

Abstract
Aims
The aim of this study was to assess the tolerability, pharmacokinetics and inhibitory effect on erythrocyte soluble catechol-O-methyltransferase (S-COMT) activity following repeated doses of opicapone.
Methods
This randomized, placebo-controlled, double-blind study enrolled healthy male subjects who received either once daily placebo or opicapone 5, 10, 20 or 30 mg for 8 days.
Results
Opicapone was well tolerated. Its systemic exposure increased in an approximately dose-proportional manner with an apparent terminal half-life of 1.0 to 1.4 h. Sulphation was the main metabolic pathway. Opicapone metabolites recovered in urine accounted for less than 3% of the amount of opicapone administered suggesting that bile is likely the main route of excretion. Maximum S-COMT inhibition (Emax) ranged from 69.9% to 98.0% following the last dose of opicapone. The opicapone-induced S-COMT inhibition showed a half-life in excess of 100 h, which was dose-independent and much longer than plasma drug exposure. Such a half-life translates into a putative underlying rate constant that is comparable with the estimated dissociation rate constant of the COMT–opicapone complex.
Conclusion
Despite its short elimination half-life, opicapone markedly and sustainably inhibited erythrocyte S-COMT activity making it suitable for a once daily regimen.
Keywords: catechol-O-methyltransferase, COMT inhibition, opicapone, pharmacodynamics, pharmacokinetics
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Currently available catechol-O-methyltransferase (COMT) inhibitors used as adjunctive therapy in levodopa-treated Parkinson's disease patients have clinical limitations. Due to liver toxicity, the use of tolcapone requires liver function monitoring. Entacapone is considered to be safe, but its efficacy is limited and it requires frequent dosing. Opicapone is a novel third generation COMT inhibitor designed to provide high COMT inhibitory potency and avoid cell toxicity.
WHAT THIS STUDY ADDS
Based on the observation that the duration of COMT inhibition after opicapone administration is dose-independent and that it reflects an underlying kinetic process that is consistent with the dissociation rate constant of the COMT-opicapone complex, we propose that the sustained COMT inhibition, far beyond the observable point of clearance of circulating drug, is due to the long residence time of the reversible COMT–opicapone complex. The results clearly suggest that opicapone has a unique pharmacodynamic profile adequate for a once daily regimen, which in the treatment of Parkinson's disease patients could represent an advantage over entacapone and tolcapone.
Introduction
Despite decades of clinical use, levodopa still remains the most effective symptomatic treatment in Parkinson's disease (PD) [1, 2]. The therapeutic effect of levodopa depends on its biotransformation to dopamine in the brain. However, levodopa undergoes rapid and extensive metabolism by peripheral aromatic L-amino acid decarboxylase (AADC) and catechol-O-methyltransferase (COMT) and only 1% of an oral dose of levodopa actually reaches the brain [3, 4]. Therefore, levodopa is usually co-administered with an AADC inhibitor (carbidopa or benserazide), which increases levodopa bioavailability, but still approximately 90% of a levodopa dose is converted by COMT to 3-O-methyldopa, which competes with levodopa at the level of the blood–brain barrier for transport [5–8]. Thus, an additional strategy to inhibit peripheral levodopa metabolism further and increase the delivery of levodopa to the brain is the administration of a COMT inhibitor [9, 10]. Only two COMT inhibitors (tolcapone and entacapone) are currently available for clinical use, and both have some clinical limitations. The use of tolcapone requires liver function monitoring and thus is limited to fluctuating patients poorly controlled with other therapies [11]. Entacapone is considered to be safe [12], but its efficacy is limited and it requires frequent dosing [3]. Therefore, there is a need for more efficacious and safer COMT inhibitors.
Opicapone (2,5-dichloro-3-[5-(3,4-dihydroxy-5-nitrophenyl]-1,2,4-oxadiazol-3-yl)-4,6-dimethylpyridine 1-oxide, also known as BIA 9–1067) (Figure 1) is a novel third generation COMT inhibitor currently under phase III clinical trials by BIAL – Portela & Cª, S.A. (S. Mamede do Coronado, Portugal) for use as adjunctive therapy in levodopa-treated PD patients. Opicapone was designed as a hydrophilic 1,2,4-oxadiazole analogue with a pyridine N-oxide residue at position 3 to provide high COMT inhibitory potency and avoid cell toxicity [13]. Opicapone is endowed with an exceptionally high binding affinity (sub-picomolar Kd) [14] that translates into a slow complex dissociation rate constant and a long duration of action in vivo [15, 16]. In liver and brain homogenates from rats administered opicapone, tolcapone and entacapone by gastric tube, opicapone was shown to have a stronger and more sustained COMT inhibitory effect than the comparing COMT inhibitors, tolcapone and entacapone. One hour after administration, COMT inhibition was 99% with opicapone vs. 82% with tolcapone and 68% with entacapone. Nine hours after administration, entacapone showed no COMT inhibition and tolcapone produced minimal inhibitory effect (16%), whereas opicapone continued to inhibit COMT activity by 91% [15]. Opicapone was well tolerated in studies in several animal species (data on file – BIAL – Portela & Cª, S.A.). In an entry into man study in healthy male volunteers, single doses of opicapone ranging from 10 mg to 1200 mg were very well tolerated [17]. The adverse event (AE) profile did not differ from that of placebo and the clinical safety tests showed no signs for concern. The extent of systemic exposure to opicapone increased in an approximately dose-proportional manner. Despite the relatively short half-life (t1/2) of opicapone (0.8 to 3.2 h), a dose-dependent and long-lasting COMT inhibitory effect was observed. Maximum erythrocyte soluble COMT (S-COMT) inhibition (Emax) ranged from 34.5% (opicapone 10 mg) to 100% (opicapone 1200 mg), and an inhibition of 25.1 to 76.5% remained 24 h post-dose. On the basis of these promising results, it was decided to proceed to further clinical trials with opicapone. In patients with PD, opicapone as adjunctive therapy to a combination of levodopa/AADC inhibitor increased levodopa systemic exposure, decreased 3-O-methyldopa exposure, decreased S-COMT activity and improved patients' motor performance in clinical studies [18, 19].
Figure 1.
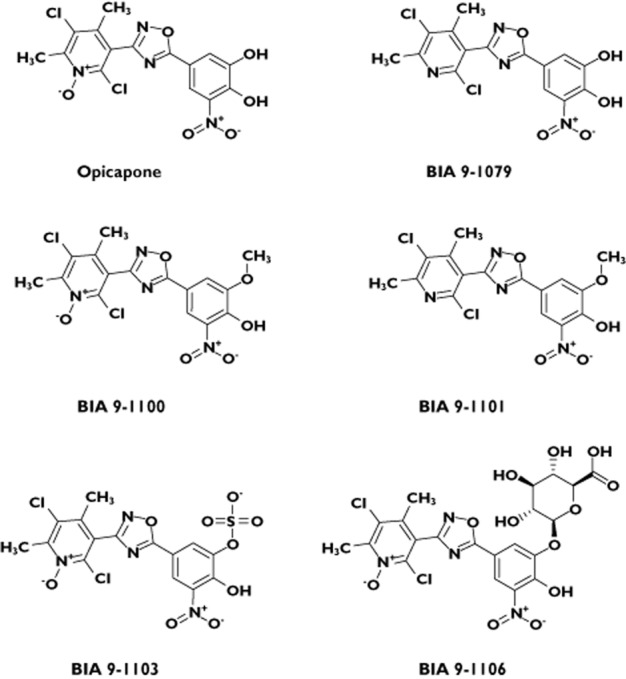
Structural formula of opicapone and its metabolites BIA 9–1079, BIA 9–1100, BIA 9–1101, BIA 9–1102, BIA 9–1103 and BIA 9–1106
The present study investigated the tolerability, pharmacokinetic and pharmacodynamic (COMT inhibition) profiles of once daily oral doses of opicapone to young healthy male volunteers.
