

Abstract
Auriculocondylar syndrome (ACS) is a rare craniofacial disorder with mandibular hypoplasia and question-mark ears (QMEs) as major features. QMEs, consisting of a specific defect at the lobe-helix junction, can also occur as an isolated anomaly. Studies in animal models have indicated the essential role of endothelin 1 (EDN1) signaling through the endothelin receptor type A (EDNRA) in patterning the mandibular portion of the first pharyngeal arch. Mutations in the genes coding for phospholipase C, beta 4 (PLCB4) and guanine nucleotide binding protein (G protein), alpha inhibiting activity polypeptide 3 (GNAI3), predicted to function as signal transducers downstream of EDNRA, have recently been reported in ACS. By whole-exome sequencing (WES), we identified a homozygous substitution in a furin cleavage site of the EDN1 proprotein in ACS-affected siblings born to consanguineous parents. WES of two cases with vertical transmission of isolated QMEs revealed a stop mutation in EDN1 in one family and a missense substitution of a highly conserved residue in the mature EDN1 peptide in the other. Targeted sequencing of EDN1 in an ACS individual with related parents identified a fourth, homozygous mutation falling close to the site of cleavage by endothelin-converting enzyme. The different modes of inheritance suggest that the degree of residual EDN1 activity differs depending on the mutation. These findings provide further support for the hypothesis that ACS and QMEs are uniquely caused by disruption of the EDN1-EDNRA signaling pathway.
Main Text
Neural crest cells (NCCs) are a transient embryonic population whose derivatives make major contributions to the skeletal and connective tissue of the face, the cardiac outflow tract, the peripheral and enteric nervous systems, and melanocytes. The endothelin system, consisting of three peptide ligands (endothelins 1, 2, and 3, encoded by EDN1 [MIM 131240], EDN2 [MIM 131241], and EDN3 [MIM 131242], respectively) and two G-protein-coupled seven transmembrane domain receptors (endothelin receptors type A and B, encoded by EDNRA [MIM 131243] and EDNRB [MIM 131244], respectively), plays key roles in the development of various NCC derivatives. In humans, mutations in EDN3 and EDNRB cause Waardenburg syndrome, type 4 (WS4 [MIM 277580 and 613265]), a disorder involving enteric and melanocytic NCCs and comprising Hirschsprung disease, pigmentation defects, and hearing loss (reviewed in Pingault et al.1). Studies in animal models have highlighted the importance of the EDN1-EDNRA signaling pathway in the development of the lower jaw. During early stages of craniofacial morphogenesis, ectomesenchymal NCCs migrate from the dorsal neural tube at cranial levels and populate the pharyngeal arches (PAs), where they receive cues from surrounding tissues that promote patterning and differentiation (reviewed in Cordero et al.2). The first PA is divided into maxillary and mandibular prominences, from which the skeleton of the upper and lower jaws will arise, respectively, whereas the external ear is derived from a series of swellings that surround the cleft between the first and second PAs (reviewed in Passos-Bueno et al.3). Edn1 is expressed from the epithelium of the mandibular prominence of the first PA and caudal PAs,4 where the EDN1 peptide signals to underlying ectomesenchyme by stimulating EDNRA on cranial NCCs. Mice with a targeted deletion of Edn1, Ednra, or endothelin-converting enzyme 1 (Ece1, encoding an enzyme involved in EDN1 cleavage) display severe defects in mandibular development, in which changes in skeletal morphology are consistent with a homeotic transformation of the lower jaw into an upper jaw, as well as defects of the cardiovascular system.5–10 In an ENU screen in zebrafish, a substitution in the mature Edn1 peptide was identified in the mutant line sucker, which has severe defects in the skeletal elements of the lower jaw.11 Mutations in human EDN1 or EDNRA are currently absent from publically available databases.
