

Abstract
Background: Inhibition of inosine monophosphate dehydrogenase (IMPDH) by mycophenolic acid (MPA) can inhibit proliferation and induce apoptosis in cancer cells. This study investigated the underlying molecular mechanisms of MPA’s anticancer activity. Methods: A gastric cancer cell line (AGS) was treated with MPA and gene expression at different time points was analyzed using Illumina whole genome microarrays and selected genes were confirmed by real-time RT-PCR. Results: Transcriptomic profiling identified 1070 genes with ≥2 fold changes and 85 genes with >4 fold alterations. The most significantly altered biological processes by MPA treatment include cell cycle, apoptosis, cell proliferation and migration. MPA treatment altered at least ten KEGG pathways, of which eight (p53 signaling, cell cycle, pathways in cancer, PPAR signaling, bladder cancer, protein processing in ER, small cell lung cancer and MAPK signaling) are cancer-related. Among the earliest cellular events induced by MPA is cell cycle arrest which may be caused by six molecular pathways: 1) up-regulation of cyclins (CCND1 and CCNE2) and down-regulation of CCNA2 and CCNB1, 2) down-regulation of cyclin-dependent kinases (CDK4 and CDK5); 3) inhibition of cell division related genes (CDC20, CDC25B and CDC25C) and other cell cycle related genes (MCM2, CENPE and PSRC1), 4) activation of p53, which activates the cyclin-dependent kinase inhibitors (CDKN1A), 5) impaired spindle checkpoint function and chromosome segregation (BUB1, BUB1B, BOP1, AURKA, AURKB, and FOXM1); and 6) reduction of availability of deoxyribonucleotides and therefore DNA synthesis through down-regulation of the RRM1 enzyme. Cell cycle arrest is followed by inhibition of cell proliferation, which is mainly attributable to the inhibition of the PI3K/AKT/mTOR pathway, and caspase-dependent apoptosis due to up-regulation of the p53 and FAS pathways. Conclusions: These results suggest that MPA has beneficial anticancer activity through diverse molecular pathways and biological processes.
Keywords: MPA, drug repurposing, regulatory networks, microarray
Introduction
Inosine monophosphate dehydrogenase (IMPDH) is the rate-limiting enzyme for the de novo synthesis of guanosine nucleotides [1,2], which play crucial roles in cell proliferation and other cellular functions [3]. In many tumor cells, the expression of IMPDH, particularly IMPDH2, is significantly up-regulated [4,5]. Therefore, IMPDH is potentially a biomarker and target for cancer therapy. Mycophenolate mofetil (MMF) is the morpholinoethyl ester prodrug of mycophenolic acid (MPA), which is a potent uncompetitive inhibitor of IMPDH. It has been used for the prevention of acute graft rejection in transplantation [6,7]. MPA prevents graft rejection through blocking T and B lymphocyte proliferation and clonal expansion, and prevents the generation of cytotoxic T cells and other effector T cells. Therefore, it has long been hypothesized that MPA may also inhibit cancer cell proliferation. Indeed, a number of studies have reported the inhibitory role of MPA on cancer cell proliferation and induction of apoptosis in cancer cells [8-13]. We have recently evaluated the anticancer activity of MPA in 13 different cancer lines including stomach, colon, pancreas, liver, ovary and cervix cancer and leukemia [14]. Our results suggested that five cell lines (AGS, NCI-N87, HCT-8, A2780 and BxPC-3) were highly sensitive to MPA with IC50 <0.5 μg/ml, four cell lines (Hs746T, PANC-1, HepG2 and MCF-7) are very resistant to MPA with IC50 >20 μg/ml and the four other cell lines (KATO III, SNU-1, K562 and HeLa) have intermediate sensitivity. We and others also demonstrated the in vivo anticancer activity of MPA using xenograft mouse models [14].
Our comprehensive studies indicated that MPA can effectively induce cell cycle arrest and consequently inhibits cancer cell proliferation and eventually leading to cell death through caspase-dependent apoptosis. Our analyses using a targeted proteomics approach identified several proteins that may be implicated in MPA-induced cell cycle arrest, reduced proliferation and increased apoptosis [14]. However, our understanding of the molecular mechanism underlying MPA’s anticancer activity is incomplete. In this study, global transcriptomic profiling was carried out to construct the overall molecular network underlying MPA’s antitumor activity.
Materials and methods
Cell culture and reagents
Two gastric cancer cell lines (AGS and Hs746T) were obtained from the American Type Culture Collection (ATCC). Both cell lines were grown in RPMI 1640 medium containing 10% fetal bovine serum, 100 units/ml of penicillin and 100 μg/ml of streptomycin at 37°C with 5% CO2. MPA was purchased from VWR. Approximately 5x104 cells were seeded in 6-well plates and cultured overnight before MPA is added to the culture medium at a final concentration of 2 μg/ml. Cells were collected after 24, 48 and 72 hours of treatment.
Microarray experiments
Total RNA was extracted from AGS cells using a magnetic beads RNA extraction kit (Jinfiniti Biosciences, Augusta, GA). Gene expression profiling was performed using the human Illumina HumanHT-12 v4 BeadChip (Illumina, San Diego, CA). An aliquot of 200 ng of total RNA was converted into double stranded cDNA (ds-cDNA) by using the Illumina TargetAmp-Nano labeling kit with an oligo-dT primer containing a T7 RNA polymerase promoter (Genset, St. Louis, MO). In vitro transcription was performed on the above ds-cDNA using the Enzo RNA transcript labeling kit. Biotin-labeled cRNA was purified by using an RNeasy affinity column (Qiagen), and fragmented randomly to sizes ranging from 35-200 bases by incubating at 94°C for 35 min. The hybridization solutions contained 100 mM MES, 1 M Na+, 20 mM EDTA, and 0.01% Tween 20. The final concentration of fragmented cRNA was 0.05 μg/μl in hybridization solution. Target for hybridization was prepared by combining 40 μl of fragmented transcript with sonicated herring sperm DNA (0.1 mg/ml), BSA and 5 nM control oligonucleotide in a buffer containing 1.0 M NaCl, 10 mM Tris.HCl (pH7.6), and 0.005% Triton X-100. Target was hybridized for 16h at 45°C in an Illumina hybridization oven. Chips were then washed at 50°C with stringent solution, then again at 30°C with non-stringent washes. Arrays were then stained with streptavidin-Cy3. The microarray data are MIAME compliant and have been deposited in NCBI Gene Expression Omnibus and are accessible through GEO Series accession number GSE46671 (GEO reviewer link: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=hdkfdoisykeuybo&acc=GSE46671).
Microarray data analysis
All statistical analyses were performed using the R language and environment for statistical computing (www.r-project.org). The lumi package was used to preprocess microarray data. Differential expression analyses were conducted using the limma package from the Bioconductor project [15]. We used the false discovery rate (FDR) to adjust for multiple testing [16] B-statistics were calculated for each gene. A combination of adjusted p-value and absolute value of fold change (FC) were used for selecting the differentially expressed genes.
Cluster analysis was performed for grouping differentially expressed genes exhibiting similar expression patterns using the HPCluster program [17]. The Bioconductor package “GOstats” was used for testing the association of Gene Ontology terms (biological processes) to the differentially expressed genes [18]. A p-value based on the hypergeometric test was computed to assess whether the number of genes associated with the term is larger than expected. The p-values obtained were adjusted for multiple testing using the FDR. The microarray data was also analyzed using prebuild KEGG pathways. We used “spia” (signaling pathway impact analysis) package to identify the pathways impacted by the observed changes in the gene expression [19]. Network analysis was performed to construct and visualize molecular interaction networks using the STRING database [20]. The networks were imported in simple interaction format to Cytoscape [21].
Real-time RT-PCR analysis
An aliquot of total RNA (2 μg per sample) were arrayed in 96-well plates and then converted to cDNA using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). The cDNA products were used for quantitative real-time PCR using ready-to-use TaqMan gene expression assays from the Applied Biosystems. Five reference genes with relatively constant expression (GAPDH, ESD, GUSB, IPO8 and MRPL19) were used for normalizing RNA concentration. Real-time PCR was performed with 96x96 Fluidigm Expression chips.