Methods
Study design
This study (trial registration EudraCT no. 2008-001288-12) was a dose-escalation, single centre (Biotrial SA, Rueil-Malmaison, France), double-blind, placebo-controlled, randomized study in which once daily doses (5, 10, 20 and 30 mg) of opicapone were administered for 8 days to four sequential groups of healthy young male subjects. The starting dose (5 mg) and the once daily regimen were defined according to the results of a previous single dose study [17]. Within each group (n = 8), two subjects were randomized to placebo and six to opicapone. Subjects were randomly assigned to opicapone or placebo within each dose group according to a randomization list generated by computerized methods. Both the investigation staff and the study subjects were blinded. The investigational products (opicapone or placebo capsules of identical appearance) were manufactured by BIAL – Portela & Cª, S.A.
Healthy male subjects, aged between18 and 45 years, were allowed to participate in the study if they provided written informed consent and if at screening they were considered healthy on the basis of medical history, physical examination, clinical laboratory safety tests, vital signs and 12-lead electrocardiogram (ECG), and tested negative for drugs of abuse, alcohol, HIV and hepatitis C and B. Subjects were excluded from participation if they smoked 10 or more cigarettes per day, had history of drug abuse or alcoholism in the previous 1 year, consumed more than 50 g ethanol day–1, or had donated blood in the previous 3 months. Participation included a screening evaluation within 28 days before a 14 day residence period at the clinical research unit and a follow-up visit 7 to 10 days after last administration. Volunteers were admitted to the unit on the day prior to the start of receiving the study medication and remained in the unit under clinical supervision for at least 144 h after last dose. On days 1 (first dose) and day 8 (last dose), subjects remained fasted for at least 10 h before and until 4 h post-dose. For all other administrations (days 2 to 7), subjects remained fasted for at least 8 h before and until 2 h post-dose. The medication was administered between 8.00 and 10.00 h, and subjects had free access to a maximum of 2.5 l of water per day but were not allowed to drink for 2 h before and after the dosing on days 1 and 8.
No concomitant medications were allowed during the study, unless required for the treatment of AEs. Subjects had to abstain from strenuous physical activity, smoking and consumption of grapefruit juice, alcohol and xanthine-containing beverages from 3 days before the first dose until 144 h after the last dose.
The study was conducted in accordance with the Helsinki Declaration, ICH Good Clinical Practice recommendations and applicable local regulations. An Independent Ethics Committee (CPP – Comité de Protection des Personnes, Saint-Germain, France) reviewed and approved the study protocol and the subject information. Written informed consent was obtained for each study participant.
Safety assessments
AEs were monitored throughout the study. Vital signs (blood pressure and heart rate) were measured and an ECG was recorded at frequent intervals during admission. Haematology, biochemistry and urinalysis tests were performed at admission, discharge from the inpatient period (day 14) and at the follow-up visit.
Pharmacokinetic and pharmacodynamic assessments
Blood samples for pharmacokinetic analysis of opicapone and metabolites were drawn by direct venipuncture or intravenous catheter into potassium ethylenediaminetetraacetic acid (EDTA) tubes at the following time points: day 1 – before first dose and then at 0.25, 0.50, 0.75, 1, 1.5, 2, 3, 4, 6, 8, 10, 12 and 16 h post-dose, days 2 to 7 – pre-dose only and day 8 – before last dose and then at 0.25, 0.50, 0.75, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 16, 24, 36, 48, 72, 96, 120 and 144 h post-dose. After collection, blood samples were centrifuged at approximately 1500 g for 10 min at 4°C, and the resulting plasma was then separated into four aliquots of 1 ml which were stored at −70°C until required for analysis.
Urine samples for pharmacokinetic analysis of opicapone and metabolites were collected at the following intervals: day 1 – before first dose and then at 0–4, 4–8 and 12–24 h post-dose, day 8 – before last dose and then at 0–4, 4–8, 8–12, 12–24, 24–36, 36–48, 48–60, 60–72, 72–84, 84–96, 96–108, 108–120, 120–132 and 132–144 h post-dose. From each collection period, four aliquots of 1 ml were prepared and stored at −80°C until required for analysis.
The same blood samples used for pharmacokinetic assays served for the pharmacodynamic assessments. After centrifugation and removal of plasma, the supernatant (uppermost erythrocyte layer) was removed and the tubes containing the erythrocytes were placed in ice. Then, a volume of cold 0.9% NaCl solution equal to double that of erythrocytes was added. The erythrocytes were centrifuged (at 4°C and approximately 1500 g for 10 min) and the washing procedure was repeated three times. Then, two accurately pipetted 500 ml aliquots of washed erythrocytes were prepared and stored at −70°C until required for analysis.
Bioanalysis of opicapone and metabolites
Determination of plasma and urine concentrations of opicapone and metabolites BIA 9–1100 (methylated, inactive), BIA 9–1101 (methylated, inactive), BIA 9–1079 (reduced, active), BIA 9–1103 (sulphated, inactive) and BIA 9–1106 (glucuronide, inactive) (Figure 1) were carried out in compliance with good laboratory practice (GLP) by liquid chromatography with tandem mass detection (LC-MS/MS) using validated methods with a lower limit of quantification (LLOQ) of 10 ng ml−1 in plasma and 50 ng ml−1 in urine. Two separate methods were used to quantify opicapone and metabolites (see supplement supporting information). The methods were validated in accordance with FDA guidance for industry [20].
S-COMT assay
Determination of S-COMT activity was carried out in compliance with GLP at BIAL's Pharmacological Laboratory (S. Mamede do Coronado, Portugal) according to a validated method [21, 22]. In brief, pre-washed erythrocytes samples were haemolyzed with three volumes of ice cold water. After vortexing, the samples were left standing on ice for 10 min and then centrifuged for 20 min at 2–8°C at 20 000 g. The supernatant was used for S-COMT assay, which was carried out immediately after sample preparation. The incubation mixture contained 300 μl enzyme preparation, 375 μl incubation medium and 75 μl of 5 mm adrenaline as S-COMT substrate. The samples were incubated in a water bath at 37°C for 60 min. The tubes were transferred to ice and the reaction was stopped by adding 75 μl of ice cold 4 m perchloric acid. The samples were incubated in a refrigerator at 2–8°C for 2 h and then centrifuged at 2–8°C (9000 g, 4 min) and 600 μl aliquots of the supernatant, filtered on 0.22 μm pore size Spin-X filter tubes (Costar), were centrifuged at 2–8°C (1400 g, 4 min) and 150 μl used for the assay of metanephrine by means of high pressure liquid chromatography with electrochemical detection, as described elsewhere [21, 22]. S-COMT activity was expressed as the amount of metanephrine (in pmol) formed by the action of erythrocyte S-COMT upon epinephrine, per mg of haemoglobin in the sample, per h (pmol mg−1 Hb h−1).