Auriculocondylar syndrome (ACS [MIM 602483 and 614669]) is a rare craniofacial disorder involving first and second PA derivatives and has key features of micrognathia, temporomandibular joint and condyle anomalies, microstomia, prominent cheeks, and question-mark ears (QMEs).12 QMEs consist of a defect between the lobe and the upper two-thirds of the pinna, range from a mild indentation in the helix to a complete cleft between the lobe and helix, and have been reported in individuals without mandibular defects (isolated QMEs [IQMEs] [MIM 612798]). Mutations in phospholipase C, beta 4 (PLCB4 [MIM 600810]) and guanine nucleotide binding protein (G protein), alpha inhibiting activity polypeptide 3 (GNAI3 [MIM 139370]), have been identified in the majority of ACS cases.12–14 Heterozygous missense substitutions, some of which represent hot spots, have been found within the catalytic domain of each protein and are predicted to result in dominant-negative effects on the wild-type version of each protein or other proteins. In addition, two cases have been ascribed to homozygous or compound-heterozygous loss-of-function mutations in PLCB4.12,14 It has been hypothesized that PLCB4 and GNAI3 function as signal transducers within the EDN1-EDNRA pathway. This is based on (1) the known involvement of G proteins and phospholipase C enzymes in endothelin signaling in other contexts, (2) the genetic interaction between edn1 and plcb3 (an ortholog of PLCB4) in zebrafish craniofacial development,15 (3) the demonstration that known EDNRA target genes DLX5 (MIM 600028) and DLX6 (MIM 600030) are downregulated in osteoblasts from PLCB4- or GNAI3-mutated ACS individuals, and (4) the interpretation of the abnormal mandibular skeleton in ACS as a mandibular-to-maxillary homeotic transformation, as for that of Edn1- or Ednra-null mice.13
After a screen for PLCB4 or GNAI3 mutations in a series of 11 ACS and IQME index cases, we were unable to identify a mutation or deletion at either locus in only three (cases 10, 11, and 12 in Gordon et al.12 and hereafter referred to as families F1, F2, and F3, respectively). Their clinical features are summarized in Table 1. F1 consists of consanguineous healthy parents, one unaffected child, and three ACS-affected siblings (Figures 1A and 2). Although the F1 father’s earlobes are mildly anteverted, this feature was not considered related to his children’s auricular phenotype. A computed-tomography (CT) scan of F1 individual II:3 revealed hypoplasia of the mandibular ramus and a thickened zygomatic process of the right temporal bone (Figure 1A). F1 individual II:4, previously unreported, presented with micrognathia, QMEs, microstomia, full cheeks, several small hamartomatous pedicles on the ventral surface of the tongue, a bilateral paramedian submucosal cleft of the velum, and a bifid uvula with ectopic tissue beside it (Figure 1A). Families F2 and F3 each consist of individuals with dominantly inherited IQMEs: an affected mother and daughter in F2 and affected individuals through four generations in F3 (Figures 1B, 1C, and 2). To identify coding variants responsible for ACS and IQMEs in families F1–F3, we performed whole-exome sequencing on two affected siblings from F1 (II:1 and II:3), under the assumption that each shares recessive alleles by descent, and one individual from each of F2 and F3 (I:2 and III:2, respectively), under the assumption that they harbor variants in the same gene or in genes plausibly linked to the EDN1-EDNRA pathway. Approval for human genetic research was obtained from the Comité de Protection des Personnes Ile-de-France II, and participants gave their written informed consent for genetic studies. Agilent SureSelect libraries were prepared from 3 μg of genomic DNA sheared with a Covaris S2 Ultrasonicator as recommended by the manufacturer. Exome capture was performed with the 51 Mb SureSelect Human All Exon Kit V5 (Agilent technologies) via a multiplex approach with molecular barcodes for traceable identity of samples. Sequencing was carried out on a pool of barcoded exome libraries with either a SOLiD5500XL (Life technologies) or a HiSeq2500 (Illumina) machine. 75+35 bp paired-end reads were generated with the SOLiD5500XL, and 76+76 bp paired-end reads were generated with the HiSeq2500. After demultiplexing, paired-end sequences were aligned to the reference human genome (hg19, UCSC Genome Browser) with LifeScope (Life Technologies) for SOLiD data and with the Burrows-Wheeler Aligner17 for Illumina data. The mean depth of coverage obtained for each sample was greater than 80×, and more than 90% of the exome was covered at least 15×. Downstream processing was carried out with the Genome Analysis Toolkit (GATK),18 SAMtools,19 and Picard according to documented best practices. Variant calls were made with the GATK Unified Genotyper. All calls with read coverage ≤2× or a Phred-scaled SNP quality score of ≤20 were removed from consideration. An in-house software system (PolyWeb) developed by the Plateforme Bioinformatique of Université Paris Descartes was used for annotating variants (based on Ensembl release 71)20 and filtering variants according to relevant genetic models in each case. We excluded known variants listed in the public databases dbSNP (build 135), the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project Exome Variant Server (release ESP6500SI-V2), and 1000 Genomes (release date May 21, 2011) and variants previously identified in more than 3,000 in-house exomes. We then selected variants affecting splice sites or coding regions (nonsynonymous substitutions, insertions, or deletions). Upon applying these criteria to each exome of the two affected F1 siblings and selecting for homozygous variants shared by both individuals, a single variant remained: c.271A>G (p.Lys91Glu) in EDN1 (RefSeq accession number NM_001955.4). Analysis of the distribution of variants in the exome data indicated that EDN1 falls within a shared homozygosity run of 9–12 Mb in the two siblings. After the same criteria as above were applied to each F2 and F3 exome (but only genes in which both individuals harbored a filtered heterozygous variant were selected), 27 variants in 12 genes were identified. From this list, only those variants predicted to be deleterious by both PolyPhen and SIFT programs were retained. Variants in two genes met these criteria: PLXNB3 (MIM 300214) and EDN1. The two variants in PLXNB3 (RefSeq NM_005393.2) were c.4141C>T (p.Leu1381Phe) (F2 individual) and c.4751G>A (p.Gly1584Asp) (F3 individual), but PLXNB3 was not considered further given the absence of data supporting a role for this gene in craniofacial development, its localization on the X chromosome, and the existence of recurrent frameshift variants listed in the NHLBI Exome Variant Server. The EDN1 mutations identified were c.191T>A (p.Val64Asp) for the F2 individual and c.249T>G (p.Tyr83∗) for the F3 individual. Each EDN1 mutation was validated by Sanger sequencing and segregated according to phenotype and mode of inheritance in each individual available for study in F1, F2, and F3 (Figure 2). In family F1, the unaffected parents and child were heterozygous for the mutation, whereas the three affected siblings were homozygous, indicating that the allele encoding the p.Lys91Glu substitution behaves in a recessive manner. Alternatively, in families F2 and F3, the mutations were present in affected members of each generation, indicating that the alleles encoding the p.Val64Asp and p.Tyr83∗ substitutions behave dominantly. Finally, we tested EDN1 directly in a previously published simplex case of an ACS individual born to consanguineous parents (patient 2 in Guion-Almeida et al.,16 hereafter referred to as S1; see Table 1). In addition to showing typical ACS features, he has tonsillar appendages16 and previously unreported sublingual appendices. He has mild bilateral ptosis and mild bilateral ectropion. As an adult, S1 has mild intellectual disability, obstructive respiratory distress, and sleep apnea. At age 23 years, hyperintensities in centrum semiovale and subcortical white matter were observed upon T2-weighted and fluid-attenuated inversion recovery MRI. Sanger sequencing revealed a private, homozygous c.230C>A (p.Pro77His) mutation in EDN1 (Figure 2). S1’s unaffected mother was heterozygous for the variant, consistent with the recessive nature of this allele.
