) molecules at pA = pA,c produce WLC = 0.15 ± 0.06, 0.09 ± 0.05, and 0.07 ± 0.04, respectively. Similar results are obtained for m = 6, where pA,c = 0.2 (Fig. 3 B). Thus, the threshold of WLC = 0.05 is not a robust criterion for the prediction of pA,c. Although the results for WLC are sensitive to the number of molecules in the simulation in the region , the three simulation sizes show that WLC converges to Wg as pA exceeds pA,c.
It has been previously noted that in a nonreversible system with single molecule and site types, the occurrence of rings in the sol phase is negligible (13). Therefore, we felt that the occurrence of rings in the largest cluster might serve as a good criterion for the appearance of a gel phase. Indeed, our simulations show that rings consistently appear in the largest cluster whenever pA > pA,c. In Fig. 3 C, we show that this result is robust for the three simulation sizes and valencies m = 3 and m = 6. For a linear or branched polymer (i.e., with no rings), the number of bonds is equal to the total number of monomers minus one; thus, for a large polymer or cluster, the ratio of bonds to monomers approaches 1 (or 2 if computing bonds/molecules of A, Fig. 3 D). Consistently, each molecule A that belongs to the largest cluster has an average of two bonds to B molecules at pA = pA,c (Fig. 3 D). When the number of bonds per A exceeds 2, a cluster must contain rings. Fig. 3 D therefore shows that this condition robustly predicts pA,c for the three simulation sizes and valencies ranging from m = 2 to m = 6. Physically, one would expect an increasingly stable gel phase with an increasing number of rings: the larger the number of rings the less likely that a single bond dissociation will result in fragmentation of the cluster. In addition, the occurrence of rings is rare in the region pA < pA,c. In fact, our solution coincides with the F-S expressions for distribution of cluster sizes in systems of branched polymers in the sol phase (Figs. S2 and S3).
We conclude that in systems where equal reactivity is applied consistently to both sol and gel phases, the occurrence of rings in the sol phase is negligible. Indeed, the occurrence of rings in the largest cluster can be used as a reliable indicator of the sol-gel transition for the numerical simulations (Fig. 3, C and D). In contrast, the fraction of molecules in the largest cluster is sensitive to the number of molecules in the simulation and does not, therefore, offer a robust criterion for prediction of the sol-gel transition (Fig. 3, A and B).
Efficiency of the clustering algorithm
The computational cost of purely stochastic algorithms scales with the number of events. Therefore, no matter how coarsely a trajectory is ultimately sampled (including only once when a steady-state solution is all that is sought), the full trajectory of events must be simulated. Our method, on the other hand, randomly distributes a deterministically established fraction of bound sites assigned at each of a number of time points, predetermined as sufficient to characterize the system behavior. If a trajectory sampled at a series of discrete time points is sufficient for a simulation of multivalent kinetics, our method will be significantly more efficient than purely stochastic approaches. This is because our algorithm bypasses repeated stochastic dissociations and associations within the intervals between sampled time points; it establishes only the bonds that exist at the sampled times.
Solvers optimized for systems that do not allow rings have increased computational cost due to rejection of events as the system approaches the sol-gel transition (percolation threshold) (31). In contrast, Step 3 of our method was developed so that it is most efficient when the probability of bonds exceeds the percolation threshold. We use the system described in the previous section to illustrate the efficiency of the solver. In Fig. S4 we show that once the percolation threshold is met, the computational cost decreases exponentially with the valency of the system, for a fixed number of bonds. This computational advantage arises from the algorithm described in Step 3, and because equal reactivity is consistently applied in both sol and gel phases, ring formation is allowed and no reactions are rejected.
Analysis of an experimental PE system with cooperative synergistic interactions
Nck is an adaptor protein with both SH2 and SH3 domains that link, respectively, phosphotyrosine motifs on the cytoplasmic face of the plasma membrane to proline-rich motifs (PRMs) in actin-nucleation-promoting factors such as N-Wasp. In particular, the phosphotyrosines on nephrin in the membranes of kidney podocytes interact with Nck, which in turn recruits N-Wasp to organize the delicate foot processes that comprise the filtration barrier in the kidney (2). In addition to recruiting Nck to the membrane, tryrosine phosphorylation at multiple sites on nephrin may help promote the formation of nephrin-Nck-N-Wasp clusters. Because of the importance of this PE system, and because it has been characterized in vitro (1), we chose it to demonstrate the applicability of our algorithm to an experimental system.