Analyses
Pharmacokinetic analysis
The pharmacokinetic parameters of opicapone and metabolites were derived by non-compartmental analysis from the individual plasma concentration–time profiles and included the maximum observed plasma concentration (Cmax), time at which Cmax was observed (tmax), area under the plasma concentration–time curve (AUC) calculated using the trapezoidal method from zero to the last quantifiable drug concentration (AUC(0,t)) and from zero to infinity (AUC(0,∞)), the accumulation index (R0) and the apparent terminal half-life (t1/2). In urine, the following pharmacokinetic parameters were estimated: maximum drug concentration (Cmax-u), time to Cmax-u (tmax-u), area under the urine excretion curve from time 0 to the last observed concentration (AURC(0,t)), cumulative amount of drug excreted in urine, and percentage of drug recovered in urine in relation to the amount of opicapone administered (REC%). Summary statistics were prepared for each parameter.
Pharmacodynamic analysis
The following pharmacodynamic parameters were derived from the individual S-COMT activity profiles: maximum inhibition of COMT activity post-dose (Emax), time to occurrence of Emax (tEmax) and area under the effect–time curve (AUEC). The value observed before the first dose was taken as the baseline value (E0). Descriptive statistics were prepared for each parameter. Emax and change in AUEC were compared between dose groups by using a one way analysis of variance (anova) with dose as fixed effect. If global treatment effect was significant, a multiple comparisons Dunnett's test was performed to compare each dose group with placebo. Estimates of the differences between each group and placebo with the associated standard error and 95% CI were calculated.
Pharmacodynamic modelling
The time course of COMT inhibition in the erythrocytes, including a pre-dose effect, was characterized using a simple empirical model, Ea = f(t), expressed in Equation 1.
| (1) |
This equation describes the percent enzyme activity (Ea) as a function of time and is composed of two distinct parts. The first part expresses the onset of enzyme inhibition and is described by the Hill equation D·th/(L + th). Instead of its common use to quantify drug–response relationships, the Hill equation was herein used to model the sigmoidal nature of the onset of pharmacological response over time. Parameter D reflects the dose-dependent extent of COMT inhibition whereas the Hill coefficient h and parameter L define the shape of the curve corresponding to the onset of enzyme inhibition. In experimental designs using multiple dose regimens, one may find that the initial value of the enzyme activity (at t = 0) is lower than baseline, due to the effect of a previous dose intake. Hence, the parameter P is set to the value of the enzyme inhibition (% of baseline) at time zero. In the absence of any previous dose, i.e. on day 1, P is set to 100 and Equation 1 simplifies. The second part of the equation describes the recovery of enzyme activity, as a two phase exponential function. The curve parameters h, D, L, k1 and k2 were adjusted to the experimental data points by non-linear least-squares, using the software Prism v5.03 (GraphPad Software, Inc.).
Adverse events
AEs were coded according to the Medical Dictionary for Regulatory Activities (MedDRA, version 11.0). For the laboratory safety data, clinically significantly abnormal values were considered as AEs.
Statistical analysis
The pharmacokinetic and pharmacodynamic parameters were calculated by using WinNonlin (Version 5.2.1, Pharsight Co, Palo Alto, CA, USA). Statistical analysis used SAS software 8.2 release (SAS Institute Inc., Cary, NC, USA). Statistical tests were performed two-sided with the level of significance set at 5%.
Results
Populations
A total of 34 male subjects were enrolled in the study and constituted the safety population (see supplementary Table S1). Their mean ± SD (range) demographic data were as follows: age = 32.9 ± 7.3 (range 20–45) years, height = 175.7 ± 6.5 (163–189) cm and body mass index (BMI) = 24.3 ± 2.5 (19.3–29.4) kg m−2. Twenty-one (61.8%) subjects were Caucasian, seven (20.6%) were Black and six (17.6%) were of other ethnicity. No relevant differences in demographic characteristics were found between the dose groups. Two subjects were discontinued, one on day 6, due to a serious AE (hemiparesis in a subject receiving placebo) and another on day 2, due to consent withdrawal (receiving 30 mg opicapone). Thirty-two subjects completed the study and six subjects in each opicapone dose group were available for pharmacokinetic analysis. S-COMT assessment could not be performed in one subject who received placebo and thus the S-COMT analysis population set consisted of 31 subjects (six subjects in each opicapone dose group and seven subjects in the placebo group). The first and last enrolment of subjects took place on 24 April and 19 September 2008, respectively.
Pharmacokinetics
Plasma concentrations of BIA 9–1100 and BIA 9–1101 were below the LLOQ at any time point and at any dose. Plasma concentrations of BIA 9–1106 were below the LLOQ at all time points in subjects receiving opicapone 5 and 10 mg, and at most time points in subjects receiving 20 and 30 mg. Thus no results are presented for BIA 9–1100, BIA 9–1101 and BIA 9–1106.
Figure 2 displays the plasma concentration–time profiles and Table 1 presents the plasma pharmacokinetic parameters of opicapone and its metabolites BIA 9–1079 (active) and BIA 9–1103 (inactive), following single and repeated oral doses of opicapone.
Figure 2.

Mean plasma concentration–time profiles of opicapone and its metabolites BIA 9–1079 (active) and BIA 9–1103 (inactive) following the first (day 1) and last (day 8) doses of a 8 day once daily regimen with opicapone (n = 6 per dose group).  , 5 mg;
, 5 mg;  , 10 mg;
, 10 mg;  , 20 mg;
, 20 mg;  , 30 mg
, 30 mg
Table 1.