Table 1.
Clinical Features of Individuals Studied in This Report
|
Identity in Present Report |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| F1:II-1 | F1:II-3 | F1:II-4 | F2:I-2 | F2:II-2 | F3:III-2 | F3:IV-1 | F3:IV-2 | S1:II-1 | |
| Identity in previous publication | case 10 (Gordon et al.12) | case 10 (Gordon et al.12) | − | case 11 (Gordon et al.12) | case 11 (Gordon et al.12) | case 12 (Gordon et al.12) | case 12 (Gordon et al.12) | case 12 (Gordon et al.12) | patient 2 (Guion-Almeida et al.16) |
| Inheritance | AR | AR | AR | AD | AD | AD | AD | AD | AR |
| Diagnosis | ACS | ACS | ACS | IQMEs | IQMEs | IQMEs | IQMEs | IQMEs | ACS |
| Gender | male | female | female | female | female | female | male | female | male |
| QMEs | + | + | + | + | + | + | + | + | + |
| Micrognathia and/or retrognathia | + | + | + | − | − | − | − | − | + |
| Oral anomalies | bifid uvula, laryngeal cleft | glossoptosis | bifid and ectopic uvula, lingual hamartomas, submucosal velar cleft | − | − | − | − | − | ectopic tonsillar and lingual appendages |
| Other phenotypes | vein of Galen dilation | bilateral conductive hearing loss with narrow auditory canals | − | − | reduced thyroid function | − | − | acute lymphoblastic leukemia | ptosis, ectropion, intellectual disability, sleep apnea, white-matter hyperintensities |
| EDN1 mutation | c.271A>G | c.271A>G | c.271A>G | c.191T>A | c.191T>A | c.249T>G | not tested | c.249T>G | c.230C>A |
| Protein change | p.Lys91Glu | p.Lys91Glu | p.Lys91Glu | p.Val64Asp | p.Val64Asp | p.Tyr83∗ | − | p.Tyr83∗ | p.Pro77His |
Abbreviations are as follows: ACS, auriculocondylar syndrome; AD, autosomal dominant; AR, autosomal recessive; and IQME, isolated question-mark ear.
Figure 1.
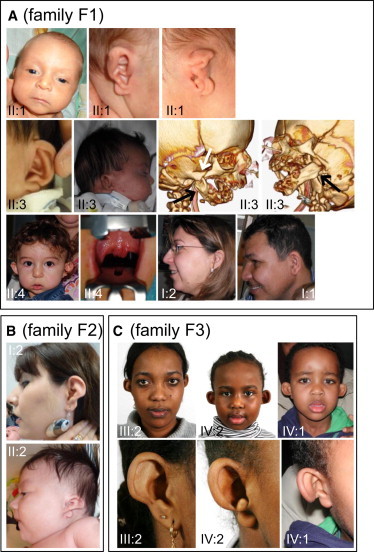
Craniofacial Features of ACS and IQME Individuals
Families F1 (A), F2, (B), and F3 (C). The numbering of individuals refers to the pedigrees in Figure 2. In (A), right and left lateral CT scans of individual II:3 are presented (black arrows indicate proximal mandibular hypoplasia, and the white arrow indicates the thickened zygomatic process of the right temporal bone), and an intraoral view of individual II:4 depicts the bifid uvula with adjacent ectopic tissue.
Figure 2.