Western blotting
Cells were harvested and resuspended in PBS. After centrifugation at 2000 rpm for 5 min, the pellet was lysed in ice cold M-PER Mammalian Protein Extraction Reagent containing 1% Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Scientific) for 30 min. The supernatant was collected after 10 min of centrifugation at 12000 rpm, equaled by spectrophotometry, denatured with sample loading buffer for 10 min at 95°C and stored at 4°C for future use. Proteins were separated by 10% SDS-polyacrylamide gels and transferred to PVDF membrane, and incubated with primary antibodies of interest at 4°C overnight. The appropriate horseradish-conjugated secondary antibody at a dilution of 1:10000 in blocking buffer (3% BSA-TBST) was added and incubated for 1h at room temperature. All antibodies were diluted in 3% BSA-TBST. Antibody for RRM1 was purchased from Cell Signaling.
Results
MPA alters expression of genes in multiple cancer-relevant pathways
The AGS cell line was used as a model to investigate the molecular mechanisms of MPA on cancer cells. A concentration of 2 μg/ml MPA was selected to treat AGS cells for 0, 12, 24, 48 and 72 hours. Gene expression was analyzed using Illumina microarrays. A total of 1070 genes were altered by ≥2 fold by MPA treatment and 85 genes were altered by >4 fold at one or more time points (Table 1). The significantly altered genes by MPA treatment were functionally annotated to identify the molecular pathways and biological functions. Among the prebuilt KEGG pathways, at least ten pathways are significantly altered by MPA and eight of these pathways (p53 signaling, cell cycle, pathways in cancer, PPAR signaling, bladder cancer, protein processing in ER, small cell lung cancer and MAPK signaling) are well-known to be cancer-related (Table 2).
Table 1.
Summary of differentially expressed genes
| 12H | 24H | 48H | 72H | |
|---|---|---|---|---|
| >4 fold up | 8 | 50 | 48 | 27 |
| 2-4 fold up | 185 | 335 | 315 | 265 |
| 2-4 fold down | 150 | 206 | 225 | 212 |
| >4 fold down | 5 | 9 | 11 | 9 |
Only those genes with false discovery rate (FDR) p values <0.05 are included in this table.
Table 2.
Major KEGG pathways altered in MPA treated AGS cells
| Pathway Name | Gene # | Gene # changed | pFDR-12 | Status-12 | pFDR-24 | Status-24 | pFDR-48 | Status-48 | pFDR-72 | Status-72 |
|---|---|---|---|---|---|---|---|---|---|---|
| p53 signaling pathway | 68 | 22 | 6E-11 | I | 7E-11 | I | 8E-11 | I | 9E-11 | I |
| Cell cycle | 123 | 22 | 5E-06 | I | 5E-06 | I | 4E-06 | I | 3E-06 | I |
| Pathways in cancer | 328 | 35 | 4E-04 | I | 3E-04 | A | 3E-04 | A | 3E-04 | I |
| Small cell lung cancer | 87 | 15 | 5E-04 | A | 2E-04 | A | 7E-05 | A | 1E-04 | A |
| PPAR signaling pathway | 71 | 10 | 2E-03 | I | 1E-03 | I | 1E-03 | I | 2E-03 | I |
| HTLV-I infection | 259 | 27 | 2E-03 | I | 2E-03 | I | 2E-03 | I | 2E-03 | I |
| Bladder cancer | 42 | 9 | 7E-03 | I | 7E-03 | A | 6E-03 | A | 7E-03 | A |
| Protein processing in ER | 163 | 18 | 1E-02 | I | 6E-03 | I | 8E-03 | I | 8E-03 | I |
| MAPK signaling pathway | 266 | 24 | 1E-02 | I | 3E-02 | I | 2E-02 | I | 2E-02 | I |
| Pathogenic E. coli infection | 54 | 9 | 2E-02 | I | 1E-02 | I | 1E-03 | I | 1E-03 | I |
I: inhibited; A: activated; pFDR: FDR p value for each time point (12, 24, 48 and 72 hours).
The most significantly altered biological processes by MPA treatment are shown in Table 3. The processes highly relevant to cancer include cell cycle, cell death, cell proliferation, cell migration, small molecule metabolic process and response to stress. For example, 14 cell proliferation genes are altered by >4-fold (p = 0.0027) and 129 cell proliferation genes are altered by 2-4 fold (p = 1.5x10-8). Similarly, 13 cell death genes are altered by >4-fold (p = 0.022) and 142 cell death genes are altered by 2-4 fold (p = 5.2x10-8). These results suggest that MPA treatment can significantly alter a number of cancer-related cell functions including induction of cell cycle arrest and apoptosis as well as inhibition of cell proliferation.
Table 3.
Major biological processes modified in MPA treated AGS cells
| Processes | ≥2 fold (1070) | >4 fold (85) | ||||
|---|---|---|---|---|---|---|
| UP: 620; Down: 450 | UP: 69; Down: 16 | |||||
|
|
|
|||||
| # Genes | p-value | OR | # Genes | p-value | OR | |
| Angiogenesis | 23 | 0.183643 | 1.25 | 4 | 0.05803 | 2.88 |
| Lipid metabolic process | 84 | 0.001398 | 1.46 | 10 | 0.01807 | 2.34 |
| Response to stress | 219 | 4.47E-09 | 1.64 | 23 | 0.00079 | 2.47 |
| Cell cycle | 112 | 6.61E-06 | 1.64 | 6 | 0.52851 | 1.04 |
| Cell death | 142 | 5.22E-08 | 1.72 | 13 | 0.02280 | 2.04 |
| Cell proliferation | 129 | 1.45E-08 | 1.81 | 14 | 0.00272 | 2.64 |
| Cell migration | 59 | 0.00419 | 1.49 | 10 | 0.00130 | 3.52 |
| Regulation of phosphate metabolic process | 77 | 0.000128 | 1.63 | 9 | 0.01561 | 2.52 |
| Cellular response to stress | 92 | 3.85E-05 | 1.63 | 6 | 0.33588 | 1.30 |
| Small molecule metabolic process | 195 | 1.09E-06 | 1.52 | 21 | 0.00187 | 2.35 |
| Regulation of angiogenesis | 12 | 0.098609 | 1.57 | 2 | 0.12597 | 3.36 |
| Blood vessel morphogenesis | 30 | 0.06548 | 1.38 | 6 | 0.00784 | 3.73 |
| Cell motility | 64 | 0.002427 | 1.51 | 11 | 0.00061 | 3.65 |
| Regulation of protein metabolic process | 115 | 3.40E-07 | 1.75 | 10 | 0.05236 | 1.92 |
Molecular changes underlying MPA-induced cell cycle arrest
The expression patterns of cell cycle genes altered by MPA treatment are shown in Figure 1A. These genes are clustered in three distinct groups. The cluster 1 genes are gradually up-regulated during MPA treatment and the cluster 2 genes are gradually and increasingly down-regulated over the course of treatment, while the cluster 3 genes are quickly down-regulated at the 12h but then up-regulated at the 48h time point. The most well-known cell cycle genes include cyclins, cyclin-dependent kinases (CDK), cell division cycle (CDC) genes and various cell cycle regulators.
Figure 1.