The subject of this analysis is the recently published study of liquid-liquid demixing phase separation involving the multivalent domains of phosphorylated nephrin, Nck, and N-Wasp (1). Based on physical characterization of multivalent interactions of model systems, and on theoretical descriptions of multivalent polymers (20), it was postulated that these phase separations are accompanied by sol-gel transitions within the more concentrated of the two coexisting phases. Accordingly, we use the experimentally observable phase separation as an indicator of the sol-gel transition in this system. Nck SH3 domains interact with the important actin-nucleation promoter N-Wasp through the latter’s PRM domains. Although affinities have not yet been reported in this system, analogies to other SH3-PRM systems suggest that they will have dissociation constant values on the order of tens to hundreds of micromolar.
First, we investigate the potential importance of a synergistic interaction to help explain the general shape of the sol-gel transition diagram for Nck and N-Wasp in the absence of nephrin. Multivalent molecules may be organized in supramodules, whereby the specific sequence of sites and linker domains may promote synergistic interactions (6) between a single binding site in one molecule and multiple domains in its binding partner. For example, it has been shown that a single PRM in Pak1 may interact with any one of the three SH3 domains of Nck. However, if both the second and third SH3 domains of Nck are present, a significantly stronger synergistic interaction between Nck and Pak1 occurs (27).
The Nck-N-Wasp interaction is modeled as an interaction between a molecule with three SH3 domains (SH33) and a molecule with five PRMs (PRM5). The SH33 molecule is treated as cooperative. We label the SH3 domains as s1, s2, and s3. The synergistic interaction is depicted by the arrow labeled (s) in Fig. 4 A and is represented as follows: if s2 establishes a bond with any one PRM, and s3 of the same molecule is free, the latter may cooperatively bind to the same PRM with binding and unbinding-rate constants ks+ and ks−. To break the bond, both unbinding steps must occur. A similar sequence of reactions may occur if s3 establishes the first link. These interactions can be thought of as a two-stage binding. Alternatively, the s2 and s3 sites may establish two independent links to separate PRM domains (Fig. 4 A, (i)). In Fig. 4, B and C, we explore the impact of synergy strength by varying ks+/ks−.We use the occurrence of rings in the largest cluster (absolute number of molecules) as an indicator of the presence of a gel phase (Fig. S5 shows the fraction of SH33 and PRM5 in the largest cluster). For a fixed pairwise affinity for the binding between a single SH3 domain and a PRM motif (arbitrarily set to KD = 90 μM), the ratio ks+/ks− tunes the shape of the sol-gel transition interface. When ks+/ks− ≪ 1 (i.e., synergy is negligible), the transition boundary overlaps with that predicted by the analytical F-S theory (Fig. 4 B, yellow curve), but the fit to the experimental data are poor. In contrast, increasing the value of ks+/ ks− shifts the sol-gel transition interface toward higher concentrations of SH33 molecules. When ks+/ ks− = 50, corresponding to a high degree of synergy, the shape of the predicted region of gel phase closely follows the pattern in the experimental data (Fig. 4 C).
Figure 4.
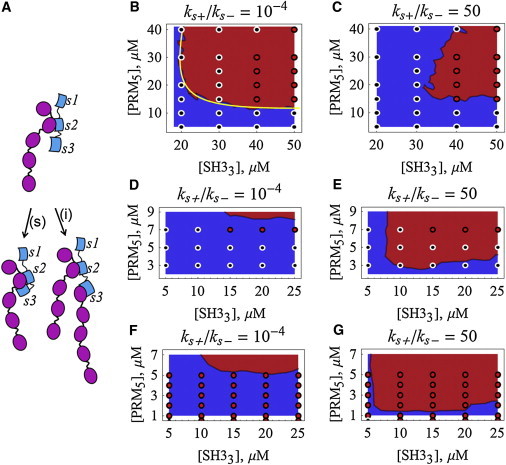
Impact of synergistic interactions in sol-gel transition diagrams. (A) The second and third SH3 domains of Nck may act independently (i) or synergistically (s). Sol-gel transition diagrams for systems with trivalent molecule SH33 (of Nck) binding to pentavalent molecule PRM5 of (N-Wasp), without (B and C) or with (D–G) the inclusion of Nck binding to nephrin. Plots in D–G are simulated with 9 μM of phosphorylated tyrosine sites distributed among bivalent (D and E) or trivalent (F and G) molecules. The intensity of the synergistic interaction between the last two SH3 domains is represented by the ratio ks+/ks−. The results shown are averages of 20 simulations for each condition, with 50 molecules/μM (B and C) or 250 molecules/μM (D–G). The occurrence of rings in the largest cluster is used to identify the occurrence of a gel phase. Experimental data for Nck and N-Wasp are represented by the circles (red, gel phase present; black, gel phase absent) (1). The yellow line in B corresponds to the F-S theoretical prediction when ks+ = 0. To see this figure in color, go online.
