

Abstract
Cardiac amyloidosis is an infiltrative cardiomyopathy with a grave prognosis. Its clinical manifestations include restrictive cardiomyopathy, diastolic heart failure, conduction defects, and arrhythmias. Isolated cardiac involvement and significant conduction disturbances are reported very infrequently. We report a rare case of isolated cardiac involvement in primary amyloidosis, in a 76-year-old man who initially presented with sick sinus syndrome that necessitated permanent pacemaker insertion. Subsequent symptoms of heart failure led to additional evaluation, including an endomyocardial biopsy that revealed primary cardiac amyloidosis. Medical therapy improved the patient's symptoms, and he was discharged from the hospital in stable condition. In addition to discussing the patient's case, we review the relevant medical literature.
Key words: Amyloidosis/complications/drug therapy/pathology, cardiomyopathies/complications/pathology, heart failure/diagnosis/etiology, prognosis, treatment outcome
Cardiac amyloidosis is an infiltrative cardiomyopathy caused by the deposition of proteinaceous material, called amyloid, in the extracellular space of the heart. Clinical manifestations can include restrictive cardiomyopathy, diastolic heart failure, conduction defects, and arrhythmias.1
We describe a rare case of primary cardiac amyloidosis in an elderly man who initially presented with sick sinus syndrome that necessitated permanent pacemaker insertion. Subsequent symptoms of heart failure prompted additional evaluation that established a diagnosis of primary cardiac amyloidosis.
Case Report
In March 2012, a 76-year-old man presented with a 2-year history of gradually progressive dyspnea, orthopnea, lower-extremity swelling, and abdominal distention. His relevant medical history included obstructive sleep apnea and paroxysmal atrial fibrillation. Three years before the current presentation, he had experienced an episode of syncope. The diagnosis was sick sinus syndrome that was presumptively attributed to age-related degenerative causes, and a permanent pacemaker was implanted. The patient had no history of myocardial infarction or hypertension.
At presentation, the patient had a blood pressure of 89/49 mmHg; heart rate, approximately 70 beats/min with irregularly irregular rhythm; respiratory rate, 20 breaths/min; and oxygen saturation, 97% on room air.
Physical examination revealed a jugular venous pressure of 15 cm H2O and bilateral pitting edema extending from the feet up to the knees. Cardiac examination yielded soft heart sounds with a pansystolic murmur best heard in the left lower parasternal region, suggesting tricuspid regurgitation. Respiratory examination revealed bibasilar fine crackles. The patient's distended abdomen with shifting dullness indicated ascites.
Results of laboratory tests, including renal function, liver function, and a complete blood count, were not unusual. An electrocardiogram (ECG) showed atrial fibrillation with a ventricular rate of 70 to 80 beats/min, right bundle branch block, and normal voltage complexes. Transthoracic echocardiograms showed globally abnormal biventricular systolic function, a left ventricular ejection fraction of 0.35, and dilation of both atria and the right ventricle. There was concentric and severe left ventricular hypertrophy with interventricular septal thickness of 2.1 cm (Fig. 1). Diastolic dysfunction of the restrictive type was noted, and there was moderate tricuspid valve regurgitation with a pulmonary artery systolic pressure of 40 mmHg. The ventricular wall showed increased echogenicity with a speckled pattern, suggesting amyloidosis.
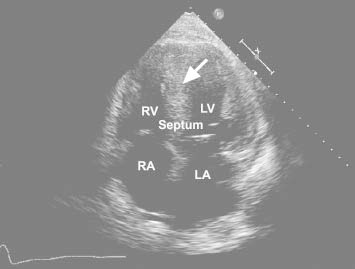
Fig. 1 Transthoracic echocardiogram (apical 4-chamber view) shows features typical of amyloidosis: thickening of all myocardial walls with increased echogenicity (arrow).