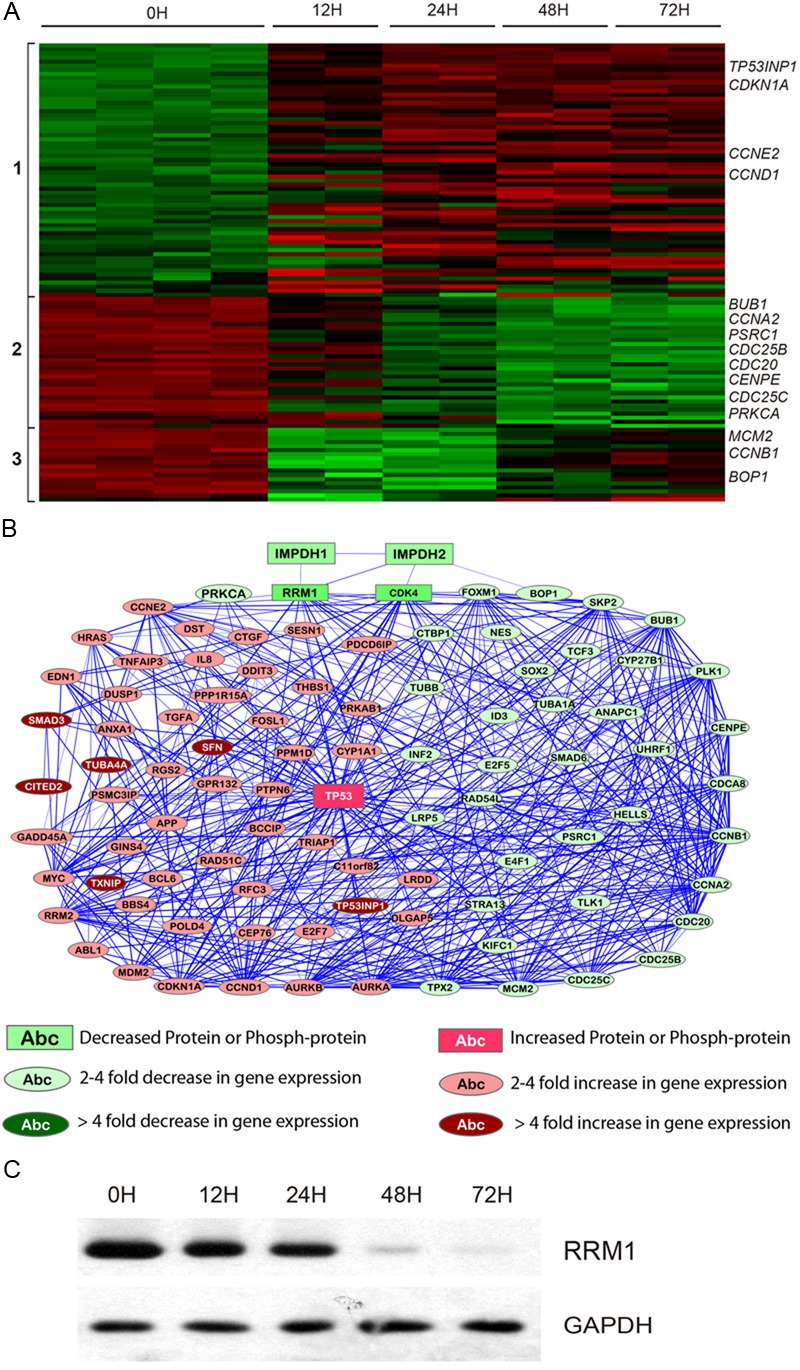
Molecular changes related to cell cycle arrest induced by MPA treatment. A: Heatmap for genes differentially expressed after MPA treatment for 0h, 12h, 24h, 48h and 72h. Each sample is represented in a column and each gene is represented in a row. Increased expression is indicated as red and decreased expression is indicated as green. Representative genes are shown on the right panel and gene clusters are indicated on the left. B: A network illustrating the connectedness of the genes (>2-fold differences) involved in cell cycle regulation. C: Western blot for RRM1.
Cyclins are a family of proteins that control the progression of cells through the cell cycle by activating cyclin-dependent kinases (CDK) [22,23]. Several different cyclins are active in different phases of the cell cycle and they cause the CDKs to phosphorylate different substrates. Cyclin D1 encoded by the CCND1 gene and Cyclin E encoded by the CCNE2 gene are essential for the control of the cell cycle at the G1/S transition. Cyclin A (CCNA2) regulates transition from G1 to S phase. Cyclin B (CCNB1) is essential for the control of the cell cycle at the G2/M transition (mitosis). Our microarray data suggest that CCND1 and CCNE2 are increased, while CCNA2 and CCNB1 are decreased in MPA treated AGS cells.
The expression of the CDK4 and CDK5 genes was significantly decreased (Table 4), consistent with our previous observation on reduction of CDK4 protein expression by MPA treatment [14]. Several cell division related genes (CDC20, CDC25B and CDC25C) and cell cycle related genes (MCM2, CENPE and PSRC1) are also decreased by MPA treatment (Table 4).
Table 4.
Selective genes differentially expressed after MPA treatment
| Genes | R12h | R24h | R48h | R72h | p-12h | p-24h | p-48h | p-72h |
|---|---|---|---|---|---|---|---|---|
| ABCA12 | 1.0 | 2.3 | 5.4 | 4.2 | 9.7E-01 | 3.7E-09 | 8.1E-15 | 2.6E-13 |
| ABCB9 | 2.3 | 3.4 | 4.3 | 2.9 | 6.2E-09 | 3.7E-12 | 1.4E-13 | 5.1E-11 |
| ATF3 | 5.6 | 6.3 | 7.0 | 6.8 | 3.8E-13 | 2.0E-14 | 6.5E-15 | 1.1E-14 |
| CDH10 | 1.5 | 3.1 | 4.8 | 7.4 | 2.6E-03 | 8.3E-10 | 2.8E-12 | 5.0E-14 |
| CEACAM1 | 2.8 | 6.0 | 5.3 | 2.9 | 1.9E-10 | 4.2E-15 | 1.3E-14 | 6.0E-11 |
| CITED2 | 2.2 | 2.2 | 4.6 | 3.5 | 1.5E-06 | 6.8E-07 | 1.9E-11 | 7.8E-10 |
| CYR61 | 2.7 | 3.5 | 4.9 | 3.5 | 8.1E-09 | 5.3E-11 | 8.3E-13 | 7.6E-11 |
| GRIN2C | 5.9 | 8.3 | 9.6 | 10.6 | 6.4E-14 | 4.3E-16 | 1.3E-16 | 5.8E-17 |
| NOS3 | 1.1 | 1.6 | 4.7 | 6.5 | 8.3E-01 | 4.3E-05 | 4.8E-14 | 1.9E-15 |
| NPPB | 2.9 | 8.9 | 17.0 | 11.6 | 1.4E-07 | 4.0E-13 | 3.1E-15 | 6.5E-14 |
| RN7SK | 3.6 | 12.1 | 81.6 | 176.9 | 3.7E-11 | 1.2E-16 | 6.2E-22 | 1.5E-23 |
| RNA28S5 | 1.0 | 1.2 | 15.9 | 40.4 | 9.2E-01 | 3.6E-01 | 3.1E-15 | 2.9E-17 |
| SLC30A1 | 5.3 | 6.3 | 3.8 | 3.4 | 7.6E-13 | 2.1E-14 | 6.3E-12 | 4.4E-11 |
| SLC30A1 | 4.6 | 5.5 | 3.9 | 4.0 | 2.4E-13 | 6.5E-15 | 3.5E-13 | 3.7E-13 |
| SLC31A2 | 2.8 | 4.1 | 2.9 | 3.4 | 4.9E-11 | 4.4E-14 | 7.3E-12 | 1.0E-12 |
| SMAD3 | 1.6 | 4.3 | 3.6 | 2.7 | 1.5E-04 | 1.2E-12 | 1.5E-11 | 1.0E-09 |
| TP53I3 | 5.4 | 12.0 | 11.0 | 7.7 | 1.2E-13 | 4.2E-17 | 5.7E-17 | 8.6E-16 |
| TP53INP1 | 3.2 | 4.2 | 4.3 | 4.0 | 7.1E-11 | 6.0E-13 | 4.5E-13 | 1.8E-12 |
| TXNIP | 2.7 | 3.1 | 5.5 | 6.1 | 2.5E-09 | 9.3E-11 | 7.5E-14 | 3.1E-14 |
| ARRB1 | 0.18 | 0.20 | 0.19 | 0.18 | 3.0E-14 | 8.1E-15 | 5.1E-15 | 2.8E-15 |
| CHN2 | 0.38 | 0.37 | 0.27 | 0.32 | 2.4E-09 | 4.1E-10 | 3.5E-12 | 6.0E-11 |
| CLDN2 | 0.53 | 0.37 | 0.23 | 0.22 | 5.9E-07 | 2.2E-10 | 2.0E-13 | 1.7E-13 |
| D2HGDH | 0.71 | 0.29 | 0.20 | 0.23 | 1.2E-03 | 3.6E-12 | 2.3E-14 | 2.0E-13 |
| ESRRG | 0.29 | 0.28 | 0.26 | 0.29 | 1.5E-08 | 4.7E-09 | 2.7E-09 | 1.3E-08 |
| HSPA5 | 0.22 | 0.18 | 0.25 | 0.28 | 1.8E-12 | 2.8E-14 | 1.4E-12 | 8.2E-12 |
| IGFBP5 | 0.14 | 0.10 | 0.10 | 0.09 | 1.2E-13 | 9.6E-16 | 1.0E-15 | 8.6E-16 |
| INF2 | 0.25 | 0.22 | 0.24 | 0.24 | 4.9E-11 | 4.4E-12 | 1.1E-11 | 1.7E-11 |
| LRP8 | 0.43 | 0.31 | 0.20 | 0.27 | 1.9E-08 | 4.6E-11 | 2.0E-13 | 7.2E-12 |
| NAV2 | 0.18 | 0.17 | 0.13 | 0.12 | 1.3E-12 | 1.7E-13 | 1.0E-14 | 6.6E-15 |
| PAGE4 | 0.63 | 0.34 | 0.14 | 0.10 | 2.9E-04 | 9.0E-10 | 2.3E-14 | 1.9E-15 |
| PDGFRA | 0.41 | 0.28 | 0.23 | 0.16 | 7.3E-09 | 5.1E-12 | 4.7E-13 | 1.1E-14 |
| PLA2G2A | 0.68 | 0.41 | 0.29 | 0.29 | 3.6E-03 | 5.6E-08 | 2.3E-10 | 4.4E-10 |
| PPARG | 0.56 | 0.33 | 0.28 | 0.35 | 2.2E-06 | 4.2E-11 | 2.3E-12 | 1.3E-10 |
| PRKCA | 0.31 | 0.35 | 0.28 | 0.27 | 3.7E-11 | 8.4E-11 | 2.8E-12 | 3.5E-12 |
| SNCAIP | 0.31 | 0.18 | 0.12 | 0.14 | 1.7E-10 | 7.3E-14 | 1.3E-15 | 5.4E-15 |
| AURKA | 0.73 | 0.68 | 0.64 | 0.54 | 1.3E-03 | 1.2E-04 | 1.6E-05 | 1.4E-07 |
| AURKB | 0.96 | 0.82 | 0.50 | 0.51 | 9.1E-01 | 1.8E-01 | 3.3E-06 | 5.0E-06 |
| FOXM1 | 0.79 | 0.65 | 0.42 | 0.54 | 1.3E-01 | 1.6E-03 | 1.5E-07 | 2.1E-05 |
| BOP1 | 0.48 | 0.34 | 0.41 | 0.57 | 3.4E-07 | 3.9E-10 | 8.6E-09 | 9.8E-06 |
| BUB1 | 0.66 | 0.52 | 0.39 | 0.42 | 1.8E-04 | 2.7E-07 | 6.4E-10 | 2.7E-09 |
| BUB1B | 0.61 | 0.66 | 0.62 | 0.56 | 3.0E-05 | 1.9E-04 | 2.8E-05 | 2.5E-06 |
| SRC | 0.97 | 0.82 | 0.67 | 0.56 | 9.2E-01 | 1.4E-01 | 9.0E-04 | 1.1E-05 |
R12h, R24h, R48h and R72h denote gene expression ratios at the respective time point over untreated control (0h). p-12h, p-24h, p48h and p72h denote p-values for the comparison between untreated control (0h) and the respective time point.