We now turn our attention to the addition of a third component to the system, nephrin (Fig. 4, D–G). The interaction between Nck and nephrin occurs via the single SH2 domain of the former with the phosphorylated tyrosine (pY) domains of the latter, with affinity 1 μM (2). By distributing 9 μM of pY sites among bivalent molecules (pY2, as for rat nephrin; Fig. 4, D and E) or trivalent molecules (pY3, as for human or mouse nephrin; Fig. 4, F and G), the sol-gel transition interface is brought to much lower concentrations of SH33 and PRM5 than in the absence of nephrin (Fig. 4, B and C), as shown both in our simulations and in experiments (note the different x and y scales in the plots of Fig. 4); the trivalent nephrin or pY3 (Fig. 4, F and G) displays greater potency in promoting the gel phase than the bivalent nephrin or pY2 (Fig. 4, D and E), as would be expected and as was found both in our simulations and in experiments (1). Thus, in vivo, multiple phosphorylations may be expected to do more than simply recruit Nck to the membrane-bound nephrin, but would also be expected to organize dense foci of N-Wasp-mediated actin-nucleation activity.
The two-component SH33-PRM5 system shows that the synergistic interaction reduces the region of concentrations where the gel phase can occur (compare Fig. 4, B and C). However the opposite trend occurs when the pY2 or pY3 scaffolding molecules are added (Fig. 4, D–G). This behavior likely reflects the tradeoff, for the synergistic interaction, between increased effective affinity and decreased valency, because one strong cooperative interaction uses up two SH3 domains. For the two-component system of Fig. 4 C, the decreased valency of the synergistic interaction dominates and the gel region shrinks compared to its size in Fig. 4 B. The addition of pY2 or pY3 to the system effectively boosts the valency of the overall system so that this factor is no longer limiting; thus, in Fig. 4, D–G, increased affinity is most important, producing a dramatic increase in the size of the gel region when synergy is included in the simulations. Consistently, the synergy increases the percentage of SH33 molecules that are bound and the stoichiometry (SH33/PRM5) of the largest cluster (Figs. S6 and S7).
Although the trends predicted by the models follow the trends in experiments, the simulations in Fig. 4, C, E, and G, are not perfect reproductions of the experiment. The lack of precise agreement with experiment may reflect our use of estimates for the relevant affinities. We did not attempt an extensive parameter search over a range of reasonable values of affinities or ks+/ks−. Furthermore, additional cooperative or synergistic interactions may need to be considered. It is also possible that these discrepancies arise from the limitations of the algorithm. However, this analysis points to the importance of cooperative synergistic interactions and the complex interplay between valency and affinity in this important PE system.
Discussion
Multimolecular weak-affinity multivalent interactions are ubiquitous in cell biological systems and can lead to formation of PEs within the cytoplasm (1,3,4,8,33,34). However, these interactions are difficult to model and analyze because of the combinatorial explosion of the possible number of states and species (8). Such interactions have been successfully treated with fully stochastic network-free particle-based methods (25,26), which probabilistically assign binding or unbinding events in a complete trajectory for the assembly of PEs. In this work, we describe a multistep hybrid deterministic/stochastic approach toward predicting the size, composition, and assembly kinetics of PEs. To do so, we compute the properties of the ensemble of binding sites rather than the kinetic details of a single trajectory. We make use of the concept, first demonstrated over 50 years ago by F-S theory, that the fractions of occupied binding sites determine cluster-size distribution. Thus, by building on the F-S theory, we can avoid using a purely stochastic particle-based method. Instead, we deterministically compute the fractional site occupancy for each time point, and then stochastically distribute the bonds between molecules containing the multivalent binding sites. For purely stochastic simulations, every binding and unbinding event must be traversed during a dynamic simulation to generate a complete trajectory; this is true even when only the steady-state properties of the PE are desired. For our algorithm, the deterministic step precalculates the probability of a site being occupied at each point in time, thus dramatically improving simulation efficiency; for steady-state properties, it suffices to determine only a single set of site occupancies calculated from the binding-site concentrations and affinities. The same concept may be extended to systems with cooperativity, as validated against numerical simulations performed with existing fully stochastic software tools (25,26) (Fig. 2).