LA = left atrium; LV = left ventricle; RA = right atrium; RV = right ventricle
In view of the above findings, investigation for amyloidosis was initiated. Results of serum electrophoresis showed a mild increase in gamma globulin with a polyclonal band. Results of a urinalysis showed no Bence-Jones protein. Subsequent tests, including an abdominal fat-pad biopsy and a bone marrow aspiration and biopsy, were negative for amyloid. An endomyocardial biopsy was then performed. Hematoxylin and eosin stain disclosed interstitial and perimyocytic deposition of pale, finely fibrillar eosinophilic material that enveloped individual myocytes (Fig. 2A). Congo red stain disclosed apple-green birefringence under polarized light, characteristic of amyloid. Immunohistochemical stains were positive for AL (primary) amyloidosis (Fig. 2B).
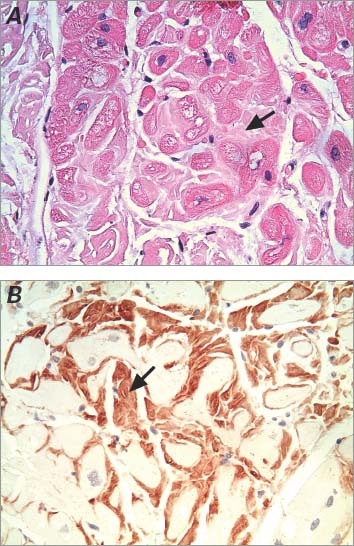
Fig. 2 Photomicrographs show typical findings of cardiac amyloidosis on an endomyocardial biopsy specimen. Arrows indicate the deposition of amyloid. A) Histologic section shows interstitial and perimyocytic deposition of amorphous, light-pink, finely fibrillar eosinophilic material (amyloid) enveloping individual myocytes (H & E, orig. ×400). B) Immunohistochemistry stain confirms the deposition of amyloid (orig. ×400).
The patient was treated for acute decompensated heart failure with a diuretic, a β-blocker, and an angiotensin-converting enzyme inhibitor. His atrial fibrillation reverted to sinus rhythm after therapy with dofetalide, and warfarin was started for anticoagulation. His symptoms improved substantially; he was discharged from the hospital after 2 weeks, in stable condition.
Discussion
Cardiac amyloidosis is a progressive, infiltrative cardiomyopathy that results from the extracellular deposition of a proteinaceous material called amyloid. Deposits of different types of proteins form the basis for the classification of amyloidosis. Monoclonal light chains constitute the amyloid protein in AL amyloidosis, transthyretin (TTR) in senile amyloidosis, and serum amyloid A protein in AA (secondary) amyloidosis.1
Amyloid deposition and infiltration of the cardiac muscle causes the myocardium to become firm, rubbery, and noncompliant, resulting in restrictive cardiomyopathy. Amyloidosis can cause focal thickening of cardiac valves and infrequently leads to valvular dysfunction. Rarely, infiltration of the conduction system can result in heart block and sick sinus syndrome.2
Clinical evidence of cardiac involvement is seen frequently in AL amyloidosis (in up to 50% of cases), variably in senile type, and very rarely in AA amyloidosis. Cardiac amyloidosis is diagnosed more frequently in men, and patients usually present after the age of 40 years. In contrast with other types of amyloidosis, cardiac involvement in AL amyloidosis often leads to rapidly progressive cardiac dysfunction. It is a multisystemic disease. Isolated cardiac involvement, as in our patient, is rare and was described in only 4% of a series of 232 patients who had AL amyloidosis.3 Despite a high prevalence of conduction abnormalities on surface ECG,4 symptomatic conduction-system disease involvement is infrequently reported. In a study of 696 patients who had cardiac amyloidosis, only 3.9% needed pacemakers.2 Among patients who received a pacemaker, about half had complete heart block, 18% had isolated sinus node dysfunction, and another 18% had sinoatrial, atrioventricular, or His-Purkinje disease.