The cip/kip family (CDK interacting protein/Kinase inhibitory protein) prevents the progression of the cell cycle. Because of their instrumental role in prevention of tumor formation, these genes are known as tumor suppressors. The cip/kip family includes p21, p27 and p57, which halt cell cycle in G1 phase, by binding to, and inactivating, cyclin-CDK complexes. The p2-1 protein encoded by the CDKN1A gene is a potent cyclin-dependent kinase inhibitor (C-KI). The expression of this gene is tightly controlled by the tumor suppressor protein p53, through which this protein mediates the p53-dependent cell cycle G1 phase arrest in response to a variety of stress stimuli. Although the microarrays used in this study do not contain a probe for the p53 gene (TP53), a gene encoding a p53-induced protein (TP53INP1) was significantly increased, consistent with increased expression of p53 protein after MPA treatment [14].
Several kinases implicated in cell cycle control are significantly altered by MPA treatment. A prominently down-regulated gene is the PRKCA gene that plays an important role in cell cycle check point and many other cellular functions (Table 4). Furthermore, the BUB1 and BUB1B genes are down-regulated by MPA treatment (Figure 1 and Table 4). These results are consistent with our proteomic results indicating a dramatic reduction of the BUB1 protein after MPA treatment [14]. BUB1 and BUB1B encode two related mitotic checkpoint serine/threonine-protein kinases, which are involved in spindle checkpoint function. Many cancers have impaired spindle checkpoint function. The AURKA and AURKB genes encode the Aurora A and Aurora B kinases, which play important roles in mitosis. Aurora A kinase regulates spindle assembly and stability, while Aurora B kinase regulates chromosome segregation and cytokinesis. These two genes were significantly down-regulated by MPA treatment (Table 4).
Consistent with the protein expression changes in our previous study, the FOXM1 gene is significantly down-regulated by MPA treatment (Table 4). FOXM1 is known to play a key role in cell cycle progression. Endogenous FOXM1 expression peaks at S and G2/M phases [24]. FOXM1 knockout mice are neonatal lethal as a result of the development of polyploidy cardiomyocytes and hepatocytes, highlighting the role of FOXM1 in mitotic division. FOXM1 regulates expression of a large array of G2/M-specific genes including PLK1 and CENPE that are down-regulated by MPA (Figure 1). It plays an important role in maintaining chromosomal segregation and genomic stability [25].
The BOP1 gene encodes the ribosome biogenesis protein involved in rRNA processing, thereby controlling the cell cycle [26]. It is required for the maturation of the 25S and 5.8S ribosomal RNAs and it may serve as an essential factor in ribosome formation. The BOP1 complex is involved in ribosome biogenesis and altered chromosome segregation. The expression of BOP1 gene is significantly reduced by MPA treatment (Table 4). Consistent with the gene expression changes, the BOP1 protein is also significantly down-regulated by MPA treatment [14].
Network analysis revealed that many of the cell cycle genes altered by MPA are connected to the RRM1 gene. Although RRM1 gene expression is not altered by MPA, Western blot showed that the RRM1 protein is drastically reduced by MPA treatment (Figure 1C). However, it is still unknown why the RRM1 protein is significantly reduced, while the gene expression is not changed by MPA treatment. RRM1 is the large subunit of ribonucleoside-diphosphate reductase that is essential for the production of deoxyribonucleotides prior to DNA synthesis in S phase of dividing cells.
Molecular mechanisms underlying MPA-induced inhibition of cell proliferation
The 143 proliferation genes with >2-fold differential expression can be clustered into four groups (Figure 2A) and the functional network of the proliferation genes is presented in Figure 2B. The cluster 1 genes are slightly up-regulated at the 24h time point and then more highly up-regulated at later time points, while the cluster 2 genes are already highly up-regulated at the 12h time point but the up-regulation is lower at later time points. The cluster 3 genes are gradually down-regulated starting at the 24h time point, while the cluster 4 genes are already highly down-regulated at the 12h time point but then the expression goes up at later time points. MPA treatment increased the expression of a number of proliferation-inhibitory genes such as TXNIP, NOS3, NPPB, SMAD3 and transcription repressors ATF3, E2F7 and CITED2 and down-regulated several proliferation-promoting genes such as IGFBP5 and PPARG (Figure 2A and Table 4). Six most altered genes implicated in cell proliferation (IGFBP5, PPARG, PRKCA, TXNIP, PDGFRA and ATF3) were selected for real-time RT-PCR confirmation (Figure 3). All six genes were confirmed to be altered in AGS cells which are sensitive to MPA treatment, while only two of these genes (ATF3 and TXNIP) were altered in Hs746T cell that are resistant to MPA treatment (Figure 4).
