

Abstract
Foveal hypoplasia and optic nerve misrouting are developmental defects of the visual pathway and only co-occur in connection with albinism; to date, they have only been associated with defects in the melanin-biosynthesis pathway. Here, we report that these defects can occur independently of albinism in people with recessive mutations in the putative glutamine transporter gene SLC38A8. Nine different mutations were identified in seven Asian and European families. Using morpholino-mediated ablation of Slc38a8 in medaka fish, we confirmed that pigmentation is unaffected by loss of SLC38A8. Furthermore, by undertaking an association study with SNPs at the SLC38A8 locus, we showed that common variants within this gene modestly affect foveal thickness in the general population. This study reveals a melanin-independent component underpinning the development of the visual pathway that requires a functional role for SLC38A8.
Main Text
Foveal hypoplasia is a developmental defect found in a number of eye conditions, including aniridia (MIM 106210), achromatopsia (MIM 216900), and retinopathy of prematurity, but to date, it has only been identified in association with optic nerve misrouting in cases of ocular (MIM 300500) or oculocutaneous (MIM 203100) albinism.1 Mutations in multiple genes have been reported to be responsible for the different forms of nonsyndromic and syndromic albinism, and each of these results in a defect in melanin biosynthesis or melanocyte differentiation.2,3 However, we recently described FHONDA (foveal hypoplasia, optic-nerve-decussation defects, and anterior segment dysgenesis) syndrome, which combines these features in the absence of albinism.4 Individuals with FHONDA syndrome all have a poorly defined foveal avascular zone, absent or abnormal foveal and/or macular reflexes, and absent foveal pits, consistent with a diagnosis of foveal hypoplasia (Figure 1). Visual-evoked potential (VEP) analysis has shown that these individuals have optic nerve misrouting, in which an increased number of axons cross the optic chiasm to innervate the contralateral cortex (Figure 1). In addition, people with FHONDA syndrome have either posterior embryotoxon or Axenfeld anomaly, indicating dysgenesis of the anterior segment.4,5 However, they do not display any of the pigmentation defects associated with albinism or ocular albinism; these include hypopigmentation of the skin and hair (from careful clinical examination and from inspection of baby pictures or comparison with siblings), reduced pigmentation of the iris and retina, and iris transillumination.4,6
Figure 1.
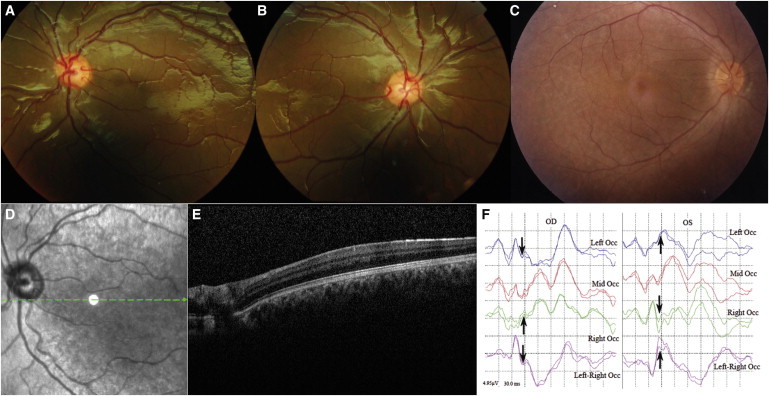
Foveal Hypoplasia and Chiasmal Misrouting Are Present in Individuals with Recessive SLC38A8 Mutations
(A and B) Fundus photographs of the left (A) and right (B) eyes of individual IV:1 from family F5 at 12 years of age show retinal vessels within the normally avascular macula region, signifying foveal hypoplasia.
(C) A normal control fundus is included for comparison. Note that the fundus pigmentation in (A)–(C) is normal and represents natural variation. An optical-coherence-tomography scan of the left eye of the same child at the age of 11 years confirmed foveal hypoplasia.
(D) A fundus image showing the position of the scan (green arrow) spans the presumptive foveal region (white circle).
(E) Scan results show normal retinal morphology but the absence of a foveal pit.
(F) Flash VEP results of individual IV:1 from family F1 show contralateral asymmetry of VEP, demonstrating chiasmal misrouting. The arrow shows the N2 peak, which is similar to that seen in albinos. Abbreviations are as follows: OD, right eye; and OS, left eye. Note that the time on the x axis begins at −15 ms.