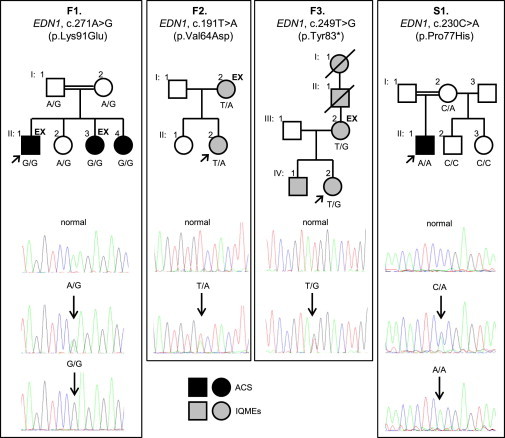
EDN1 Mutations and Their Segregation in ACS- or IQME-Affected Families F1–F3 and Simplex Case S1
“EX” indicates those individuals submitted to exome sequencing. The relevant EDN1 genotype is indicated for all those individuals whose DNA was available for testing.
EDN1, first identified in 1988 as a potent vasoconstrictor,21 is translated as a 212 amino acid preproprotein that undergoes a series of proteolytic processing events: cleavage by a signal peptidase to produce proEDN1, cleavage by furin at two sites in proEDN1 to liberate the 38 amino acid bigEDN1, and cleavage of bigEDN1 by ECE enzymes to produce the mature, bioactive EDN1 peptide of 21 amino acids (Figure 3). Furin, a member of the proprotein convertase family, cleaves a wide range of target proteins at basic amino acid consensus sites, several of which are altered in human diseases (reviewed in Thomas28 and Nakayama29). Furin cleavage occurs immediately C-terminally to each of two four-residue consensus sites in proEDN1: Arg-Ser-Lys-Arg (with amino acid position nomenclature P-4, P-3, P-2, and P-1, respectively) (Figure 3). The p.Lys91Glu substitution in family F1 affects the lysine at position P-2 in the C-terminal proEDN1 furin recognition site (Figure 3). A basic amino acid (either lysine or arginine) occurs at the P-2 position of EDN1 in all sequenced vertebrates (according to the Multiz alignment in the UCSC Genome Browser), whereas the F1 substitution (glutamic acid) is an acidic amino acid. The sensitivity of the furin recognition sites in endothelins to disruption is highlighted by substitutions at the P-4 and P-1 positions in proEDN3 in WS4 cases and in mouse lines with WS4-like phenotypes1,22,26,27 (Figure 3). Furthermore, a mutation resulting in substitution of the P-2 lysine of the furin recognition site in ectodysplasin A (encoded by EDA [MIM 300451]) has been identified in X-linked hypohidrotic ectodermal dysplasia (MIM 305100).30,31 In vitro assays of the ability of furin to cleave mutagenized substrates have indicated that the P-4 and P-1 sites are critical for cleavage, whereas the P-2 site is either as sensitive to disruption as P-4 and P-130 or less sensitive but nonetheless required for maximal furin activity.32–35 The above studies strongly support a causal role for the p.Lys91Glu substitution in the phenotype of affected F1 family members.
Figure 3.
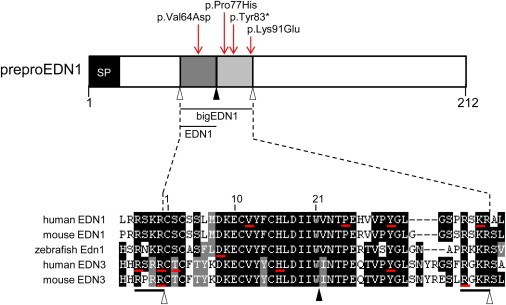
PreproEDN1 Domain Structure and Positions of Amino Acids Affected by Mutations
In the diagram at the top, bigEDN1 is shown in two shades of gray (the mature 21 amino acid peptide is the darker shade). Open arrowheads indicate furin cleavage sites. A filled arrowhead indicates the ECE cleavage site. Red arrows indicate the positions of substitutions identified in this report. The lower part of the figure is a multiple-sequence alignment of big endothelins and surrounding furin cleavage sites from selected species. Sequences used in the alignment were human EDN1 (RefSeq NP_001946.3), mouse EDN1 (RefSeq NP_034234.1), zebrafish Edn1 (RefSeq NP_571594.1), human EDN3 (RefSeq NP_000105.1), and mouse EDN3 (RefSeq NP_031929.1). The numbering above the alignment refers to the 21 amino acid mature peptide. Residues underlined with a red bar are affected by mutations in human EDN1 (this report), zebrafish edn1,11 human EDN3,1,22–25 and mouse Edn326,27 (see text for details). Black bars under the alignment indicate the two four-residue furin recognition sites. SP stands for signal peptide.