Figure 2.
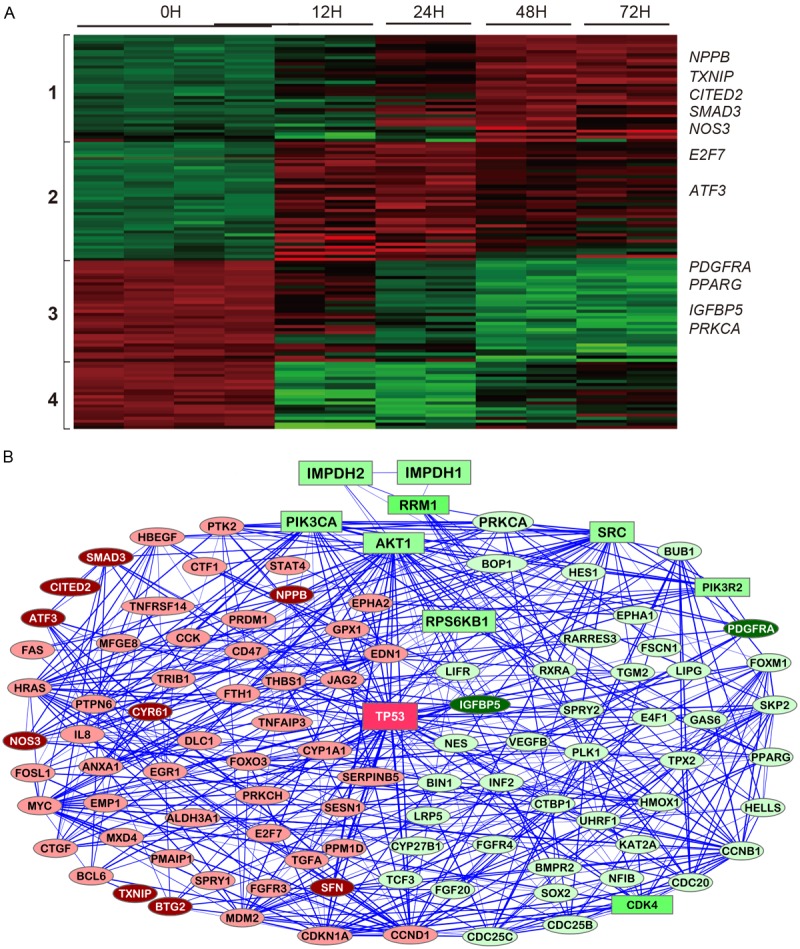
Molecular changes related to inhibition of cell proliferation induced by MPA treatment. A: Heatmap for genes differentially expressed after MPA treatment for 0h, 12h, 24h, 48h and 72h. B: A network illustrating the connectedness of the genes (>2-fold differences).
Figure 3.
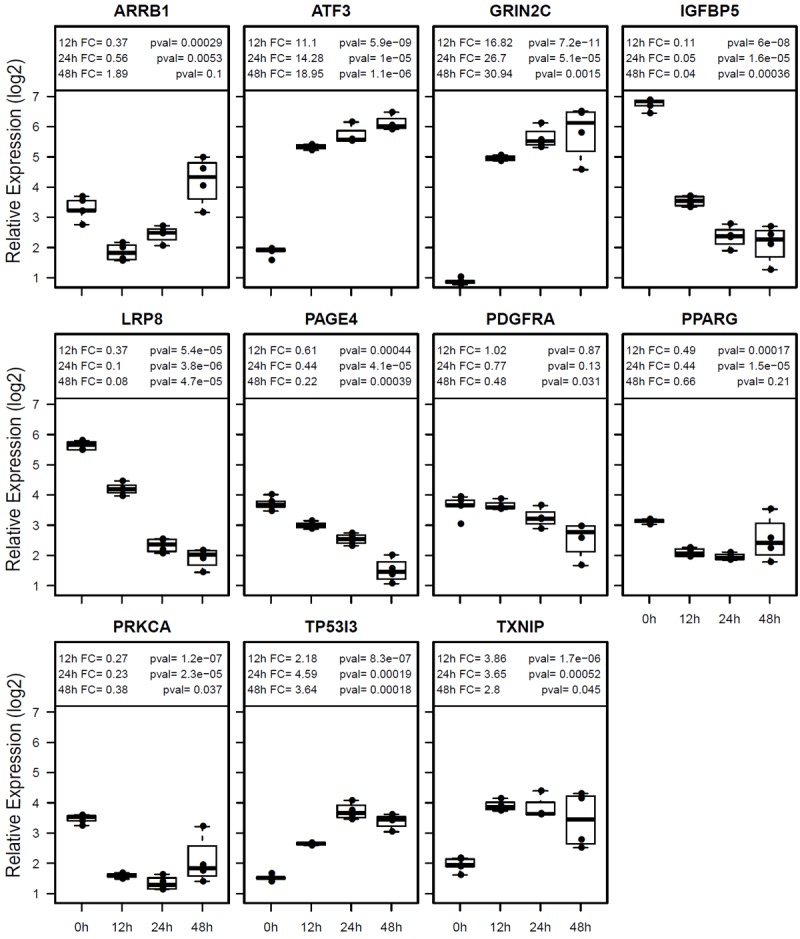
Confirmation of gene expression differences in AGS cells by real-time RT-PCR. Box blots are shown for each gene. Relative expression levels are shown in log 2 scale. FC (fold change) for each time point (12h, 24h and 48h) is compared to untreated controls (0h) with respective p-values.
Figure 4.
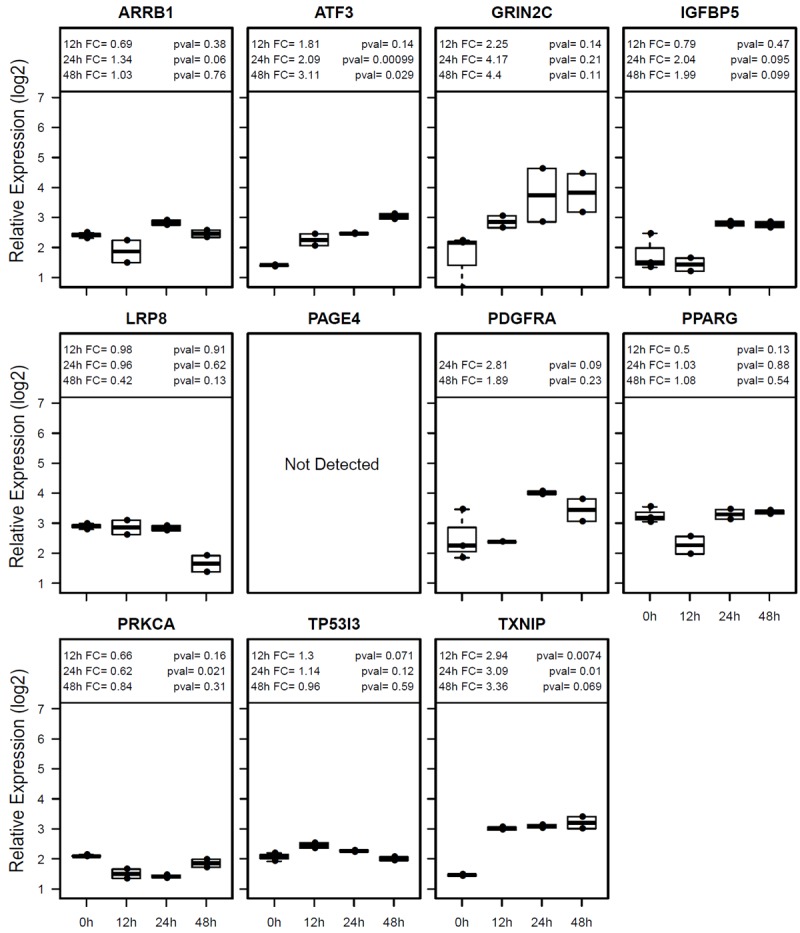
Gene expression analyses in Hs746T cells by real-time RT-PCR. Data presentation is identical to Figure 4.