Using a combination of linkage analysis and autozygosity mapping in two consanguineous families (F1 and F2; Figure 2), we mapped the gene mutated in FHONDA syndrome to a 3.1 Mb locus in chromosomal region 16q23.3–24.1 (chr16: 83,639,061–86,716,445; UCSC Genome Browser, hg19).4 To identify the mutated gene, we used Sanger sequencing to screen the coding sequence and flanking splice sites of all 33 genes within the FHONDA locus in a single affected member from both families. Primers were designed with the ExonPrimer script accessed through the UCSC Genome Browser (Table S1, available online). Genomic DNA was extracted and amplified by PCR according to standard protocols. PCR products were processed with ExoSAP-IT (Affymetrix USB), sequenced with BigDye Terminator v.3.1 (Applied Biosystems), and run on an ABI3130xl Genetic Analyzer (Applied Biosystems) according to the manufacturers’ instructions. Informed consent was obtained from all subjects tested, and ethical approval was provided by the Leeds Teaching Hospitals NHS Trust Research Ethics Committee (Ref. N. REC 03/362).
Figure 2.
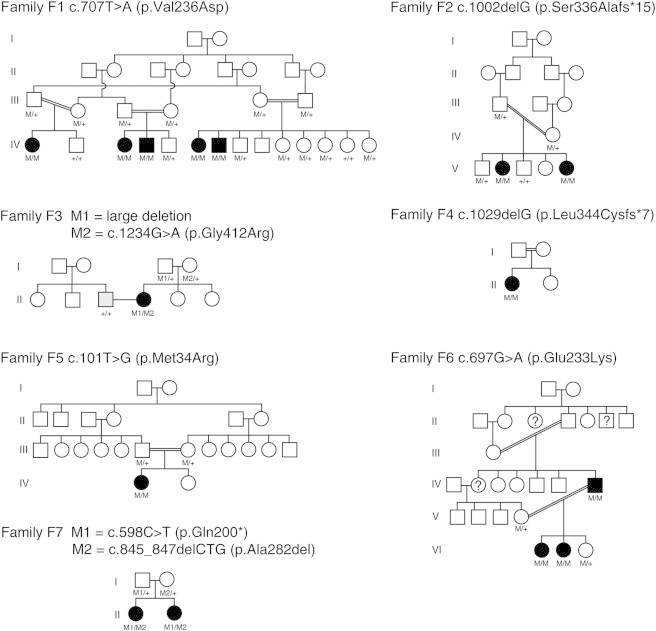
Pedigrees of Families Reported in This Study and Mutation Segregation Data
Affected individuals are shaded black. Detailed descriptions of members of F1, F2, F3, and F6 have been reported previously.4–6,8 The husband of the affected individual in F3 has oculocutaneous albinism (gray shading). In family F6, the members with a question mark have not undergone a full clinical examination, but the fact that they have esotropia, poor vision, and nystagmus suggests that they have the same condition. The mutation genotypes for all tested family members are shown below each individual—M represents the mutant allele, and + represents the wild-type allele.
This analysis revealed different homozygous mutations in SLC38A8 in both families (RefSeq accession number NM_001080442.1). We identified a homozygous missense mutation (c.707T>A [p.Val236Asp]) in family F1 and a homozygous 1 bp deletion resulting in a frameshift (c.1002delG [p.Ser336Alafs∗15]) in family F2 (Figure 3). Each mutation segregated with the disease in its respective family (Figure 2) and was excluded from ethnically matched controls and publically available databases (National Heart, Lung, and Blood Institute [NHLBI] Exome Sequencing Project Exome Variant Server [EVS] and dbSNP). Conservation analysis showed that the substituted valine amino acid is fully conserved down to zebrafish, with the exception of the Tasmanian devil (Figure S1). In addition, six different pathological prediction tools support the pathogenic nature of the p.Val236Asp substitution (Table S2).
Figure 3.
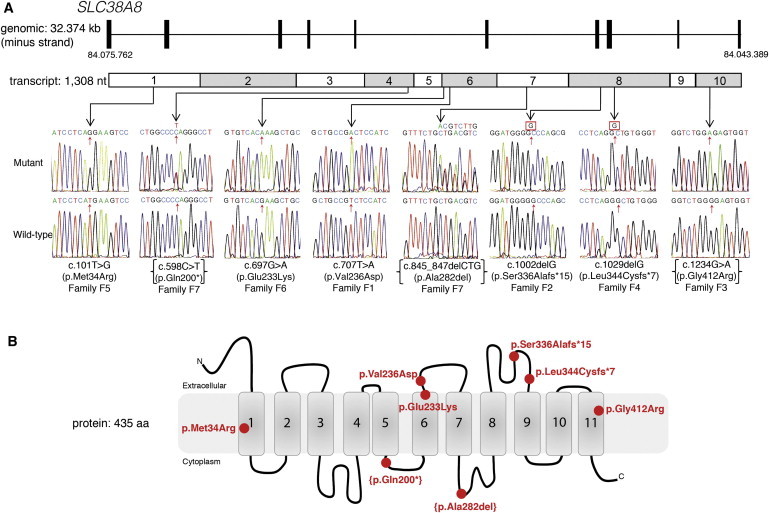
Mutations in SLC38A8 Cause Foveal Hypoplasia and Optic-Nerve-Decussation Defects
(A) A schematic representation of the SLC38A8 genomic structure and transcript shows the location and sequence traces of eight mutations identified in this study.