Mean (coefficient of variation, %) plasma pharmacokinetic parameters of opicapone and its metabolites BIA 9–1079 and BIA 9–1103 following the first (day 1) and the last (day 8) doses of an once daily oral regimen of opicapone (n = 6 per dose group)
| Analyte | Dose | Day | Cmax | tmax | AUC(0,t) | AUC(0,∞) | t1/2 | R0 |
|---|---|---|---|---|---|---|---|---|
| (mg) | (ng ml−1) | (h) | (ng ml−1 h) | (ng ml−1 h) | (h) | |||
| Opicapone | 5 | 1 | 58 (29) | 2.0 (1.5–1.4) | 124 (27) | – | NC | – |
| 8 | 95 (25) | 2.5 (1.5–4.0) | 238 (35) | 295 (42) | 1.1 (33) | NC | ||
| 10 | 1 | 127 (19) | 3.0 (1.5–6.0) | 377 (29) | – | 1.2 (17) | – | |
| 8 | 149 (22) | 2.5 (1.5–4.0) | 433 (26) | 464 (56) | 1.0 (16) | NC | ||
| 20 | 1 | 207 (30) | 3.0 (1.5–4.0) | 773 (31) | – | 1.3 (32) | – | |
| 8 | 216 (35) | 2.5 (1.1–4.0) | 745 (49) | 790 (52) | 1.2 (19) | 0.98 (21) | ||
| 30 | 1 | 343 (37) | 4.0 (2.0–6.0) | 1 432 (31) | – | 1.4 (48) | – | |
| 8 | 301 (39) | 2.0 (0.8–4.0) | 1 441 (40) | 1 481 (39) | 1.4 (32) | 0.91 (23) | ||
| BIA 9–1079 | 5 | 1 | NC | NC | NC | – | NC | – |
| 8 | NC | NC | NC | NC | NC | NC | ||
| 10 | 1 | 13 (24) | 3.5 (2.0–6.0) | 22 (91) | – | NC | – | |
| 8 | 15 (22) | 4.0 (4.0–6.0) | 38 (76) | NC | NC | NC | ||
| 20 | 1 | 24 (25) | 4.0 (3.0–4.1) | 72 (42) | – | NC | – | |
| 8 | 22 (22) | 4.0 (2.0–6.0) | 76 (73) | NC | NC | NC | ||
| 30 | 1 | 30 (29) | 4.0 (4.0–6.0) | 122 (45) | – | NC | – | |
| 8 | 36 (24) | 4.0 (3.0–4.0) | 231 (46) | NC | NC | NC | ||
| BIA 9–1103 | 5 | 1 | NC | NC | NC | – | NC | – |
| 8 | 47 (35) | 7.0 (4.0–10.0) | 3 714 (39) | 6 034 (42) | 98.5 (11) | NC | ||
| 10 | 1 | 21 (22) | 9.0 (6.0–12.0) | 320 (20) | – | 25.1 (11) | – | |
| 8 | 81 (21) | 9.0 (4.0–10.0) | 6 532 (21) | 11 166 (23) | 107.0 (9) | 5.19 (11) | ||
| 20 | 1 | 45 (35) | 8.0 (6.0–10.0) | 721 (36) | – | 26.2 (14) | – | |
| 8 | 140 (30) | 6.0 (4.0–10.0) | 11 481 (27) | 19 718 (25) | 112.0 (7) | 4.00 (10) | ||
| 30 | 1 | 83 (39) | 8.0 (8.0–12.0) | 1 368 (37) | – | 26.9 (19) | – | |
| 8 | 236 (36) | 8.0 (8.0–12.0) | 19 940 (37) | 33 925 (39) | 106 (6) | 3.85 (23) |
tmax values are median with range of values in parenthesis.
AURC(0,t), area under the plasma concentration-time curve from time 0 to last observed concentration; AUC(0,∞), AUC from time 0 to infinity; Cmax, maximum plasma concentration; NC, not calculated; R0, accumulation index; t1/2, apparent elimination half-life; tmax, time to Cmax.
Opicapone Cmax and AUC showed a dose-dependent increase. The inter-individual variability was moderate, ranging from 19% to 37% for Cmax and 27% to 31% for AUC(0,t) following the first dose and from 22% to 39% for Cmax and 39% to 56% for AUC(0,∞) following the last dose. The t1/2 was short, ranging from 1.0 to 1.4 h. R0 showed no accumulation between last and first dose. No statistical evaluation of steady-state could be performed because day 2 to day 8 trough concentrations were all below the LLOQ.
BIA 9–1079 plasma concentrations were all below the LLOQ at the opicapone 5 mg dose. At any dose, BIA 9–1079 AUC(0,∞) and t1/2 could not be reliably calculated because more than half of the values were below the LLOQ. The inter-individual variability for Cmax was moderate, ranging from 22% to 29%, and high for AUC(0,t), ranging from 42% to 91%.
BIA 9–1103 was the main metabolite. Its t1/2 was relatively long, ranging from 25.1 to 26.9 h following the first dose and from 98.5 to 112.0 h following the last dose. The R0 ranged from 3.85 (30 mg) to 5.19 (10 mg).
Urine concentrations of opicapone and metabolites BIA 9–1079, BIA 9–1100, BIA 9–1101 and BIA 9–1103 were below the LLOQ in the tested dose range. BIA 9–1106 was the only metabolite detected in urine and its urine pharmacokinetic parameters are presented in Table 2. Urine drug recovery occurred predominantly over the first 12 h of the urine collection period and the amount of BIA 9–1106 recovered in urine accounted for less than 3% of the amount of opicapone administered.
Table 2.
Mean (coefficient of variation, %) urine pharmacokinetic parameters of BIA 9–1106 following the first (day 1) and the last (day 8) doses of an once-daily oral regimen of opicapone (n = 6 per dose group)
| Analyte | Dose | Day | Cmax-u | tmax-u | AURC(0,t) | AmtCUM | REC |
|---|---|---|---|---|---|---|---|
| (mg) | (nmol h−1) | (h) | (nmol) | (nmol) | (%) | ||
| Opicapone | 5 | 1 | 34 (24) | 2.0 (2.0–6.1) | 114 (32) | 160 (27) | 1.3 (27) |
| 8 | 52 (23) | 2.0 (2.0–2.0) | 205 (21) | 282 (25) | 2.3 (25) | ||
| 10 | 1 | 78 (19) | 2.0 (2.0–6.1) | 270 (22) | 426 (22) | 1.8 (22) | |
| 8 | 95 (10) | 2.0 (2.0–6.1) | 305 (29) | 461 (22) | 1.9 (22) | ||
| 20 | 1 | 151 (33) | 2.0 (2.0–6.1) | 728 (34) | 992 (24) | 2.1 (24) | |
| 8 | 184 (28) | 2.0 (2.0–6.0) | 772 (48) | 1014 (37) | 2.1 (37) | ||
| 30 | 1 | 255 (39) | 2.0 (2.0–6.1) | 1411 (22) | 1616 (17) | 2.2 (17) | |
| 8 | 234 (24) | 6.0 (2.0–6.1) | 1216 (17) | 1485 (16) | 2.1 (16) |
tmax-u values are median with range of values in parenthesis. Amtcum, cumulative amount of drug excreted in urine; AURC(0,t), area under the urine excretion curve from time 0 to last observed concentration; Cmax-u, maximum drug concentration in urine; REC, percent of drug recovered in urine; tmax-u, time to Cmax-u.
Pharmacodynamics
Inhibition of S-COMT
Figure 3 depicts S-COMT activity over time in the placebo and opicapone dose groups and Table 3 presents the statistical comparison of the main pharmacodynamic parameters following opicapone 5 mg, 10 mg, 20 mg and 30 mg vs. placebo.
Figure 3.