Active endothelin peptides are stabilized by two intramolecular disulphide bridges—one between Cys1 and Cys15 and one between Cys3 and Cys11 (numbering from the start of the mature 21 amino acid peptide; see Figure 3). The valine substituted by aspartic acid in family F2 corresponds to Val12 in mature EDN1 and is adjacent to the third cysteine. A valine at this position is highly conserved in EDN1, EDN2, and EDN3 in vertebrates. In an alanine-scanning mutagenesis study of EDN1, a p.Val12Ala substitution resulted in a peptide with binding capacity 16% of that of wild-type EDN1 in a cell-based binding assay and with agonist activity 29% of that of wild-type EDN1 in a rabbit vena cava organ-bath-culture contractility assay.36 Note that a valine-to-alanine substitution is a less chemically dissimilar change than the valine-to-aspartic-acid substitution identified in F2 (Grantham scores of 64 versus 152, respectively37); thus, the effect of the F2 mutation might be more severe than that predicted by the alanine-scanning study. Notably, the missense substitution in Edn1 in the zebrafish craniofacial mutant sucker falls at Asp8 of the mature peptide11 (Figure 3). Considering EDN3, substitutions affecting Thr2 (two different alterations) and His16 in the mature peptide have been identified in individuals with WS41,23,24 (Figure 3). Collectively these studies highlight the sensitivity of mature endothelin peptides to substitutions and support the pathogenicity of the EDN1 missense substitution in family F2. The p.Tyr83∗ substitution in family F3 falls within bigEDN1 and is C-terminal to the mature peptide in the region encoded by the third of the five exons of EDN1, presumably leading to a null allele via nonsense-mediated decay. p.Pro77His, the homozygous substitution in individual S1, also falls within bigEDN1 and is four amino acids C-terminal to the site of cleavage by ECE enzymes (Figure 3). A proline at this position is highly conserved in endothelin paralogs. In a mutagenesis study of selected residues in the C-terminal portion of bigEDN1, substitution of Pro77 (Pro25 numbering from the start of bigEDN1) to alanine resulted in a significant reduction in the conversion of bigEDN1 to EDN1, as assayed in the media of cells transfected with plasmids encoding wild-type or altered preproEDN1 and ECE1.38 In addition, p.Pro25Ala-transfected cells had less total recombinant protein (bigEDN1 plus EDN1) measured in the media than did cells transfected with wild-type preproEDN1, suggesting that this proline might play a role in bigEDN1 stability. These data argue that the p.Pro77His substitution in individual S1 is pathogenic.
The craniofacial phenotypes observed in the EDN1-mutated individuals reported here are consistent with, albeit less severe than, those of Edn1-homozygous-null mice, which demonstrated mandibular and auricular hypoplasia and cleft palate.5 As discussed below, the F1 and S1 EDN1 alleles might be hypomorphic, accounting for the fact that their phenotype was less severe in the homozygous state than that of Edn1-homozygous-null mice. In ACS, the mandible is most severely affected proximally, and hypoplasia of the condyle and the ramus are typical findings. A CT scan of individual II:3 in family F1 also indicated proximal mandibular hypoplasia. Interestingly, some mouse models with partially reduced dosage of various components of the EDN1 pathway display more severe defects in proximal regions of the mandible than in distal regions, as demonstrated, for example, in mice with combined heterozygous loss of Edn1, Dlx5, and Dlx639 and in Ednra−/− mice in which a knockin of the Ednrb cDNA at the Ednra locus rescues only distal regions of the mandibular phenotype.40 The morphology of the mandible in Edn1−/− and Ednra−/− mice is consistent with a homeotic transformation of the lower jaw into an upper-jaw-like structure,9,10 and a similar transformation has been proposed in individuals with mutations in PLCB4 and GNAI3.13 Soft-tissue changes on the floor of the oral cavity in Ednra−/− mice were suggestive of ectopic palatal rugae.10 Notably, ectopic sublingual, uvular, and tonsillar appendages were observed here in F1 and S1 individuals and previously in cases with PLCB4 and GNAI3 mutations,12,13 although it remains unclear whether these structures are related to a mandibular-to-maxillary identity switch. Edn1-knockout mice also display cardiac defects with incomplete penetrance.6 No congenital heart defects were identified upon echocardiography in individuals II:1 and II:3 of family F1 (the other individuals in this report did not present with symptoms of cardiac defects). Individual II:1 in family F1 presented with an aneurismal dilation of the vein of Galen, associated with two arteriovenous fistulae, but it remains unclear whether this was related to his EDN1 mutation.
The different mode of inheritance for the F1 and S1 mutant EDN1 alleles compared to the F2 and F3 alleles might be explained by differences in the amount of residual functional protein in each case. We suggest a model in which heterozygous-null EDN1 alleles result in IQMEs and in which hypomorphic alleles result in an ACS phenotype in homozygotes and no phenotype in heterozygotes. According to this model, homozygous EDN1 loss of function would lead to a more severe phenotype than classical ACS. This model also suggests that, surprisingly, the external ear is more sensitive than the mandible to perturbations of EDN1 signaling in humans. However, it remains possible that EDN1 mutations also underlie isolated mandibular defects. Interestingly, homozygous EDN3 mutations typically lead to WS4, comprising Hirschsprung disease, hearing loss, and pigmentation defects, whereas heterozygous carriers might display hearing loss and/or pigmentation defects only.1,24 Rare cases of heterozygous EDN3 mutations in WS4 or isolated Hirschsprung disease have also been reported.41,42 This could indicate the influence of modifying alleles on the phenotype associated with EDN3 mutations. It remains to be seen whether EDN1 mutations are also influenced by genetic background. Indeed, incomplete penetrance has been observed in several ACS cases with PLCB4 or GNAI3 mutations. A larger number of ACS and IQME cases harboring EDN1 mutations will aid our understanding of the molecular basis of the phenotypic variability reported here.