(B) A schematic diagram of SLC38A8 shows the location of the alterations within the protein domains. The compound-heterozygous changes are in parentheses.
Screening of SLC38A8 in a further 12 individuals with foveal hypoplasia in the apparent absence of albinism identified mutations in five additional cases (Figure 3). In a third family (family F3), we found an apparently homozygous mutation (c.1234G>A [p.Gly412Arg]) in a nonconsanguineous Northern European female with foveal hypoplasia, optic nerve misrouting, and Kartagener syndrome (MIM 244400).6 This missense mutation is predicted to be damaging by six prediction tools and is fully conserved in all orthologs except gorilla, whose predicted protein sequence deviates from all other species for the last 40 amino acids (Figure S1). Unexpectedly, mutation screening in the female’s parents only confirmed the mother as a heterozygous mutation carrier and failed to identify the mutation in her father (Figure 2). However, we confirmed paternity by genotyping ten fluorescently labeled microsatellite markers (Figure S2), thus indicating that this individual inherited a large deletion from her father and is actually hemizygous for the SLC38A8 missense mutation. Interestingly, a known gene nearby (DNAAF1 [MIM 613190]) is mutated in Kartagener disease, and a large 640 kb deletion encompassing both genes has previously been reported.7 We suspect that the presence of a similar large deletion in this individual is the most likely explanation for the co-occurrence of these two rare disorders in a nonconsanguineous family.
In a female simplex case with foveal hypoplasia and consanguineous Pakistani parents (family F4), we identified a homozygous frameshift deletion (c.1029delG [p.Leu344Cysfs∗7]). Unfortunately, no additional family DNA samples were available, so we were unable to perform segregation analysis for this mutation. In another female simplex case with foveal hypoplasia and optic-nerve-decussation defects, we identified a homozygous missense mutation (c.101T>G [p.Met34Arg]). This female is from a consanguineous Turkish family (family F5), and SNP microarray genotyping showed that her largest region of homozygosity is ∼19 Mb spanning SLC38A8. As expected, both of her parents were found to be heterozygous for the mutation, but DNA was unfortunately not available for her unaffected sibling, so further segregation analysis was not possible (Figures 2 and 3). Conservation analysis showed that the methionine residue substituted in this individual is not conserved and is frequently replaced with leucine (Figure S1). However, both methionine and leucine are similarly sized hydrophobic amino acids, whereas the substituted arginine residue is positively charged and is therefore likely to disrupt the hydrophobic edge of the transmembrane helix. Furthermore, four of the six pathogenic prediction tools class this substitution as deleterious (Table S2).
Screening in a large highly consanguineous Indian family (family F6) affected by foveal hypoplasia variably associated with additional developmental eye defects8 led to the identification of another homozygous missense mutation (c.697G>A [p.Glu233Lys]). The mutated gene in this family had previously been mapped to an ∼4 Mb region overlapping the FHONDA locus (maximum LOD score = 2.3 for marker rs254347).9 The mutation segregates with the disease phenotype in all available family members, and the substituted glutamic acid residue is fully conserved in all species. However, the pathogenicity prediction tools gave mixed responses: three predicted benign outcomes, and three predicted deleterious effects (Table S2). All affected members of this family have bilateral foveal hypoplasia, but IV:7 also has bilateral microphthalmia and a unilateral retinochoroidal coloboma. Similarly, his 9-year-old daughter (VI:1) also has bilateral microphthalmia, but his 6-year-old daughter (VI:2) only has foveal hypoplasia. Not all members of this family have undergone a full clinical examination, but the fact that three further members suffer from esotropia, poor vision, and nystagmus suggests that they have the same condition (Figure 2). Finally, we identified compound-heterozygous mutations in two affected sisters from a Northern European family (F7). The first mutation is a nonsense change (c.598C>T [p.Gln200∗]), and the second is an in-frame deletion causing the removal of a single amino acid (c.845_847delCTG [p.Ala282del]). The deleted amino acid is fully conserved in mammals, and both parents are heterozygous for one of the mutations. One of the sisters has FHONDA syndrome, but the other only has foveal hypoplasia and optic nerve misrouting and does not have any anterior segment abnormalities.