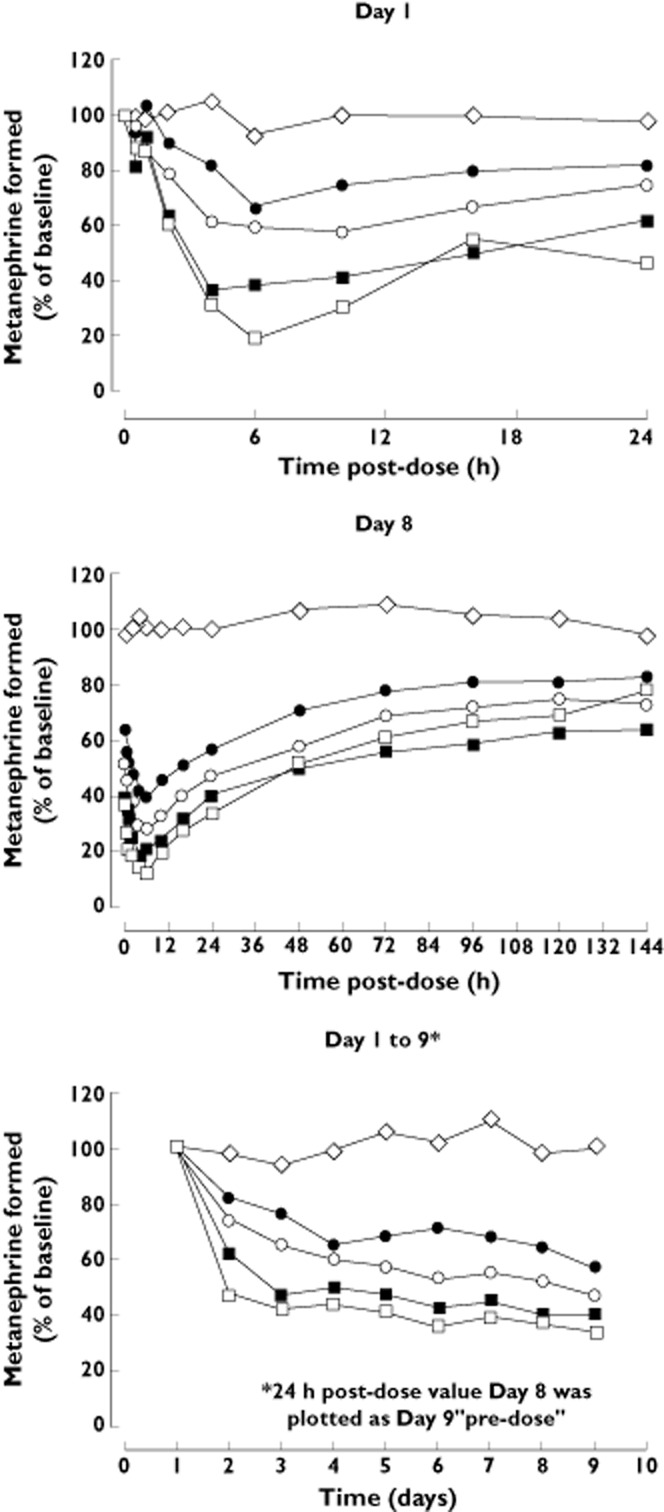
S-COMT activity (% of baseline, pre-dose day 1) vs. time curve following the first (day 1) and last (day 8) doses of a 8 day once daily regimen with opicapone (n = 6 per dose group of opicapone; n = 7 for placebo).  , placebo;
, placebo;  , 5 mg;
, 5 mg;  , 10 mg;
, 10 mg;  , 20 mg;
, 20 mg;  , 30 mg
, 30 mg
Table 3.
Emax and AUEC of S-COMT inhibition in relation to placebo following the first (day 1) and the last (day 8) doses of an once-daily oral regimen of opicapone (n = 6 per opicapone dose group; n = 7 for placebo)
| Dose | Day | Emax | AUEC0–24 | ||||
|---|---|---|---|---|---|---|---|
| (mg) | (%) | 95% CI | P value | (%.h) | 95% CI | P value | |
| 5 | 1 | 38.1 | −26, 103 | NS | 565 | −87, 1218 | NS |
| 8 | 69.9 | 46, 94 | <0.0001 | 1329 | 865, 1793 | <0.0001 | |
| 10 | 1 | 48.6 | −16, 113 | NS | 811 | 159, 1464 | <0.05 |
| 8 | 82.8 | 59, 107 | <0.0001 | 1601 | 1137, 2065 | <0.0001 | |
| 20 | 1 | 52.7 | −12, 117 | NS | 1119 | 467, 1772 | <0.001 |
| 8 | 89.9 | 66, 114 | <0.0001 | 1768 | 1305, 2232 | <0.0001 | |
| 30 | 1 | 57.7 | −7, 122 | NS | 1329 | 676, 1981 | <0.0001 |
| 8 | 98.0 | 74, 122 | <0.0001 | 1945 | 1483, 2410 | <0.0001 | |
AUEC(0,24 h), area under the effect (S-COMT activity inhibition) vs. time curve over 24 h (i.e. dosing interval); CI, confidence interval; Emax, maximum S-COMT inhibition; NS, not statistically significant.
A dose-dependent inhibitory effect of opicapone upon S-COMT activity was observed. Maximum S-COMT inhibition (Emax) occurred between 3.8 h and 7.7 h after the first and 3.7 h and 5.3 h after the last dose (tEmax). In relation to placebo, Emax ranged from 38.1% (5 mg) to 57.7% (30 mg) following the first dose of opicapone and from 69.9% (5 mg) to 98.0% (30 mg) following the last dose of opicapone. On day 8, Emax of S-COMT activity inhibition reached statistical significance vs. placebo at all opicapone dose levels tested (P < 0.0001, Dunnett's test). AUEC(0,24 h) was statistically different from placebo for opicapone 10 mg (P < 0.05), 20 mg (P < 0.001) and 30 mg (P < 0.0001) on day 1 and for all opicapone doses on day 8 (P < 0.0001).
The inhibitory effect of opicapone on S-COMT activity was long lasting. In relation to E0, S-COMT activity presented a decrease of 42.8%, 52.4%, 56.8% and 64.9% 24 h after the last dose of opicapone, in the 5 mg, 10 mg, 20 mg and 30 mg opicapone groups, respectively (−2.9% in the placebo group). At 144 h post-dose, a decrease in S-COMT activity still remained: 16.3%, 21.3%, 31.4% and 20.3% in the 5 mg, 10 mg, 20 mg and 30 mg opicapone groups, respectively (+0.5% in the placebo group).
Pharmacodynamic modelling
We have modelled the enzyme inhibition time course, on day 8, using the empirical Equation 1, by adjusting the model parameters to the experimental data with non-linear least-squares fitting. Our goal was to estimate the half-life of the enzyme activity recovery.
The onset kinetics of the pharmacodynamic effect is affected by a number of pharmacokinetic events that are likely affected by the dose taken and physiological status. Hence, the parameters L, D and h, which affect the shape and magnitude of the onset of enzyme inhibition over time, were independently adjusted for each dose. As a result of pre-dose inhibition levels on day 8, the enzyme activity starts at lower values than the baseline level. Hence, the parameter P in Equation 1 was set to the value of the pre-dose COMT activity (as a percentage of baseline), at each dose.