MPA induces changes in genes implicated in apoptosis
Figure 5 shows the heatmap and functional network for all 142 cell death genes with 2-4 fold differences and 13 genes with >4-fold differences after MPA treatment. Four distinct clusters of genes are recognized. The cluster 1 genes are quickly and highly up-regulated at the 12h and 24h time points but then down-regulated at the 48h and 72h time points, while up-regulation becomes apparent only at the 48h time point for the cluster 2 genes. The cluster 3 genes are down-regulated starting at 24h time point, while the cluster 4 genes are quickly and highly down-regulated at the 12h and 24h time points but then slightly up-regulated at later time points. Eight of the most altered genes (TP53I3, TXNIP, PPARG, ARRB1, PAGE4, SNCAIP, IGFBP5 and PRKCA) were confirmed by real-time RT-PCR (Figure 3), while only one of these genes (TXNIP) was altered in the MPA-resistant Hs746T cells (Figure 4).
Figure 5.
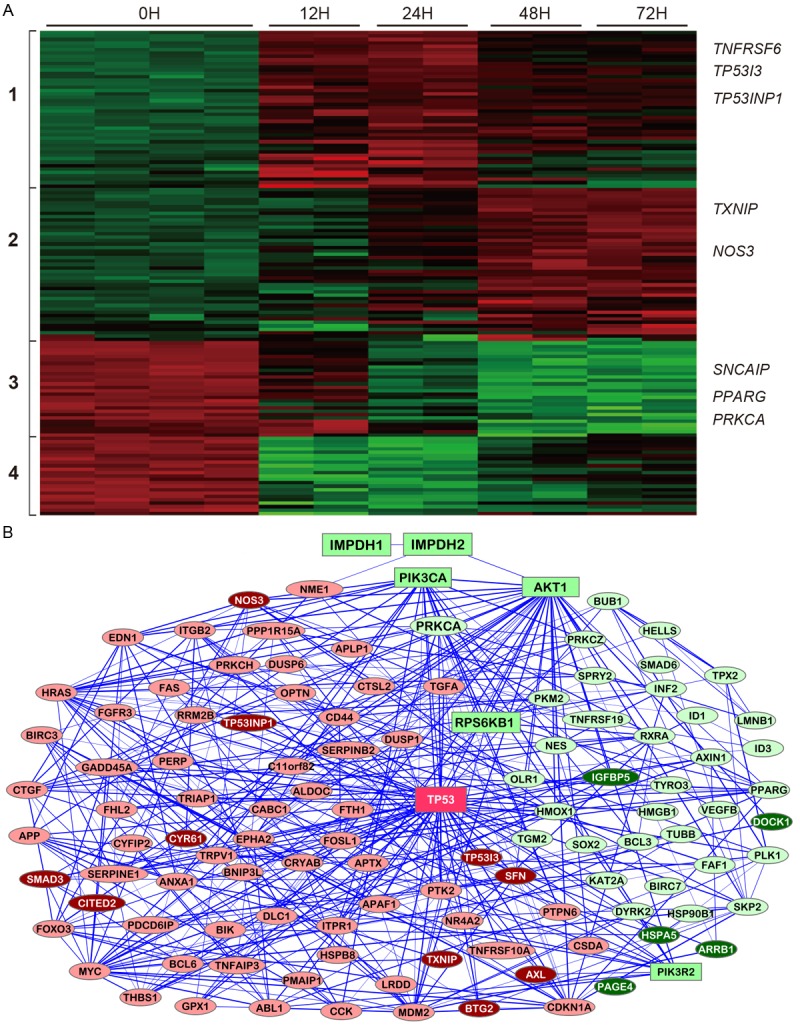
Molecular changes related to induction of apoptosis induced by MPA treatment. A: Heatmap for genes differentially expressed after MPA treatment for 0h, 12h, 24h, 48h and 72h. B: A network illustrating the connectedness of the genes (>2-fold differences).
TP53I3 is the most up-regulated gene related to cell death (Table 4) and encodes a protein similar to oxidoreductases, which are enzymes involved in cellular responses to oxidative stresses and irradiation. This gene, which is induced by the tumor suppressor p53, is thought to be involved in p53-mediated cell death. TP53INP1 (tumor protein 53-induced nuclear protein 1) is a tumor suppressor, whose expression is down-regulated in cancers from different organs. It is a p53 target gene involved in cell death, cell-cycle arrest and cellular migration. TP53INP1 could act as a tumor suppressor by inducing cell death by caspase-dependent autophagy [27]. TP53NP1 is quickly up-regulated (~4-fold) and remains highly expressed during the course of MPA treatment (Table 4).
The thioredoxin-interacting protein (TXNIP) is a critical node in the terminal unfolded protein response (UPR). When unfolded proteins accumulate to irremediably high levels within the endoplasmic reticulum (ER), the UPR signaling pathways become hyperactivated to cause programmed cell death [28]. The TXNIP gene is progressively up-regulated by MPA treatment as suggested by the microarray data and confirmed by real-time RT-PCR (Figure 3).
NOS3 (NO synthase-3) is the third most up-regulated cell death gene by MPA treatment (Table 4). NOS-3 overexpression increases oxidative/nitrosative stress, p53 and CD95 expression, caspase-8 activation and cell death in HepG2 cells. NOS-3 overexpression or NO donor injection into hepatoma-derived tumors reduced the size and increased p53 and cell death receptor expression in nude mice [29].
Prostate-associated gene 4 (PAGE4) is a member of the CT-X family of cancer/testis antigens that is highly expressed in testis and the fetal prostate and may also play an important role both in benign and malignant prostate diseases. PAGE4 is a DNA binding protein. Silencing PAGE4 expression induces cell death via apoptosis and knockdown of the PAGE4 gene attenuates tumor growth in prostate cancer xenograft mouse models. Furthermore, overexpression of PAGE4 protects cells from stress-induced death [30]. PAGE4 is drastically down-regulated by MPA treatment.
The Fas receptor is a death receptor on the surface of cells. The expression of the gene encoding Fas (TNFRSF6) is already significantly increased at 12h of MPA treatment (Figure 5 and Table 4).
Discussion
Previous studies have demonstrated that MPA can exert potent antitumor activities including cell cycle arrest, inhibition of proliferation, and induction of apoptosis [14]. Although the anticancer activities of MPA are now well established, it is still unclear how this drug exerts its functions. In this study, we used transcriptomic technologies to elucidate the molecular and biological pathways activated or inhibited by MPA. Transcriptomic profiling is a powerful technology to assess global molecular changes. The major advantage of gene expression microarray is its ability to rapidly and economically evaluate the expression pattern of almost all genes expressed in the cells of interest. Transcriptomic comparison of AGS cells treated with MPA for 0, 12, 24, 48 and 72 hours revealed over 1000 significantly altered genes (≥2-fold and p < 0.05). To validate our findings, we used real-time RT-PCR and confirmed the expression differences for genes selected for validation. Pathway analyses of the significantly altered genes allowed us to reveal the three main biological processes that were observed in the functional studies. Pathway analyses also identified ten molecular pathways that help account for MPA’s biological functions.
Global assessment during the course of MPA treatment allowed us to deduce the cascade of molecular and biological events depicted in Figure 6. We have previously shown that cell cycle arrest already occurred at the 12h time point, while alterations in cell proliferation and apoptosis only became apparent at the 24h time point [14]. MPA-induced cell cycle arrest appears to be the first and most important mechanism underlying MPA’s anticancer activity. Our microarray data presented in this manuscript as well as the confirmation data with RT-PCR and proteomic data from our previous targeted proteomic analyses [14] suggest that MPA can induce cell cycle arrest through six major pathways: 1) up-regulation of cyclins (CCND1 and CCNE2) and down-regulation of CCNA2 and CCNB1, 2) down-regulation of cyclin-dependent kinases (CDK4 and CDK5); 3) inhibition of cell division related genes (CDC20, CDC25B and CDC25C) and other cell cycle related genes (MCM2, CENPE and PSRC1), 4) activation of p53, which activates the cyclin-dependent kinase inhibitors (CDKN1A) and many other genes, 5) impaired spindle checkpoint function and chromosome segregation (BUB1, BUB1B, BOP1, AURKA, AURKB, and FOXM1); 6) reduction of availability of deoxyribonucleotides and therefore DNA synthesis through down-regulating the RRM1 enzyme. The drastically decreased RRM1 protein expression is of particular interest. RRM1 is an enzyme involved in the production of deoxyribonucleotides prior to DNA synthesis. As MPA’s main function is to specifically inhibit the IMPDH enzyme, the rate-limiting enzyme for the de novo synthesis of guanosine nucleotides, RRM1 protein reduction by MPA treatment should exacerbate its effect on DNA synthesis resulted from the reduced availability of guanosine nucleotides.