All mutations were excluded from ethnically matched controls by sequencing. Control samples were European Collection of Cell Cultures (ECACC) Human Random Control Panel DNA, ECACC Human Ethnic Diversity Panel DNA, or unaffected, unrelated members of families who had been recruited for genetics studies. None of the mutations were present in dbSNP or the NHLBI EVS except for p.Gln200∗ (rs149592537), which has a frequency of 1/12,999 in the NHLBI EVS and 1/4,545 in dbSNP and is therefore likely to be a pathogenic allele identified in carriers. The seven individuals without SLC38A8 mutations might indicate possible genetic heterogeneity. However, it is possible that our screening strategy missed some mutations, especially large deletions such as the one identified in family F3. Indeed, SNP microarray genotyping in one of these individuals showed a large region of homozygosity around SLC38A8, suggesting that mutations were missed (data not shown). Unfortunately, because the clinical information for these cases is limited, especially regarding pigmentation and iris transillumination, we cannot exclude the possibility that they might actually be undiagnosed cases of albinism.
No iris transillumination or reduced pigmentation was evident in the skin, hair, or eyes of any of the SLC38A8-mutation-affected individuals, including 12 affected members of five Asian families and three from two European families. However, pigmentation defects in albinism can be subtle (see GeneReviews in Web Resources). This has led to speculation that FHONDA syndrome is actually mild albinism,1 especially given that anterior segment dysgenesis is observed in albinism.10,11 Medaka fish (Oryzias latipes) have been shown to be a reliable model system for investigating pigmentation defects,12 so we examined the effect of morpholino-mediated ablation of Slc38a8 in these fish. All experiments were conducted in strict accordance with the institutional guidelines for animal research and approved by the Department of Public Health, Animal Health, Nutrition and Food Safety of the Italian Ministry of Health in accordance with the law on animal experimentation (article 7, D.L. 116/92, protocol number 00001/08/IGB, approval date October 22, 2008). Furthermore, all animal treatments were reviewed and approved in advance by the ethics committee of the Institute of Genetics and Biophysics Animal House (Naples).
Morpholinos were designed against both medaka SLC38A8 orthologs present in the UCSC Genome Browser (October 2005 v.1.0): ENSORLT00000016172 on chromosome 6 (19,760,744–19,770,301 bp) and ENSORLT00000011189 on chromosome 3 (20,486,490–20,491,975 bp), along with corresponding mismatch controls (Table S3). The Cab strain of wild-type medaka fish was used, and embryos were staged according to Iwamatsu.13 Morpholinos (Gene Tools) were injected into 1-cell fertilized embryos according to established protocols.14 Optimal morpholino concentrations were determined on the basis of morphological criteria. Specificity and inhibitory efficiency of each morpholino were determined as previously detailed.15
Both orthologs of SLC38A8 were simultaneously targeted with either morpholinos targeting the 5′ UTR or the splice donor site of exon 4. Off-target effects of the morpholino injections were excluded by repeated experiments with mismatched morpholinos or by coinjection with a p53 morpholino.16 None of the resulting morphant embryos were found to have any pigmentation defects of the eyes or tegument, as determined by morphological analysis of both the retinal pigment epithelium and tegument melanophores (Figure 4 and Figure S3). Structural abnormalities were restricted to the eye. Compared to controls, the majority of the knockdown embryos (87%–92%) had microphthalmia, a significant number (29%–60%) had lens defects, and a smaller number (16%–36%) had fissure coloboma (Figure 4). Given that medaka fish do not have a fovea, one would not expect to see a fully overlapping phenotype between humans and fish, but it is interesting to note that members of family F6 have microphthalmia and coloboma.
Figure 4.
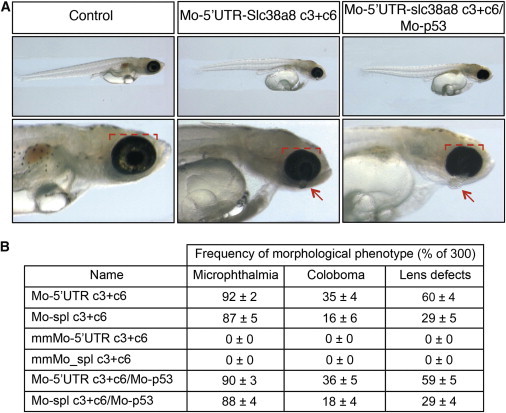
Morpholino Knockdown of Slc38a8 in Medaka Fish Results in Microphthalmia, Coloboma, and Lens Defects, but No Skin- or Ocular-Pigmentation Abnormalities
(A) Bright-field stereomicroscope images show lateral views of representative fish for the controls and Mo-5′UTR-slc38a8 c3+c6 and Mo-5′UTR-Slc38a8 c3+c6/Mo-p53 mutants. The red bracket highlights the reduced eye size in morphants, and the arrows highlight displaced lenses. Ocular pigmentation identical to that of the controls can clearly be seen in the mutant fish.