The second part of the equation describes the exponential decay of enzyme inhibition (recovery). The exponential terms could only be determined for the time-course obtained after the last dose, as it required data collected for a longer period of time. In that situation, it was found that the decay of the inhibition was better modelled by a two-phase exponential curve than with a simpler model that considered a monoexponential recovery phase (F-test, F statistics = 148.0, P < 0.0001). A faster rate of recovery is observed during the initial 24 h, followed by a slower and longer recovery phase. The initial rate constant (k1) was also found to depend on the dose taken and is likely to be affected by the level of exposure to circulating drug. Therefore, k1 was independently adjusted for each dose. On the other hand, the full set of curves (from 5 to 30 mg) could be simultaneously fitted using a single global value of k2, thus suggesting that the late stage rate of recovery from enzyme inhibition follows an exponential decay that is independent from the dose. From the current data, a best fit was obtained with a value of k2 = 0.0053 ± 0.0014 h−1, which translates into a half-life of 130.4 ± 33.3 h (average ± SD).
As mentioned before, the time course data collected over the period of 24 h after the first dose (day 1) was insufficient to determine the value of k2, the rate constant of the late exponential decay of inhibition. Therefore, these curves were modelled using the same k2 value determined for the day 8 dataset. The parameter P was fixed at a value of 100 and only parameters D, L, h and k1 were adjusted, independently, for each dose. Figure 4 illustrates the fitting of Equation 1 to the experimental data points (day 1 and day 8) after 5, 10, 20 and 30 mg opicapone.
Figure 4.

S-COMT activity (% of baseline, pre-dose day 1, closed symbols) and mean plasma concentration of opicapone (open symbols) versus time, following the first (day 1) and last (day 8) doses of a 8 day once daily regimen with opicapone (n = 6 per dose group of opicapone; n = 7 for placebo). Curves represent best fits of equation 1 with non-linear regressions, using a global value for k2 (see text)
Tolerability
Eight subjects reported a total of eight treatment-emergent AEs: headache (n = 3, opicapone 5 mg), vomiting (n = 1, opicapone 10 mg; n = 1, opicapone 30 mg), orthostatic hypotension (n = 1, opicapone 20 mg), muscle spasms (n = 1, opicapone 30 mg) and hemiparesis (n = 1, placebo). All AEs, but hemiparesis, were mild in intensity and recovered without sequella. Hemiparesis was considered as a serious AE and led to study discontinuation. There were no clinically significant out of range values in urinalysis, haematology or biochemistry tests. No clinically relevant abnormalities were observed in vital signs or ECG parameters.
Discussion
The present study aimed to evaluate the tolerability, pharmacokinetics and pharmacodynamics (S-COMT activity inhibition) of oral doses of opicapone, a novel COMT inhibitor, 5 mg, 10 mg, 20 mg and 30 mg administered once daily for 8 days. All drug-related AEs were mild in severity and clinical safety tests did not raise any safety concerns.
The effect of COMT inhibitors is usually evaluated by assaying the erythrocyte S-COMT activity [23]. In the current study, a dose-dependent and long lasting S-COMT inhibitory effect was observed following single and repeated once daily oral doses of opicapone in the tested dose range. Following the last dose of opicapone, Emax ranged from 69.9% (5 mg) to 98.0% (30 mg) (P < 0.0001 at all opicapone doses tested). AUEC(0,24 h) was also statistically different from placebo for all opicapone doses tested (P < 0.0001).
Opicapone shows an outstanding long duration of COMT inhibition in human erythrocytes. Despite the low plasma exposure and short clearance half-life of the inhibitor, the levels of erythrocyte COMT inhibition are sustained far beyond the observable point of plasma drug clearance (6–10 h), for all doses tested. The time course of the enzyme inhibition after dosing shows that, after the initial onset of the pharmacodynamic effect, the level of COMT inhibition follows a two stage exponential decay. A faster rate of recovery is observed during the initial 24 h after dose, followed by a much slower, dose-independent recovery phase, with a half-life (t1/2) in excess of 100 h.
The duration of the pharmacological effect is, in most cases, dependent of the rate of clearance of the drug from its site of action, but also on the half-life of the drug–receptor complex [24–26], particularly when it supersedes that of the drug exposure itself. The dissociative half-life is given by t1/2 = 0.693/koff, where koff is the effective rate of the enzyme–inhibitor dissociation process.
The binding affinity between human COMT and opicapone (amongst other molecules) has been recently estimated by means of computer simulations, using a high accuracy statistical mechanics method [14]. As the inhibitor reaches the site of action, it may form a tight complex with COMT (EI in Figure 5), with an alleged sub-picomolar dissociation constant (Kd = 0.19 pm). It has been shown that opicapone is slowly O-methylated by COMT and released in the form of inactive metabolites [15]. Even though this reaction is slow (catalytic rate constant kcat = 10−4 s−1, t1/2 ≍ 1.5 h), it may likely represent the primary fate of the enzyme–opicapone complex, as it is significantly faster than the dissociation rate constant of the Michaelis complex (koff = 1.9 × 10−6 s−1). It should be noted, however, that the form of the enzyme thus released (E*) is not a functional one. Upon transfer of the methyl group to the COMT inhibitor, the co-substrate becomes demethylated, S-adenosyl-homocysteine (AdoHcy), and must be exchanged for a fresh S-adenosyl-L-methionine (AdoMet), before the full catalytic cycle of the enzyme is completed. This process is expected to be relatively slow, as the co-substrate is buried deeply inside the protein [27] and may require an extensive rearrangement of the enzyme's three dimensional structure. We hypothesize that, while the amount of opicapone in circulation is still significant, a second molecule of the inhibitor may effectively bind to the transient complex COMT-AdoHCy (fast process) before the slow AdoHcy/AdoMet exchange process takes place, hence leading to a non-productive ternary complex COMT-AdoHcy-OPC (EI* in Figure 5). Such complex lacks the ability to catalyze the O-methylation of the bound inhibitor but will possess a long dissociative half-life of about 100 h (t1/2 = 0.693/koff), assuming that the inhibitor molecules will not significantly rebind and will diffuse away to be finally eliminated. The koff value (1.9 × 10−6 s−1) estimated for the ternary complexes COMT-AdoMet-OPC (EI) is herein used as an approximation of the dissociation rate constant of the non-productive complex COMT-AdoHCy-OPC (EI*).
Figure 5.
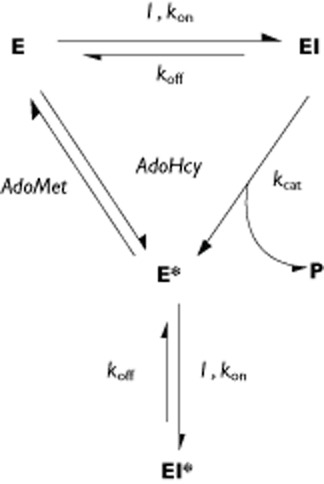
Proposed mechanism of COMT inhibition by nitrocatecholic compounds. E, I, EI and P are, respectively, COMT–AdoMet, inhibitor, COMT–AdoMet–inhibitor and O-methylated product. E* is the binary complex between COMT and AdoHcy and EI* is the non-productive ternary complex COMT–AdoHcy–inhibitor
As a consequence, the kinetics of recovery of baseline COMT activity, from this point onwards, would no longer depend on the O-methylation rate of the inhibitor or the exposure to free drug in circulation. Instead, it would become limited by the very slow dissociation rate of the complex. It is noteworthy that the observed long half-life of opicapone-induced COMT inhibition in human erythrocytes, 130.4 ± 33.3 h (average ± SD), translates into a putative underlying dissociative rate constant kdiss = 1.5 × 10−6 ± 0.4 × 10−6 s−1, which is remarkably comparable with the estimated dissociation rate constant koff of the COMT-OPC complex (1.9 × 10−6 s−1) [14].