In conclusion, we have identified EDN1 as a third locus for the ACS and IQME spectrum of disorders. Among the PLCB4, GNAI3, and EDN1 loci, we have determined the molecular bases of all phenotypically convincing ACS and IQME cases screened at Hôpital Necker to date (a total of 19 index cases from the present report, Gordon et al.,12 Rieder et al.,13 and Kido et al.,14 and unpublished data). This suggests that any further genetic heterogeneity in ACS will be rare. The small number of IQME cases tested is not yet sufficient to exclude the possibility of other loci for this anomaly. The finding that recessive EDN1 mutations result in ACS argues that the phenotype caused by PLCB4 or GNAI3 mutations is due entirely to the effect of these mutations on the EDN1-EDNRA pathway and highlights the importance of this pathway for normal craniofacial development in humans.
Acknowledgments
J.A. was supported by a Université Paris Descartes – Sorbonne Paris Cité Pôle de Recherche et d’Enseignement Supérieur grant (project number SPC/JFG/2013-031). S.L. was supported by funding from the Agence Nationale de la Recherche (project EvoDevoMut 2010). J.A. and S.L. were supported by funding from E-Rare CRANIRARE. We thank Mohammed Zarhrate, Mélanie Parisot, and Olivier Alibeu from the Plateforme Génomique at the Institut Imagine for technical assistance.
Contributor Information
Christopher T. Gordon, Email: chris.gordon@inserm.fr.
Jeanne Amiel, Email: jeanne.amiel@inserm.fr.
Web Resources
The URLs for data presented herein are as follows:
GATK Best Practices, http://www.broadinstitute.org/gatk/guide/topic?name=best-practices
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Pingault V., Ente D., Dastot-Le Moal F., Goossens M., Marlin S., Bondurand N. Review and update of mutations causing Waardenburg syndrome. Hum. Mutat. 2010;31:391–406. doi: 10.1002/humu.21211. [DOI] [PubMed] [Google Scholar]
- 2.Cordero D.R., Brugmann S., Chu Y., Bajpai R., Jame M., Helms J.A. Cranial neural crest cells on the move: their roles in craniofacial development. Am. J. Med. Genet. A. 2011;155A:270–279. doi: 10.1002/ajmg.a.33702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Passos-Bueno M.R., Ornelas C.C., Fanganiello R.D. Syndromes of the first and second pharyngeal arches: A review. Am. J. Med. Genet. A. 2009;149A:1853–1859. doi: 10.1002/ajmg.a.32950. [DOI] [PubMed] [Google Scholar]
- 4.Sato T., Kurihara Y., Asai R., Kawamura Y., Tonami K., Uchijima Y., Heude E., Ekker M., Levi G., Kurihara H. An endothelin-1 switch specifies maxillomandibular identity. Proc. Natl. Acad. Sci. USA. 2008;105:18806–18811. doi: 10.1073/pnas.0807345105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kurihara Y., Kurihara H., Suzuki H., Kodama T., Maemura K., Nagai R., Oda H., Kuwaki T., Cao W.H., Kamada N. Elevated blood pressure and craniofacial abnormalities in mice deficient in endothelin-1. Nature. 1994;368:703–710. doi: 10.1038/368703a0. [DOI] [PubMed] [Google Scholar]
- 6.Kurihara Y., Kurihara H., Oda H., Maemura K., Nagai R., Ishikawa T., Yazaki Y. Aortic arch malformations and ventricular septal defect in mice deficient in endothelin-1. J. Clin. Invest. 1995;96:293–300. doi: 10.1172/JCI118033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yanagisawa H., Yanagisawa M., Kapur R.P., Richardson J.A., Williams S.C., Clouthier D.E., de Wit D., Emoto N., Hammer R.E. Dual genetic pathways of endothelin-mediated intercellular signaling revealed by targeted disruption of endothelin converting enzyme-1 gene. Development. 1998;125:825–836. doi: 10.1242/dev.125.5.825. [DOI] [PubMed] [Google Scholar]
- 8.Clouthier D.E., Hosoda K., Richardson J.A., Williams S.C., Yanagisawa H., Kuwaki T., Kumada M., Hammer R.E., Yanagisawa M. Cranial and cardiac neural crest defects in endothelin-A receptor-deficient mice. Development. 1998;125:813–824. doi: 10.1242/dev.125.5.813. [DOI] [PubMed] [Google Scholar]
- 9.Ozeki H., Kurihara Y., Tonami K., Watatani S., Kurihara H. Endothelin-1 regulates the dorsoventral branchial arch patterning in mice. Mech. Dev. 2004;121:387–395. doi: 10.1016/j.mod.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 10.Ruest L.B., Xiang X., Lim K.C., Levi G., Clouthier D.E. Endothelin-A receptor-dependent and -independent signaling pathways in establishing mandibular identity. Development. 2004;131:4413–4423. doi: 10.1242/dev.01291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller C.T., Schilling T.F., Lee K., Parker J., Kimmel C.B. sucker encodes a zebrafish Endothelin-1 required for ventral pharyngeal arch development. Development. 2000;127:3815–3828. doi: 10.1242/dev.127.17.3815. [DOI] [PubMed] [Google Scholar]