The advantage of our numerical method, in comparison to the F-S theory, is that it can handle more than two types of binding site and more than two molecule types. Furthermore, here, for the first time that we know of, the idea of tracking the fraction of cooperative molecules in each state (15) is applied toward improving a computational method. Our method also overcomes the limitations (9,10,19) of F-S analytical theory by permitting a full treatment of ring formation (i.e., intracluster reactions) in both sol and gel phases. The same binding-rate constants are consistently applied in both the sol and gel phases, allowing intramolecular reactions. We show that rings are indeed rare within the sol phase, permitting us to use the occurrence of rings in the largest cluster to indicate that the system has reached the sol-gel transition (Fig. 3).
A higher level of accuracy can be achieved with methods that consider each molecule to be a space-filling object that diffuses and interacts with its binding partners in 3D space. Although such methods are important to assess the applicability of the theory to specific problems (22,23), the computational cost can be immense. For example, with our algorithm all the simulations used to generate the plots in Figs. 3, S2, and S3 were completed within a few hours. In contrast, a similar system with 1000 molecules computed using a spatial Monte Carlo method in 3D may require months of computer time (22,23). Three assumptions of our method are: 1), as with any nonspatial approach, the binding rates are taken to be lower than the diffusion limit; 2), for a given binding-site pair, intracluster reactions have the same binding-rate constants as intermolecular reactions; and 3), cooperative (or synergistic) molecules must interact with equal-reactivity sites. In comparison with the available nonspatial software, our method can be more efficient than time-resolved stochastic particle-based simulations, treats high valencies with simplicity, and is well suited for computation of ring formation.
A wide range of biologically relevant systems are consistent with these assumptions (1,5,6,35). Indeed, this modeling approach was designed for systems that we have called PEs (8) to denote assemblies of multivalent monomers driven by relatively weak binding interactions. These PEs are characterized by a distribution of stoichiometries and sizes. They are thus distinguished from the stoichiometric molecular assemblies that we have termed machines; these have a fixed composition and size and are characterized by strong, often highly cooperative, interactions between their subunit components. Ribosomes, viruses, flagella, the proteasome, and nuclear pores are all structures that would fall into this category.
We applied our algorithm to an experimental system consisting of interactions within a trimolecular PE consisting of nephrin, Nck, and N-Wasp. Nephrin has up to three binding sites for a single binding site on Nck; Nck has three additional binding sites that can bind up to five sites on N-Wasp. Fig. 4 shows that when we invoked cooperative synergistic interactions between the Nck SH3 domains and the PRMs on N-Wasp, we were able to reproduce the general shape of the experimentally reported (1) liquid-liquid demixing phase diagrams (which we use as a proxy for the sol-gel transition). The simulations also produced a substantial decrease in critical concentration when multiply phosphorylated nephrin was included in the system, also consistent with experiment. This analysis highlights the impact of the previously established synergistic interactions between the Nck SH3 domains and PRM binding partners (27) on the sol-gel transition diagram. The synergy results in an apparent reduction of valency, and the region of the diagram where the gel phase occurs is reduced for the bimolecular system. However, upon addition of the bivalent or trivalent nephrin, the synergistic interaction promotes gelation for lower Nck and N-Wasp concentrations than in a corresponding equal-reactivity system. This analysis demonstrates how both the shape and region of the sol-gel transition interface can be used to help elucidate the details of interactions between multivalent molecules.
Conclusions
We have described a novel (to our knowledge) and efficient method, built on classical polymer theory, for modeling the properties of PEs. It is not appropriate for modeling the assembly of stoichiometric molecular machines. Our method directly addresses the combinatorial complexity of the multivalent interactions underlying PEs, which has been a major challenge to previous modeling approaches. The utility of this method was exemplified by application to a signaling system that reveals the importance of cooperative synergistic interactions in controlling PE composition and size. This system also demonstrates how signaling through tyrosine phosphorylation and multidomain adaptor proteins can potentiate the production of large clusters of actin-nucleation promotion factors such as N-Wasp. We believe this approach can be directly applied for the modeling and analysis of cellular PEs such as signaling platforms, nucleoprotein particles, lipid rafts, and cell-cell or cell-matrix contacts.