Based on the observation that the half-life of COMT inhibition is independent of the dose and that it reflects an underlying kinetic process that is consistent with the dissociation rate constant of the COMT-opicapone complex, we propose that the sustained COMT inhibition, far beyond the observable point of clearance of circulating drug, is due to the long residence time of the reversible COMT-opicapone complex.
The S-COMT inhibitory effect by opicapone described here is much stronger than that reported for tolcapone and entacapone in healthy subjects. Emax was reported as respectively 72% and 80% for tolcapone 100 mg and 200 mg [28] and 65% for entacapone 200 mg [29]. Whereas S-COMT activity returned to baseline approximately 18 h after tolcapone [28] and 8 h after entacapone [29], the inhibitory effect of opicapone on S-COMT activity lasted longer: 24 h after the last dose of opicapone (which is the expected dosing interval for opicapone in clinical use), S-COMT activity still presented a decrease of 42.8%, 52.4%, 56.8% and 64.9% in the 5 mg, 10 mg, 20 mg and 30 mg opicapone groups, respectively. Overall, the results clearly suggest that opicapone has a unique pharmacodynamic profile adequate for a once daily regimen, which in the treatment of PD patients could represent an advantage over entacapone and tolcapone, which have a short S-COMT inhibitory effect and require multiple daily doses. Opicapone is currently under phase III clinical trials to demonstrate the efficacy and safety of opicapone once daily treatment for patients with PD and end of dose motor fluctuations in comparison with placebo [18] or placebo and entacapone [19].
On the basis of non-clinical studies with opicapone, several analytes were assayed in plasma and urine. Plasma and urine concentrations of the methylated inactive metabolites BIA 9–1100 and BIA 9–1101 were always below the LLOQ of the assay showing that humans do not produce such metabolites in noticeable amounts. Dose proportionality for Cmax and AUC was observed for opicapone, BIA 9–1079 (anime N-oxide reduced form, and found to be active in non-clinical studies against S-COMT) and BIA 9–1103 (sulphated, inactive and the major metabolite in plasma). Opicapone peak exposure occurred between 2 and 4 h (median tmax) post dose and mean t1/2 was short, ranging from 1.2 h to 1.4 h. The inter-individual variability for Cmax and AUC(0,t) was moderate, with a mean coefficient of variation (CV) ≤ 37% following the first dose and ≤49% following the last dose. No statistical evaluation of steady-state could be performed as trough concentrations (from day 2 to day 8) were all found to be below the LLOQ. However, the similarity between Cmax and AUC(0,t) values found in day 8 with those found in day 1 in the highest dosage regimens indicates that maximum exposure following a multiple dose regime is achieved following the first administrations of opicapone.
BIA 9–1079 was shown to be a minor metabolite, representing less than 20% of systemic exposure to opicapone, as assessed by AUC(0,t). Therefore, the contribution of BIA 9–1079 to the therapeutic effect following administration of opicapone will be of minor relevance. AUC(0,∞) and t1/2 could not be reliably calculated for BIA 9–1079 because at least half of the values were below the LLOQ. BIA 9–1103 was the major metabolite in plasma, suggesting that sulphation is the main metabolic pathway for opicapone. BIA 9–1103 tmax occurred between 6.0 and 9.0 h post-dose, and t1/2 ranged from 25.1 h to 26.9 h following the first dose and from 98.5 h to 112.0 h following the last dose of opicapone. BIA 9–1103 tended to accumulate with repeated dosing, as evidenced by a R0 of 3.85 to 5.19. The kidney is not the main route of excretion for opicapone and metabolites other than BIA 9–1106. BIA 9–1106 (glucuronide, inactive) was the only metabolite detected in urine, but represented less than 3% of an opicapone dose, which is consistent with the low amounts of this drug moiety found in plasma.
In conclusion, once daily oral doses of opicapone from 5 mg to 30 mg for 8 days were safe and well tolerated. Despite its short elimination half-life, opicapone markedly and sustainably inhibited erythrocyte S-COMT activity making it suitable for a once daily regimen. Sulphation is the main metabolic pathway and the bile is likely the main route of excretion for opicapone and metabolites. Globally, these promising results provide a basis for further clinical development of opicapone.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare JFR, LA, PNP, AIL, RP, MJB, LCW, TN, and PSS were employees of BIAL – Portela & Cª, S.A. in the previous 3 years and AF has received grants from BIAL – Portela & Cª – S.A. in the previous 3 years.