- 12.Gordon C.T., Vuillot A., Marlin S., Gerkes E., Henderson A., AlKindy A., Holder-Espinasse M., Park S.S., Omarjee A., Sanchis-Borja M. Heterogeneity of mutational mechanisms and modes of inheritance in auriculocondylar syndrome. J. Med. Genet. 2013;50:174–186. doi: 10.1136/jmedgenet-2012-101331. [DOI] [PubMed] [Google Scholar]
- 13.Rieder M.J., Green G.E., Park S.S., Stamper B.D., Gordon C.T., Johnson J.M., Cunniff C.M., Smith J.D., Emery S.B., Lyonnet S. A human homeotic transformation resulting from mutations in PLCB4 and GNAI3 causes auriculocondylar syndrome. Am. J. Hum. Genet. 2012;90:907–914. doi: 10.1016/j.ajhg.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kido Y., Gordon C.T., Sakazume S., Ben Bdira E., Dattani M., Wilson L.C., Lyonnet S., Murakami N., Cunningham M.L., Amiel J., Nagai T. Further characterization of atypical features in auriculocondylar syndrome caused by recessive PLCB4 mutations. Am. J. Med. Genet. A. 2013;161:2339–2346. doi: 10.1002/ajmg.a.36066. [DOI] [PubMed] [Google Scholar]
- 15.Walker M.B., Miller C.T., Swartz M.E., Eberhart J.K., Kimmel C.B. phospholipase C, beta 3 is required for Endothelin1 regulation of pharyngeal arch patterning in zebrafish. Dev. Biol. 2007;304:194–207. doi: 10.1016/j.ydbio.2006.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guion-Almeida M.L., Zechi-Ceide R.M., Vendramini S., Kokitsu-Nakata N.M. Auriculo-condylar syndrome: additional patients. Am. J. Med. Genet. 2002;112:209–214. doi: 10.1002/ajmg.10631. [DOI] [PubMed] [Google Scholar]
- 17.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Flicek P., Ahmed I., Amode M.R., Barrell D., Beal K., Brent S., Carvalho-Silva D., Clapham P., Coates G., Fairley S. Ensembl 2013. Nucleic Acids Res. 2013;41(Database issue):D48–D55. doi: 10.1093/nar/gks1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yanagisawa M., Kurihara H., Kimura S., Tomobe Y., Kobayashi M., Mitsui Y., Yazaki Y., Goto K., Masaki T. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332:411–415. doi: 10.1038/332411a0. [DOI] [PubMed] [Google Scholar]
- 22.Shamseldin H.E., Rahbeeni Z., Alkuraya F.S. Perturbation of the consensus activation site of endothelin-3 leads to Waardenburg syndrome type IV. Am. J. Med. Genet. A. 2010;152A:1841–1843. doi: 10.1002/ajmg.a.33123. [DOI] [PubMed] [Google Scholar]
- 23.Kapoor S., Bindu P.S., Taly A.B., Sinha S., Gayathri N., Rani S.V., Chandak G.R., Kumar A. Genetic analysis of an Indian family with members affected with Waardenburg syndrome and Duchenne muscular dystrophy. Mol. Vis. 2012;18:2022–2032. [PMC free article] [PubMed] [Google Scholar]
- 24.Viñuela A., Morín M., Villamar M., Morera C., Lavilla M.J., Cavallé L., Moreno-Pelayo M.A., Moreno F., del Castillo I. Genetic and phenotypic heterogeneity in two novel cases of Waardenburg syndrome type IV. Am. J. Med. Genet. A. 2009;149A:2296–2302. doi: 10.1002/ajmg.a.33026. [DOI] [PubMed] [Google Scholar]
- 25.Pingault V., Girard M., Bondurand N., Dorkins H., Van Maldergem L., Mowat D., Shimotake T., Verma I., Baumann C., Goossens M. SOX10 mutations in chronic intestinal pseudo-obstruction suggest a complex physiopathological mechanism. Hum. Genet. 2002;111:198–206. doi: 10.1007/s00439-002-0765-8. [DOI] [PubMed] [Google Scholar]
- 26.Baynash A.G., Hosoda K., Giaid A., Richardson J.A., Emoto N., Hammer R.E., Yanagisawa M. Interaction of endothelin-3 with endothelin-B receptor is essential for development of epidermal melanocytes and enteric neurons. Cell. 1994;79:1277–1285. doi: 10.1016/0092-8674(94)90018-3. [DOI] [PubMed] [Google Scholar]
- 27.Matera I., Cockroft J.L., Moran J.L., Beier D.R., Goldowitz D., Pavan W.J. A mouse model of Waardenburg syndrome type IV resulting from an ENU-induced mutation in endothelin 3. Pigment Cell Res. 2007;20:210–215. doi: 10.1111/j.1600-0749.2007.00371.x. [DOI] [PubMed] [Google Scholar]