(B) The frequency of each phenotype observed in the medaka fish is given as a percentage of 300.
To investigate the effects of Slc38a8 knockdown on optic nerve decussation, we repeated the morpholino experiment on Athonal5::GFP transgenic medaka embryos, which have GFP-labeled retinal ganglion cells.17 However, we observed no differences between the optic chiasm of wild-type medaka and the knockdown embryos (Figure S4). This result was not unexpected given that the lateral position of the eyes in medaka prevents overlap of the visual fields so that all retinal ganglion axons cross the midline at the optic chiasm.18 The significantly overlapping visual space in humans, however, makes it essential that ∼40% of the axons do not cross the midline but rather project ipsilaterally to allow binocular vision.19 Individuals with FHONDA syndrome display an increase in the number of axons projecting contralaterally,4,6 indicating a defect in the specification of the ipsilateral projections that are absent in medaka.
Examination of the clinical phenotypes of the individuals with SLC38A8 mutations showed that not all fulfill the criteria for FHONDA syndrome (Table S4). Foveal hypoplasia was present in all affected individuals, and optic-nerve-decussation defects were present in all individuals examined for this feature except one in whom the VEP analysis was inconclusive (F4). However, anterior segment abnormalities were only present in 3/7 families (F1, F2, and F7), making this a variable component of the phenotype in individuals with recessive SLC38A8 mutations. At this time, it is not possible to determine whether the microphthalmia and retinochoroidal coloboma phenotypes observed in one of our families (and in the medaka fish) are related to the SLC38A8 mutations, and further phenotype-genotype studies are thus needed. A number of cases of isolated foveal hypoplasia have been reported in the literature, and it would be interesting to screen SLC38A8 in these cases.20,21 Similarly, VEP analysis is often used for diagnosing albinism in fair-skinned populations, where pigmentation defects can be hard to identify, so it is likely that some of these cases might actually harbor SLC38A8 mutations.22
To test the hypothesis that common variants in SLC38A8 influence fovea thickness in the general population, we carried out an association study in a population cohort of Northern European ancestry from the Western Australian Pregnancy Cohort (Raine) 20-year follow-up eye study.23 As part of a comprehensive eye examination performed in this study, the foveal thickness of both eyes in each participant was imaged through dilated pupils by spectral-domain optical coherence tomography (Spectralis; Heidelberg Engineering) by an experienced operator. Retinal thickness at the fovea was automatically determined by the instrument software as the distance between the internal limiting membrane and retinal pigment epithelium. The ratio of foveal thickness to the mean parafoveal thickness and the ratio of foveal thickness to the average perifoveal thickness for each eye were also recorded. Given the high correlation between the right and left eyes (intraclass correlation coefficient of 0.872 [95% confidence interval (CI) = 0.834–0.905], 0.911 [95% CI = 0.847–0.943], and 0.733 [95% CI = 0.576–0.819] for central foveal thickness, fovea-parafovea ratio, and fovea-perifovea ratio, respectively), the mean values for each participant were used for analysis after inverse normal transformation.
DNA samples and consents for genome-wide association studies (GWASs) were available from previous assessments. Genotype data were generated with the genome-wide Illumina 660 Quad Array at the Centre for Applied Genomics (Toronto) and processed for quality control (QC). We used the EIGENSTRAT program24 to conduct principal-component analysis and constructed the first five principal components for a subset of 42,888 SNPs that were not in linkage disequilibrium with each other. Additionally, we performed the GWAS imputation of 22 autosomes in the MACH v.1.0.16 software by using the CEU (Utah residents with ancestry from northern and western Europe from the CEPH collection) samples from HapMap phase 2 (build 36, release 22). A linear regression model in R with a PLINK interface25 was used for determining associations between SNPs and foveal thickness, the ratio of foveal to parafoveal thickness, or the ratio of foveal to perifoveal thickness. The model was adjusted for age, sex, and the first two principal components that accounted for the population stratification. After QC, phenotypic and genetic data were available from 679 individuals. We focused our analysis on SNPs surrounding SLC38A8. These data suggest that variants at this locus confer an effect in normal fovea variation and suggest that SNP rs7200988 (allele A) is the most associated with foveal thickness (p = 5.77 × 10−4) (Figure S5 and Table S5).