BIAL – Portela & Cª, S.A. supported this study.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Table S1
Summary of demography characteristics
References
- 1.Morgan JC, Sethi KD. Emerging drugs for Parkinson's disease. Expert Opin Emerg Drugs. 2006;11:403–417. doi: 10.1517/14728214.11.3.403. [DOI] [PubMed] [Google Scholar]
- 2.Schapira AH, Emre M, Jenner P, Poewe W. Levodopa in the treatment of Parkinson's disease. Eur J Neurol. 2009;16:982–989. doi: 10.1111/j.1468-1331.2009.02697.x. [DOI] [PubMed] [Google Scholar]
- 3.Dingemanse J. Issues important for rational COMT inhibition. Neurology. 2000;55(11 Suppl. 4):S24–27. discussion S28–32. [PubMed] [Google Scholar]
- 4.Palma PN, Bonifácio MJ, Almeida L, Soares-Da-Silva P. Restoring dopamine levels. In: Simth HJ, Simons C, Sewell RDE, editors. Protein Misfolding in Neurodegenerative Diseases: Mechanisms and Therapeutic Strategies. Boca Raton, FL: CRC Press; 2007. pp. 415–445. [Google Scholar]
- 5.Gomes P, Soares-da-Silva P. Interaction between L-DOPA and 3-O-methyl-L-DOPA for transport in immortalised rat capillary cerebral endothelial cells. Neuropharmacology. 1999;38:1371–1380. doi: 10.1016/s0028-3908(99)00042-8. [DOI] [PubMed] [Google Scholar]
- 6.Gervas JJ, Muradas V, Bazan E, Aguado EG, de Yebenes JG. Effects of 3-OM-dopa on monoamine metabolism in rat brain. Neurology. 1983;33:278–282. doi: 10.1212/wnl.33.3.278. [DOI] [PubMed] [Google Scholar]
- 7.Nutt JG, Woodward WR, Gancher ST, Merrick D. 3-O-methyldopa and the response to levodopa in Parkinson's disease. Ann Neurol. 1987;21:584–588. doi: 10.1002/ana.410210610. [DOI] [PubMed] [Google Scholar]
- 8.Wade LA, Katzman R. 3-O-methyldopa uptake and inhibition of L-dopa at the blood-brain barrier. Life Sci. 1975;17:131–136. doi: 10.1016/0024-3205(75)90248-9. [DOI] [PubMed] [Google Scholar]
- 9.Olanow CW, Stocchi F. COMT inhibitors in Parkinson's disease: can they prevent and/or reverse levodopa-induced motor complications? Neurology. 2004;62(1 Suppl. 1):S72–81. doi: 10.1212/wnl.62.1_suppl_1.s72. [DOI] [PubMed] [Google Scholar]
- 10.Bonifácio MJ, Palma PN, Almeida L, Soares-da-Silva P. Catechol-O-methyltransferase and its inhibitors in Parkinson's disease. CNS Drug Rev. 2007;13:352–379. doi: 10.1111/j.1527-3458.2007.00020.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lang AE, Lees A. Management of Parkinson's disease: an evidence-based review. Mov Disord. 2002;17(Suppl. 4):S45–51. doi: 10.1002/mds.5555. [DOI] [PubMed] [Google Scholar]
- 12.Brooks DJ. Safety and tolerability of COMT inhibitors. Neurology. 2004;62(1 Suppl. 1):S39–46. doi: 10.1212/wnl.62.1_suppl_1.s39. [DOI] [PubMed] [Google Scholar]
- 13.Kiss LE, Ferreira HS, Torrao L, Bonifacio MJ, Palma PN, Soares-da-Silva P, Learmonth DA. Discovery of a long-acting, peripherally selective inhibitor of catechol-O-methyltransferase. J Med Chem. 2010;53:3396–3411. doi: 10.1021/jm1001524. [DOI] [PubMed] [Google Scholar]
- 14.Palma PN, Bonifácio MJ, Loureiro AI, Soares-da-Silva P. Computation of the binding affinities of catechol-O-methyltransferase inhibitors: multisubstate relative free energy calculations. J Comput Chem. 2012;33:970–986. doi: 10.1002/jcc.22926. [DOI] [PubMed] [Google Scholar]
- 15.Bonifácio MJ, Torrão L, Loureiro AI, Wright LC, Soares-da-Silva P. Opicapone: characterization of a novel peripheral long-acting catechol-O-methyltransferase inhibitior. Parkinsonism Relat Disord. 2012;18(S2):S125. [Google Scholar]
- 16.Bonifácio MJ, Sutcliffe JS, Torrão L, Wright LC, Soares-da-Silva P. Brain and peripheral levodopa pharmacokinetics in the Cynomolgus monkey following administration of opicapone, a novel catechol-O-methyltransferase inhibitor. Parkinsonism Relat Disord. 2012;18(S2):S125. [Google Scholar]
- 17.Nunes T, Rocha JF, Pinto R, Machado R, Wright LC, Falcao A, Almeida L, Soares-da-Silva P. Pharmacokinetics, pharmacodynamics and tolerability of opicapone, a novel COMT inhibitor, during first administration to healthy male subjects. Parkinsonism Relat Disord. 2012;18(S2):S126. [Google Scholar]
- 18.Lees A, Costa R, Oliveira C, Lopes N, Nunes T, Soares-da-Silva P. The design of a double-blind, placebo-controlled, multi-national phase-III trial in patients with Parkinson's disease and end-of-dose motor fluctuations: opicapone superiority vs. placebo. Mov Disord. 2012;27(Suppl. 1):S127. [Google Scholar]
- 19.Ferreira JJ, Rocha JF, Santos A, Nunes T, Soares-da-Silva P. The design of a double-blind, placebo- and active-controlled, multi-national phase-III trial in patients with Parkinson's disease and end-of-dose motor fluctuations: opicapone superiority vs. placebo and non-inferiority vs. entacapone. Mov Disord. 2012;27(Suppl. 1):S118. [Google Scholar]
- 20.FDA/CDER. Guidance for Industry: Bioavailability and Bioequivalence Studies for Orally Administered Drug Products – General Considerations. Rockville, MD: U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER); 2000. [Google Scholar]
- 21.Vieira-Coelho MA, Soares-da-Silva P. Ontogenic aspects of liver and kidney catechol-O-methyltransferase sensitivity to tolcapone. Br J Pharmacol. 1996;117:516–520. doi: 10.1111/j.1476-5381.1996.tb15220.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vieira-Coelho MA, Soares-da-Silva P. Effects of tolcapone upon soluble and membrane-bound brain and liver catechol-O-methyltransferase. Brain Res. 1999;821:69–78. doi: 10.1016/s0006-8993(99)01063-x. [DOI] [PubMed] [Google Scholar]
- 23.Ferreira JJ, Almeida L, Cunha L, Ticmeanu M, Rosa MM, Januário C, Mitu CE, Coelho M, Correia-Guedes L, Morgadinho A, Nunes T, Wright LC, Falcão A, Sampaio C, Soares-da-Silva P. Effects of nebicapone on levodopa pharmacokinetics, catechol-O-methyltransferase activity, and motor fluctuations in patients with Parkinson disease. Clin Neuropharmacol. 2008;31:2–18. doi: 10.1097/wnf.0b013e3180645cb0. [DOI] [PubMed] [Google Scholar]
- 24.Copeland RA, Pompliano D. Drug–target residence time and its implications for lead optimization. Nat Rev Drug Discov. 2006;5:1–10. doi: 10.1038/nrd2082. [DOI] [PubMed] [Google Scholar]
- 25.Tummino PJ, Copeland RA. Residence time of receptor-ligand complexes and its effect on biological function. Biochemistry. 2008;47:5481–5492. doi: 10.1021/bi8002023. [DOI] [PubMed] [Google Scholar]
- 26.Vauquelin G, Van Liefde I. Slow antagonist dissociation and long-lasting in vivo receptor protection. Trends Pharmacol Sci. 2006;27:356–359. doi: 10.1016/j.tips.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 27.Vidgren J, Svensson LA, Liljas A. Crystal structure of catechol O-methyltransferase. Nature. 1994;368:354–358. doi: 10.1038/368354a0. [DOI] [PubMed] [Google Scholar]
- 28.Dingemanse J, Jorga KM, Schmitt M, Gieschke R, Fotteler B, Zürcher G, Da Prada M, van Brummelen P. Integrated pharmacokinetics and pharmacodynamics of the novel catechol- O-methyltransferase inhibitor tolcapone during first administration to humans. Clin Pharmacol Ther. 1995;57:508–517. doi: 10.1016/0009-9236(95)90035-7. [DOI] [PubMed] [Google Scholar]
- 29.Keranen T, Gordin A, Karlsson M, Korpela K, Pentikainen PJ, Rita H, Schultz E, Seppala L, Wikberg T. Inhibition of soluble catechol-O-methyltransferase and single-dose pharmacokinetics after oral and intravenous administration of entacapone. Eur J Clin Pharmacol. 1994;46:151–157. doi: 10.1007/BF00199880. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.