- 28.Thomas G. Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat. Rev. Mol. Cell Biol. 2002;3:753–766. doi: 10.1038/nrm934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakayama K. Furin: a mammalian subtilisin/Kex2p-like endoprotease involved in processing of a wide variety of precursor proteins. Biochem. J. 1997;327:625–635. doi: 10.1042/bj3270625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Y., Molloy S.S., Thomas L., Gambee J., Bächinger H.P., Ferguson B., Zonana J., Thomas G., Morris N.P. Mutations within a furin consensus sequence block proteolytic release of ectodysplasin-A and cause X-linked hypohidrotic ectodermal dysplasia. Proc. Natl. Acad. Sci. USA. 2001;98:7218–7223. doi: 10.1073/pnas.131076098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schneider P., Street S.L., Gaide O., Hertig S., Tardivel A., Tschopp J., Runkel L., Alevizopoulos K., Ferguson B.M., Zonana J. Mutations leading to X-linked hypohidrotic ectodermal dysplasia affect three major functional domains in the tumor necrosis factor family member ectodysplasin-A. J. Biol. Chem. 2001;276:18819–18827. doi: 10.1074/jbc.M101280200. [DOI] [PubMed] [Google Scholar]
- 32.Walker J.A., Molloy S.S., Thomas G., Sakaguchi T., Yoshida T., Chambers T.M., Kawaoka Y. Sequence specificity of furin, a proprotein-processing endoprotease, for the hemagglutinin of a virulent avian influenza virus. J. Virol. 1994;68:1213–1218. doi: 10.1128/jvi.68.2.1213-1218.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krysan D.J., Rockwell N.C., Fuller R.S. Quantitative characterization of furin specificity. Energetics of substrate discrimination using an internally consistent set of hexapeptidyl methylcoumarinamides. J. Biol. Chem. 1999;274:23229–23234. doi: 10.1074/jbc.274.33.23229. [DOI] [PubMed] [Google Scholar]
- 34.Molloy S.S., Bresnahan P.A., Leppla S.H., Klimpel K.R., Thomas G. Human furin is a calcium-dependent serine endoprotease that recognizes the sequence Arg-X-X-Arg and efficiently cleaves anthrax toxin protective antigen. J. Biol. Chem. 1992;267:16396–16402. [PubMed] [Google Scholar]
- 35.Hatsuzawa K., Nagahama M., Takahashi S., Takada K., Murakami K., Nakayama K. Purification and characterization of furin, a Kex2-like processing endoprotease, produced in Chinese hamster ovary cells. J. Biol. Chem. 1992;267:16094–16099. [PubMed] [Google Scholar]
- 36.Tam J.P., Liu W., Zhang J.W., Galantino M., Bertolero F., Cristiani C., Vaghi F., de Castiglione R. Alanine scan of endothelin: importance of aromatic residues. Peptides. 1994;15:703–708. doi: 10.1016/0196-9781(94)90099-x. [DOI] [PubMed] [Google Scholar]
- 37.Grantham R. Amino acid difference formula to help explain protein evolution. Science. 1974;185:862–864. doi: 10.1126/science.185.4154.862. [DOI] [PubMed] [Google Scholar]
- 38.Brooks C., Ergul A. Identification of amino acid residues in the C-terminal tail of big endothelin-1 involved in processing to endothelin-1. J. Mol. Endocrinol. 1998;21:307–315. doi: 10.1677/jme.0.0210307. [DOI] [PubMed] [Google Scholar]
- 39.Vieux-Rochas M., Mantero S., Heude E., Barbieri O., Astigiano S., Couly G., Kurihara H., Levi G., Merlo G.R. Spatio-temporal dynamics of gene expression of the Edn1-Dlx5/6 pathway during development of the lower jaw. Genesis. 2010;48:262–373. doi: 10.1002/dvg.20625. [DOI] [PubMed] [Google Scholar]
- 40.Sato T., Kawamura Y., Asai R., Amano T., Uchijima Y., Dettlaff-Swiercz D.A., Offermanns S., Kurihara Y., Kurihara H. Recombinase-mediated cassette exchange reveals the selective use of Gq/G11-dependent and -independent endothelin 1/endothelin type A receptor signaling in pharyngeal arch development. Development. 2008;135:755–765. doi: 10.1242/dev.012708. [DOI] [PubMed] [Google Scholar]
- 41.Pingault V., Bondurand N., Lemort N., Sancandi M., Ceccherini I., Hugot J.P., Jouk P.S., Goossens M. A heterozygous endothelin 3 mutation in Waardenburg-Hirschsprung disease: is there a dosage effect of EDN3/EDNRB gene mutations on neurocristopathy phenotypes? J. Med. Genet. 2001;38:205–209. doi: 10.1136/jmg.38.3.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Svensson P.J., Von Tell D., Molander M.L., Anvret M., Nordenskjöld A. A heterozygous frameshift mutation in the endothelin-3 (EDN-3) gene in isolated Hirschsprung’s disease. Pediatr. Res. 1999;45:714–717. doi: 10.1203/00006450-199905010-00018. [DOI] [PubMed] [Google Scholar]