SLC38A8 encodes an orphan member of the SLC38 sodium-coupled neutral amino acid transporter (SNAT) family of proteins, which are widely expressed and predominantly have glutamine as their preferred substrate.26 To determine the expression of SLC38A8, we performed RT-PCR with primers spanning intron 7 on cDNA created from a panel of mRNA from human adult and fetal tissue (Clontech) according to standard protocols. SLC38A8 expression was found predominantly in neuronal tissue (adult brain, fetal brain, and spinal cord), and very weak expression was also present in the kidneys, thymus, and testes (Figure S6).
Using a custom-made rabbit polyclonal antibody (GenScript) raised against 14 amino acids located at the N terminus of SLC38A8 (QTPGSRGLPEKPHP), we investigated the protein localization of SLC38A8 by using human brain and eye tissues obtained from the Leeds Tissue Bank. SLC38A8 was shown to be located throughout the neuronal retina but had strong staining in the inner and outer plexiform layers and the photoreceptor layer (Figure S6). Similarly, in the brain we showed that SLC38A8 is localized in the cell body and axon of the majority of neuronal cells and in a subset of glial cells (Figure S6). These data are almost identical to those obtained for the recently characterized glutamine transporter SLC38A7,27 the closest homolog of SLC38A8,28 and suggest that both proteins have similar functions.
Glutamine is present at high concentrations in the brain extracellular fluid and cycles between neurons and glial cells, where it serves as an intermediate metabolite for the formation of the neurotransmitters glutamate and gamma-amino butyric acid. The location of SLC38A8 in the retina is consistent with a synaptic neurotransmitter-recycling role. However, this would not adequately explain the developmental defect observed in SLC38A8-mutation carriers. Active synapses are known to play a role in the formation and remodeling of neural circuits, including the projecting retinal ganglion cells,29 providing one possible developmental disease mechanism. Alternatively, the localization of SLC38A8 in the somata and axons of brain cells might imply an additional role or roles for this protein. Consistent with this hypothesis, there is emerging evidence that neurotransmitters play a role in the developing retina by modulating the proliferation of retinal progenitor cells even before synapses are formed.30 In turn, the number and spatiotemporal development of retinal cells are then believed to influence the extent to which projecting axons cross at the chiasm or project ipsilaterally, thus providing an alternative hypothesis to explain the defects seen in individuals with defects in SLC38A8.31
In summary, we have shown that recessive mutations in the putative glutamine transporter SLC38A8 cause foveal hypoplasia and optic nerve misrouting. We show that pigmentation is not affected by loss of SLC38A8 and thus identify a melanin-independent component essential for the development of these structures.
Acknowledgments
We thank the families who participated in this study. We thank J. Deuchars and M. Singh for advice with immunohistochemistry and A. Hindley and the Leeds Tissue Bank for providing and processing samples. We thank J. Wittbrodt for providing the ATH5::GFP medaka transgenic line and A.E. Davidson and Z. Li for supporting genetic analysis. This work was supported by a Royal Society University Research Fellowship (C.T.), University of Leeds Emma and Leslie Reid Scholarship (J.A.P.), Omani Government scholarship (M.A.-A.), Yorkshire Eye Research, the Italian Telethon Foundation (project grant TGM11SB2), the Italian Ministry of Research (PONA3_00311), RP Fighting Blindness, Fight For Sight, Foundation Fighting Blindness, National Institute for Health Research Moorfields Eye Hospital Biomedical Research Centre, and the Sir Jules Thorn Award for Biomedical Research (#JTA/09). We thank the Raine Study participants and staff for cohort coordination and data collection, particularly Jenny Mountain, Wei Ang, Craig Pennell, Hannah Forward, Charlotte McKnight, Seyhan Yazar, Alla Soloshenko, Sandra Oates, and Diane Wood. The core management of the Raine Study is funded by the University of Western Australia (UWA), Telethon Institute for Child Health Research, Raine Medical Research Foundation, UWA Faculty of Medicine, Dentistry and Health Sciences, Women’s and Infant’s Research Foundation, and Curtin University. Genotyping was funded by National Health and Medical Research Council (NHMRC) grant 572613. The Raine Eye Health Study was supported by NHMRC grant 1021105, the Lions Eye Institute, the Australian Foundation for the Prevention of Blindness, the Ophthalmic Research Institute of Australia, and the Alcon Research Institute.
Contributor Information
Chris F. Inglehearn, Email: c.inglehearn@leeds.ac.uk.