Acknowledgments
The authors thank Michael Rosen, Sudeep Banjade, and Soyeon Kim at University of Texas Southwestern for the discussions regarding the nephrin-Nck-N-Wasp system. We also thank Michael Rosen for his critical reading of an early version of the manuscript.
This work was supported by National Institutes of Health grants TRO1DK087650, R01HL097431, P41GM103313, and R01GM095485.
Contributor Information
Cibele V. Falkenberg, Email: falkenberg@uchc.edu.
Leslie M. Loew, Email: les@volt.uchc.edu.
Supporting Material
References
- 1.Li P., Banjade S., Rosen M.K. Phase transitions in the assembly of multivalent signalling proteins. Nature. 2012;483:336–340. doi: 10.1038/nature10879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jones N., Blasutig I.M., Pawson T. Nck adaptor proteins link nephrin to the actin cytoskeleton of kidney podocytes. Nature. 2006;440:818–823. doi: 10.1038/nature04662. [DOI] [PubMed] [Google Scholar]
- 3.Brangwynne C.P., Eckmann C.R., Hyman A.A. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science. 2009;324:1729–1732. doi: 10.1126/science.1172046. [DOI] [PubMed] [Google Scholar]
- 4.Carson J.H., Gao Y., Barbarese E. Multiplexed RNA trafficking in oligodendrocytes and neurons. Biochim. Biophys. Acta. 2008;1779:453–458. doi: 10.1016/j.bbagrm.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schiller H.B., Fässler R. Mechanosensitivity and compositional dynamics of cell-matrix adhesions. EMBO Rep. 2013;14:509–519. doi: 10.1038/embor.2013.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feng W., Zhang M. Organization and dynamics of PDZ-domain-related supramodules in the postsynaptic density. Nat. Rev. Neurosci. 2009;10:87–99. doi: 10.1038/nrn2540. [DOI] [PubMed] [Google Scholar]
- 7.Goldstein B., Perelson A.S. Equilibrium theory for the clustering of bivalent cell surface receptors by trivalent ligands. Application to histamine release from basophils. Biophys. J. 1984;45:1109–1123. doi: 10.1016/S0006-3495(84)84259-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mayer B.J., Blinov M.L., Loew L.M. Molecular machines or pleiomorphic ensembles: signaling complexes revisited. J. Biol. 2009;8:81. doi: 10.1186/jbiol185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flory P.J. Cornell University Press; Ithaca, NY: 1953. Principles of Polymer Chemistry. [Google Scholar]
- 10.Stockmayer W.H. Theory of molecular size distribution and gel formation in branched-chain polymers. J. Chem. Phys. 1943;11:45–55. [Google Scholar]
- 11.Cohen R.J., Benedek G.B. Equilibrium and kinetic theory of polymerization and the sol-gel transition. J. Phys. Chem. 1982;86:3696–3714. [Google Scholar]
- 12.Perelson A.S., Goldstein B. The equilibrium aggregate size distribution of self-associating trivalent molecules. Macromolecules. 1985;18:1588–1597. [Google Scholar]
- 13.Falk M., Thomas R.E. Molecular size distribution in random polyfunctional condensation with or without ring formation: computer simulation. Can. J. Chem. 1974;52:3285–3295. [Google Scholar]
- 14.Goldstein B., Faeder J.R., Hlavacek W.S. Mathematical and computational models of immune-receptor signalling. Nat. Rev. Immunol. 2004;4:445–456. doi: 10.1038/nri1374. [DOI] [PubMed] [Google Scholar]
- 15.Perelson A.S., DeLisi C., Wiegel F.W. Marcel Dekker; New York: 1984. Cell Surface Dynamics: Concepts and Models. [Google Scholar]
- 16.Markova O., Alberts J., Lenne P.F. Bond flexibility and low valence promote finite clusters of self-aggregating particles. Phys. Rev. Lett. 2012;109:078101. doi: 10.1103/PhysRevLett.109.078101. [DOI] [PubMed] [Google Scholar]
- 17.Stockmayer W.H. Molecular distribution in condensation polymers. J. Polym. Sci. 1952;9:69–71. [Google Scholar]