Figure 6.
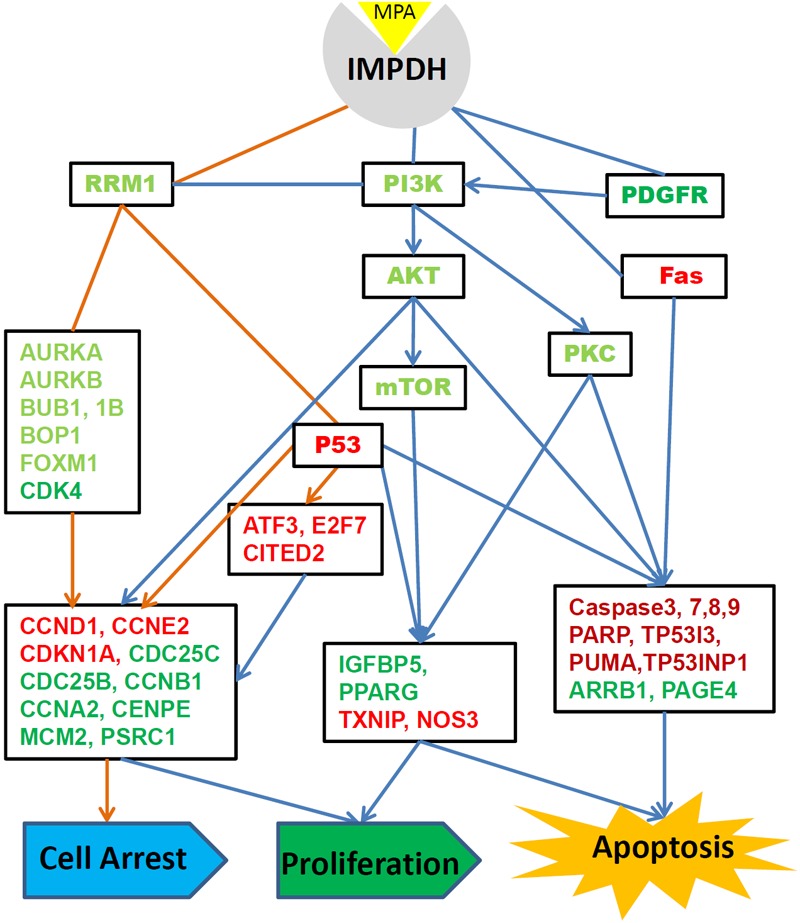
Overall molecular networks and biological functions altered by MPA treatment of AGS cells. Genes in light red and proteins in dark red are up-regulated. Genes in light green and proteins in dark green are down-regulated after MPA treatment.
MPA treatment alters the expression of over 100 genes implicated in cell proliferation. The most important events induced by MPA include the increased the expression of a number of proliferation-inhibitory genes such as TXNIP, NOS3, NPPB, SMAD3 and transcription repressors ATF3, E2F7 and CITED2 and down-regulation of proliferation-promoting genes such as IGFBP5 and PPARG. The functional network of the proliferation genes (Figure 2B) highlights the importance of the PI3K-AKT-mTOR pathway in the inhibition of AGS cell proliferation by MPA. The PI3K/AKT/mTOR pathway is activated by receptor tyrosine kinases (RTK). One of the key RTK involved in cancer is the PDGFR alpha which is encoded by the PDGFRA gene that was down-regulated by MPA treatment based on the microarray discovery data and RT-PCR confirmation data. Although the PIK3R2 gene which encodes the PI3K p85beta protein and the PIK3CA gene which encodes a PI3K p110a protein, are not significantly changed by MPA treatment, we previously demonstrated that MPA treatment slightly decreased PI3K p85beta protein expression but drastically decreased phosphorylated PI3K p110a in a time-dependent manner [14]. The expression of the AKT genes was not altered by MPA treatment and the total AKT protein only showed slight change at later time points; however, phospho-Akt (Ser473) was severely decreased by MPA treatment [14]. Furthermore, it was shown that the phospho-p70S6 kinase (Thr389), an mTOR component, was also dramatically down-regulated by MPA treatment. These results suggest that inhibition of the PI3K/AKT/mTOR pathway is a major mechanism through which MPA inhibits AGS cell proliferation. In many cancers, the PI3K/AKT/mTOR pathway is overactive, allowing proliferation of cancer cells [31-33].
The third largest group of genes altered by MPA treatment is related to regulation of apoptosis. Several molecular pathways appear to play a critical role in MPA-induced apoptosis. Two p53 induced genes (TP53I3 and TP53INP1) as well as the p53 protein [14] are up-regulated by MPA. The Fas pathway is another important cell death pathway that is activated by MPA treatment. Fas is one of the two apoptosis pathways, the other being the mitochondrial (or intrinsic) pathway [34]. Fas forms the death-inducing signaling complex (DISC) upon ligand binding. The receptor complex is internalized, allowing the adaptor molecule FADD to bind Fas [35]. FADD then binds caspase-8 that can self-activate through proteolytic cleavage. Active caspase-8 is then released from the DISC into the cytosol, where it cleaves other effector caspases, eventually leading to DNA degradation, membrane blebbing, and other hallmarks of apoptosis. Indeed, caspase-8, caspase 3 and caspase 7 are all highly activated in AGS cells after MPA treatment as shown in our previous studies [14], suggesting that the Fas-mediated apoptosis pathway is activated by MPA treatment.
In summary, our microarray data, together with RT-PCR confirmation and previous proteomic data, suggest that MPA alters a number of molecular pathways implicated in inducing cell cycle arrest, reduced proliferation and increased apoptosis. Our functional and molecular data suggest that the FDA-approved immunosuppressive drug MMF may be a good candidate that may be repurposed for anticancer therapy.
Acknowledgements
JXS is partially supported by the Georgia Research Alliance as an eminent scholar.
Disclosure of conflict of interest
The authors have declared that no competing interests exist.