Carmel Toomes, Email: c.toomes@leeds.ac.uk.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
ExonPrimer, http://ihg.gsf.de/ihg/ExonPrimer.html
GeneReviews, Lewis, R.A. (2012). Oculocutaneous Albinism Type 2, http://www.ncbi.nlm.nih.gov/books/NBK1232/
Leeds Tissue Bank, http://www.gift.leeds.ac.uk/index.html
MACH 1.0.16, http://www.sph.umich.edu/csg/yli/mach/index.html
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Michaelides M., Jeffery G., Moore A.T. Developmental macular disorders: phenotypes and underlying molecular genetic basis. Br. J. Ophthalmol. 2012;96:917–924. doi: 10.1136/bjophthalmol-2011-300994. [DOI] [PubMed] [Google Scholar]
- 2.Dessinioti C., Stratigos A.J., Rigopoulos D., Katsambas A.D. A review of genetic disorders of hypopigmentation: lessons learned from the biology of melanocytes. Exp. Dermatol. 2009;18:741–749. doi: 10.1111/j.1600-0625.2009.00896.x. [DOI] [PubMed] [Google Scholar]
- 3.Schiaffino M.V. Signaling pathways in melanosome biogenesis and pathology. Int. J. Biochem. Cell Biol. 2010;42:1094–1104. doi: 10.1016/j.biocel.2010.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Al-Araimi M., Pal B., Poulter J.A., van Genderen M.M., Carr I., Cudrnak T., Brown L., Sheridan E., Mohamed M.D., Bradbury J. A new recessively inherited disorder composed of foveal hypoplasia, optic nerve decussation defects and anterior segment dysgenesis maps to chromosome 16q23.3-24.1. Mol. Vis. 2013;19:2165–2172. [PMC free article] [PubMed] [Google Scholar]
- 5.Pal B., Mohamed M.D., Keen T.J., Williams G.A., Bradbury J.A., Sheridan E., Inglehearn C.F. A new phenotype of recessively inherited foveal hypoplasia and anterior segment dysgenesis maps to a locus on chromosome 16q23.2-24.2. J. Med. Genet. 2004;41:772–777. doi: 10.1136/jmg.2004.020040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Genderen M.M., Riemslag F.C.C., Schuil J., Hoeben F.P., Stilma J.S., Meire F.M. Chiasmal misrouting and foveal hypoplasia without albinism. Br. J. Ophthalmol. 2006;90:1098–1102. doi: 10.1136/bjo.2006.091702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Loges N.T., Olbrich H., Becker-Heck A., Häffner K., Heer A., Reinhard C., Schmidts M., Kispert A., Zariwala M.A., Leigh M.W. Deletions and point mutations of LRRC50 cause primary ciliary dyskinesia due to dynein arm defects. Am. J. Hum. Genet. 2009;85:883–889. doi: 10.1016/j.ajhg.2009.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vincent A., Kemmanu V., Shetty R., Anandula V., Madhavarao B., Shetty B. Variable expressivity of ocular associations of foveal hypoplasia in a family. Eye (Lond.) 2009;23:1735–1739. doi: 10.1038/eye.2009.180. [DOI] [PubMed] [Google Scholar]
- 9.Anandula V.R., Shetty R., Vincent A., Ramprasad V.L., Ramesh N. Gene mapping in a highly inbred consanguineous foveal hypoplasia family to cytogenetic region 16q24.1. Journal of Medical Genetics and Genomics. 2011;3:122–125. [Google Scholar]
- 10.Charles S.J., Green J.S., Grant J.W., Yates J.R., Moore A.T. Clinical features of affected males with X linked ocular albinism. Br. J. Ophthalmol. 1993;77:222–227. doi: 10.1136/bjo.77.4.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shiono T., Tsunoda M., Chida Y., Nakazawa M., Tamai M. X linked ocular albinism in Japanese patients. Br. J. Ophthalmol. 1995;79:139–143. doi: 10.1136/bjo.79.2.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fukamachi S., Shimada A., Shima A. Mutations in the gene encoding B, a novel transporter protein, reduce melanin content in medaka. Nat. Genet. 2001;28:381–385. doi: 10.1038/ng584. [DOI] [PubMed] [Google Scholar]
- 13.Iwamatsu T. Stages of normal development in the medaka Oryzias latipes. Mech. Dev. 2004;121:605–618. doi: 10.1016/j.mod.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 14.Conte I., Carrella S., Avellino R., Karali M., Marco-Ferreres R., Bovolenta P., Banfi S. miR-204 is required for lens and retinal development via Meis2 targeting. Proc. Natl. Acad. Sci. USA. 2010;107:15491–15496. doi: 10.1073/pnas.0914785107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eisen J.S., Smith J.C. Controlling morpholino experiments: don’t stop making antisense. Development. 2008;135:1735–1743. doi: 10.1242/dev.001115. [DOI] [PubMed] [Google Scholar]