- 18.Sciortino F., Zaccarelli E. Reversible gels of patchy particles. Curr. Opin. Solid State Mater. Sci. 2011;15:246–253. [Google Scholar]
- 19.Rubinstein M., Colby R.H. Oxford University Press; New York: 2003. Polymer Physics. [Google Scholar]
- 20.Semenov A., Rubinstein M. Thermoreversible gelation in solutions of associative polymers. 1. Statics. Macromolecules. 1998;31:1373–1385. [Google Scholar]
- 21.Bianchi E., Tartaglia P., Sciortino F. Fully solvable equilibrium self-assembly process: fine-tuning the clusters size and the connectivity in patchy particle systems. J. Phys. Chem. B. 2007;111:11765–11769. doi: 10.1021/jp074281+. [DOI] [PubMed] [Google Scholar]
- 22.Russo J., Tartaglia P., Sciortino F. Reversible gels of patchy particles: role of the valence. J. Chem. Phys. 2009;131:014504. doi: 10.1063/1.3153843. [DOI] [PubMed] [Google Scholar]
- 23.Tavares J.M., Teixeira P.I., Sciortino F. Equilibrium self-assembly of colloids with distinct interaction sites: thermodynamics, percolation, and cluster distribution functions. J. Chem. Phys. 2010;132:234502. doi: 10.1063/1.3435346. [DOI] [PubMed] [Google Scholar]
- 24.Sneddon M.W., Faeder J.R., Emonet T. Efficient modeling, simulation and coarse-graining of biological complexity with NFsim. Nat. Methods. 2011;8:177–183. doi: 10.1038/nmeth.1546. [DOI] [PubMed] [Google Scholar]
- 25.Feret J., Danos V., Fontana W. Internal coarse-graining of molecular systems. Proc. Natl. Acad. Sci. USA. 2009;106:6453–6458. doi: 10.1073/pnas.0809908106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Colvin J., Monine M.I., Posner R.G. RuleMonkey: software for stochastic simulation of rule-based models. BMC Bioinformatics. 2010;11:404. doi: 10.1186/1471-2105-11-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bokoch G.M., Wang Y., Knaus U.G. Interaction of the Nck adapter protein with p21-activated kinase (PAK1) J. Biol. Chem. 1996;271:25746–25749. doi: 10.1074/jbc.271.42.25746. [DOI] [PubMed] [Google Scholar]
- 28.Faeder J.R., Blinov M.L., Hlavacek W.S. Rule-based modeling of biochemical systems with BioNetGen. Methods Mol. Biol. 2009;500:113–167. doi: 10.1007/978-1-59745-525-1_5. [DOI] [PubMed] [Google Scholar]
- 29.Wyman J., Gill S.J. University Science Books; Mill Valley, CA: 1990. Binding and Linkage: Functional Chemistry of Biological Macromolecules. [Google Scholar]
- 30.Yang J., Monine M.I., Hlavacek W.S. Kinetic Monte Carlo method for rule-based modeling of biochemical networks. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2008;78:031910. doi: 10.1103/PhysRevE.78.031910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang J., Hlavacek W.S. The efficiency of reactant site sampling in network-free simulation of rule-based models for biochemical systems. Phys. Biol. 2011;8:055009. doi: 10.1088/1478-3975/8/5/055009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Monine M.I., Posner R.G., Hlavacek W.S. Modeling multivalent ligand-receptor interactions with steric constraints on configurations of cell-surface receptor aggregates. Biophys. J. 2010;98:48–56. doi: 10.1016/j.bpj.2009.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wippich F., Bodenmiller B., Pelkmans L. Dual specificity kinase DYRK3 couples stress granule condensation/dissolution to mTORC1 signaling. Cell. 2013;152:791–805. doi: 10.1016/j.cell.2013.01.033. [DOI] [PubMed] [Google Scholar]
- 34.Shen T.H., Lin H.K., Pandolfi P.P. The mechanisms of PML-nuclear body formation. Mol. Cell. 2006;24:331–339. doi: 10.1016/j.molcel.2006.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kato M., Han T.W., McKnight S.L. Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell. 2012;149:753–767. doi: 10.1016/j.cell.2012.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.