References
- 1.Morath C, Schwenger V, Beimler J, Mehrabi A, Schmidt J, Zeier M, Muranyi W. Antifibrotic actions of mycophenolic acid. Clin Transplant. 2006;20(Suppl 17):25–29. doi: 10.1111/j.1399-0012.2006.00597.x. [DOI] [PubMed] [Google Scholar]
- 2.Allison AC, Eugui EM. Mycophenolate mofetil and its mechanisms of action. Immunopharmacology. 2000;47:85–118. doi: 10.1016/s0162-3109(00)00188-0. [DOI] [PubMed] [Google Scholar]
- 3.Jackson RC, Weber G, Morris HP. IMP dehydrogenase, an enzyme linked with proliferation and malignancy. Nature. 1975;256:331–333. doi: 10.1038/256331a0. [DOI] [PubMed] [Google Scholar]
- 4.Fellenberg J, Bernd L, Delling G, Witte D, Zahlten-Hinguranage A. Prognostic significance of drug-regulated genes in high-grade osteosarcoma. Mod Pathol. 2007;20:1085–1094. doi: 10.1038/modpathol.3800937. [DOI] [PubMed] [Google Scholar]
- 5.Fellenberg J, Kunz P, Sahr H, Depeweg D. Overexpression of inosine 5’-monophosphate dehydrogenase type II mediates chemoresistance to human osteosarcoma cells. PLoS One. 2010;5:e12179. doi: 10.1371/journal.pone.0012179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Millan O, Oppenheimer F, Brunet M, Vilardell J, Rojo I, Vives J, Martorell J. Assessment of mycophenolic acid-induced immunosuppression: a new approach. Clin Chem. 2000;46:1376–1383. [PubMed] [Google Scholar]
- 7.Allison AC, Eugui EM. Mechanisms of action of mycophenolate mofetil in preventing acute and chronic allograft rejection. Transplantation. 2005;80:S181–190. doi: 10.1097/01.tp.0000186390.10150.66. [DOI] [PubMed] [Google Scholar]
- 8.Takebe N, Cheng XF, Fandy TE, Srivastava RK, Wu SL, Shankar S, Bauer K, Shaughnessy J, Tricot G. IMP dehydrogenase inhibitor mycophenolate mofetil induces caspase-dependent apoptosis and cell cycle inhibition in multiple myeloma cells. Mol Cancer Ther. 2006;5:457–466. doi: 10.1158/1535-7163.MCT-05-0340. [DOI] [PubMed] [Google Scholar]
- 9.Gortz A, Franklin TJ, Dive C, Hickman JA. Cell cycle specific induction of HL-60 cell differentiation and apoptosis by mycophenolic acid. Cell Death Differ. 1997;4:787–795. doi: 10.1038/sj.cdd.4400300. [DOI] [PubMed] [Google Scholar]
- 10.Vegso G, Sebestyen A, Paku S, Barna G, Hajdu M, Toth M, Jaray J, Kopper L. Antiproliferative and apoptotic effects of mycophenolic acid in human B-cell non-Hodgkin lymphomas. Leuk Res. 2007;31:1003–1008. doi: 10.1016/j.leukres.2006.12.019. [DOI] [PubMed] [Google Scholar]
- 11.Silva SL, Silva SF, Cavalcante RO, Mota RS, Carvalho RA, Moraes MO, Campos HH, Moraes ME. Mycophenolate mofetil attenuates Walker’s tumor growth when used alone, but the effect is lost when associated with cyclosporine. Transplant Proc. 2004;36:1004–1006. doi: 10.1016/j.transproceed.2004.03.057. [DOI] [PubMed] [Google Scholar]
- 12.Domhan S, Muschal S, Schwager C, Morath C, Wirkner U, Ansorge W, Maercker C, Zeier M, Huber PE, Abdollahi A. Molecular mechanisms of the antiangiogenic and antitumor effects of mycophenolic acid. Mol Cancer Ther. 2008;7:1656–1668. doi: 10.1158/1535-7163.MCT-08-0193. [DOI] [PubMed] [Google Scholar]
- 13.Tressler RJ, Garvin LJ, Slate DL. Anti-tumor activity of mycophenolate mofetil against human and mouse tumors in vivo. Int J Cancer. 1994;57:568–573. doi: 10.1002/ijc.2910570421. [DOI] [PubMed] [Google Scholar]
- 14.Dun B, Xu H, Sharma A, Liu H, Yu H, Yi B, Liu X, He M, Zeng L, She JX. Delineation of biological and molecular mechanisms underlying the anticancer activities of mycophenolic acid. Int J Clin Exp Pathol. 2013 [Epub ahead of print] [PMC free article] [PubMed] [Google Scholar]
- 15.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 16.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc B. 1995;57:289–300. [Google Scholar]
- 17.Sharma A, Podolsky R, Zhao J, McIndoe RA. A modified hyperplane clustering algorithm allows for efficient and accurate clustering of extremely large datasets. Bioinformatics. 2009;25:1152–1157. doi: 10.1093/bioinformatics/btp123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Falcon S, Gentleman R. Using GOstats to test gene lists for GO term association. Bioinformatics. 2007;23:257–258. doi: 10.1093/bioinformatics/btl567. [DOI] [PubMed] [Google Scholar]
- 19.Tarca AL, Draghici S, Khatri P, Hassan SS, Mittal P, Kim JS, Kim CJ, Kusanovic JP, Romero R. A novel signaling pathway impact analysis. Bioinformatics. 2009;25:75–82. doi: 10.1093/bioinformatics/btn577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C, Jensen LJ. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41:D808–815. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nigg EA. Cyclin-dependent protein kinases: key regulators of the eukaryotic cell cycle. Bioessays. 1995;17:471–480. doi: 10.1002/bies.950170603. [DOI] [PubMed] [Google Scholar]
- 23.Ortonne JP. Redefining clinical response in psoriasis: targeting the pathological basis of disease. J Drugs Dermatol. 2004;3:13–20. [PubMed] [Google Scholar]
- 24.Wierstra I, Alves J. FOXM1, a typical proliferation-associated transcription factor. Biol Chem. 2007;388:1257–1274. doi: 10.1515/BC.2007.159. [DOI] [PubMed] [Google Scholar]
- 25.Laoukili J, Kooistra MR, Bras A, Kauw J, Kerkhoven RM, Morrison A, Clevers H, Medema RH. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat Cell Biol. 2005;7:126–136. doi: 10.1038/ncb1217. [DOI] [PubMed] [Google Scholar]
- 26.Kim J, Goo SY, Chung HJ, Yang HW, Yong TS, Lee KH, Park SJ. Interaction of beta-giardin with the Bop1 protein in Giardia lamblia. Parasitol Res. 2006;98:138–144. doi: 10.1007/s00436-005-0040-8. [DOI] [PubMed] [Google Scholar]
- 27.Seillier M, Peuget S, Gayet O, Gauthier C, N’Guessan P, Monte M, Carrier A, Iovanna JL, Dusetti NJ. TP53INP1, a tumor suppressor, interacts with LC3 and ATG8-family proteins through the LC3-interacting region (LIR) and promotes autophagy-dependent cell death. Cell Death Differ. 2012;19:1525–1535. doi: 10.1038/cdd.2012.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lerner AG, Upton JP, Praveen PV, Ghosh R, Nakagawa Y, Igbaria A, Shen S, Nguyen V, Backes BJ, Heiman M, Heintz N, Greengard P, Hui S, Tang Q, Trusina A, Oakes SA, Papa FR. IRE1alpha induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 2012;16:250–264. doi: 10.1016/j.cmet.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gonzalez R, Ferrin G, Aguilar-Melero P, Ranchal I, Linares CI, Bello RI, De la Mata M, Gogvadze V, Barcena JA, Alamo JM, Orrenius S, Padillo FJ, Zhivotovsky B, Muntane J. Targeting hepatoma using nitric oxide donor strategies. Antioxid Redox Signal. 2013;18:491–506. doi: 10.1089/ars.2011.4476. [DOI] [PubMed] [Google Scholar]
- 30.Zeng Y, He Y, Yang F, Mooney SM, Getzenberg RH, Orban J, Kulkarni P. The cancer/testis antigen prostate-associated gene 4 (PAGE4) is a highly intrinsically disordered protein. J Biol Chem. 2011;286:13985–13994. doi: 10.1074/jbc.M110.210765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morgensztern D, McLeod HL. PI3K/Akt/mTOR pathway as a target for cancer therapy. Anticancer Drugs. 2005;16:797–803. doi: 10.1097/01.cad.0000173476.67239.3b. [DOI] [PubMed] [Google Scholar]
- 32.Yap TA, Garrett MD, Walton MI, Raynaud F, de Bono JS, Workman P. Targeting the PI3K-AKT-mTOR pathway: progress, pitfalls, and promises. Curr Opin Pharmacol. 2008;8:393–412. doi: 10.1016/j.coph.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 33.Cortot A, Armand JP, Soria JC. [PI3K-AKT-mTOR pathway inhibitors] . Bull Cancer. 2006;93:19–26. [PubMed] [Google Scholar]
- 34.Wajant H. The Fas signaling pathway: more than a paradigm. Science. 2002;296:1635–1636. doi: 10.1126/science.1071553. [DOI] [PubMed] [Google Scholar]
- 35.Huang B, Eberstadt M, Olejniczak ET, Meadows RP, Fesik SW. NMR structure and mutagenesis of the Fas (APO-1/CD95) death domain. Nature. 1996;384:638–641. doi: 10.1038/384638a0. [DOI] [PubMed] [Google Scholar]