- 16.Robu M.E., Larson J.D., Nasevicius A., Beiraghi S., Brenner C., Farber S.A., Ekker S.C. p53 activation by knockdown technologies. PLoS Genet. 2007;3:e78. doi: 10.1371/journal.pgen.0030078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Del Bene F., Ettwiller L., Skowronska-Krawczyk D., Baier H., Matter J.M., Birney E., Wittbrodt J. In vivo validation of a computationally predicted conserved Ath5 target gene set. PLoS Genet. 2007;3:1661–1671. doi: 10.1371/journal.pgen.0030159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoda H., Hirose Y., Yasuoka A., Sasado T., Morinaga C., Deguchi T., Henrich T., Iwanami N., Watanabe T., Osakada M. Mutations affecting retinotectal axonal pathfinding in Medaka, Oryzias latipes. Mech. Dev. 2004;121:715–728. doi: 10.1016/j.mod.2004.03.021. [DOI] [PubMed] [Google Scholar]
- 19.Petros T.J., Rebsam A., Mason C.A. Retinal axon growth at the optic chiasm: to cross or not to cross. Annu. Rev. Neurosci. 2008;31:295–315. doi: 10.1146/annurev.neuro.31.060407.125609. [DOI] [PubMed] [Google Scholar]
- 20.Curran R.E., Robb R.M. Isolated foveal hypoplasia. Arch. Ophthalmol. 1976;94:48–50. doi: 10.1001/archopht.1976.03910030014005. [DOI] [PubMed] [Google Scholar]
- 21.Oliver M.D., Dotan S.A., Chemke J., Abraham F.A. Isolated foveal hypoplasia. Br. J. Ophthalmol. 1987;71:926–930. doi: 10.1136/bjo.71.12.926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sjöström A., Kraemer M., Ohlsson J., Villarreal G. Subnormal visual acuity syndromes (SVAS): albinism in Swedish 12-13-year-old children. Doc. Ophthalmol. 2001;103:35–46. [PubMed] [Google Scholar]
- 23.Yazar S., Forward H., McKnight C.M., Tan A., Soloshenko A., Oates S.K., Ang W., Sherwin J.C., Wood D., Mountain J.A. Raine Eye Health Study: Design, Methodology and Baseline Prevalence of Ophthalmic Disease in a Birth-cohort Study of Young Adults. Ophthalmic Genet. 2013 doi: 10.3109/13816810.2012.755632. Published online January 10, 2013. [DOI] [PubMed] [Google Scholar]
- 24.Price A.L., Patterson N.J., Plenge R.M., Weinblatt M.E., Shadick N.A., Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 25.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mackenzie B., Erickson J.D. Sodium-coupled neutral amino acid (System N/A) transporters of the SLC38 gene family. Pflugers Arch. 2004;447:784–795. doi: 10.1007/s00424-003-1117-9. [DOI] [PubMed] [Google Scholar]
- 27.Hägglund M.G., Sreedharan S., Nilsson V.C., Shaik J.H., Almkvist I.M., Bäcklin S., Wrange O., Fredriksson R. Identification of SLC38A7 (SNAT7) protein as a glutamine transporter expressed in neurons. J. Biol. Chem. 2011;286:20500–20511. doi: 10.1074/jbc.M110.162404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schiöth H.B., Roshanbin S., Hägglund M.G., Fredriksson R. Evolutionary origin of amino acid transporter families SLC32, SLC36 and SLC38 and physiological, pathological and therapeutic aspects. Mol. Aspects Med. 2013;34:571–585. doi: 10.1016/j.mam.2012.07.012. [DOI] [PubMed] [Google Scholar]
- 29.Koch S.M., Dela Cruz C.G., Hnasko T.S., Edwards R.H., Huberman A.D., Ullian E.M. Pathway-specific genetic attenuation of glutamate release alters select features of competition-based visual circuit refinement. Neuron. 2011;71:235–242. doi: 10.1016/j.neuron.2011.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martins R.A., Pearson R.A. Control of cell proliferation by neurotransmitters in the developing vertebrate retina. Brain Res. 2008;1192:37–60. doi: 10.1016/j.brainres.2007.04.076. [DOI] [PubMed] [Google Scholar]
- 31.Jeffery G. Architecture of the optic chiasm and the mechanisms that sculpt its development. Physiol. Rev. 2001;81:1393–1414. doi: 10.1152/physrev.2001.81.4.1393. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.