The prevalence of conduction disturbances has probably been underestimated in all forms of amyloidosis. Symptoms such as dizziness and syncope are often attributed to other pathologic conditions, such as orthostatic hypotension, when the underlying cause could be amyloidosis. Sudden cardiac death (SCD) can occur in cases of advanced amyloidosis and could be the presenting event.5 The estimated rate of SCD in amyloidosis without evidence of heart failure is 15% to 23%.5–7 The origin of SCD in amyloidosis has been attributed to conduction abnormalities, such as high-degree atrioventricular block and ventricular arrhythmias.6
The typical ECG findings are low-voltage complexes and a pseudoinfarction pattern, seen in about half of patients. Atrial fibrillation or flutter is seen in about 20%. Low-voltage complexes in the presence of increased left ventricular mass (determined echocardiographically) are exclusive to cardiac amyloidosis and have a high sensitivity and specificity of 72% and 91%, respectively.8
Echocardiography—a valuable noninvasive diagnostic tool in cardiac amyloidosis—reveals increased ventricular wall thickness with small intracavitary chambers and enlarged atria. Diastolic dysfunction can be an early manifestation of the disease process. Increasingly severe restrictive cardiomyopathy and systolic dysfunction are late manifestations. The ventricular walls often reveal a sparkling and granular pattern that is attributed to amyloid deposition. This characteristic feature of cardiac amyloidosis, also described as speckled pattern, was shown in the 1980s to have a specificity of 81% and a sensitivity of 87%.9 Later, studies found less sensitivity, and it is recommended to use the voltage–mass relationship described above.8
The diagnosis is established by means of endomyocardial biopsy or biopsy of other tissue locations, such as abdominal fat pads, the rectum, or bone marrow. If these last specimens are negative, as in isolated cardiac amyloidosis, endomyocardial biopsy should be performed. Amyloid deposits produce apple-green birefringence under polarized light when stained with Congo red. Immunohistochemical staining further characterizes the type of amyloid, which provides prognostic information and enables targeted therapy.10
Patients with AL amyloidosis have a median survival period of 1 to 2 years; in patients with cardiac involvement, life expectancy typically decreases to 6 months after the onset of congestive heart failure.11 Treatment regimens aimed at altering the natural history of this disease are evolving, and they vary for different types of amyloidosis. High-dose intravenous melphalan with autologous hematopoietic stem-cell transplantation has enabled a median survival period of 6.4 years in patients who have AL amyloidosis without cardiac involvement, and 1.6 years in those with cardiac involvement.12 Chemotherapeutic regimens involving either melphalan with dexamethasone or the combination of cyclophosphamide, thalidomide, and dexamethasone have been used with some success.13 Newer agents such as bortezomib, bendamustine, and lenalidomide have shown promise and are being evaluated in clinical trials as part of combination regimens.14
In most patients with amyloidosis, therapy is mainly supportive and is directed toward controlling the complications, such as heart failure and arrhythmias. Digoxin can bind to amyloid protein, making patients very sensitive to the effects of digoxin despite therapeutic serum levels.15 If digoxin is indicated, such as for atrial fibrillation, patients should be closely monitored. Calcium channel blockers should also be used with caution because of their negative inotropic effect.16 Anticoagulant therapy should be considered in cases of atrial fibrillation.
Progressive distal conduction-system disease with high-degree atrioventricular block frequently occurs; if the patient needs a permanent pacemaker, consideration should be given to biventricular pacing, because right ventricular pacing could aggravate the already decreased cardiac output.7 Despite the high prevalence of SCD in amyloidosis, prophylactic implantable cardioverter-defibrillator (ICD) implantation has not yielded any benefit. In the absence of large studies, the indications for ICD implantation, pacemaker implantation, or both are largely determined by the standard guidelines for the more typical clinical conditions.
Novel approaches targeting small-molecule ligands that stabilize the native tetrameric structure of TTR and consequently prevent its fibrillogenesis are being actively investigated for prophylaxis and therapy in patients who have TTR amyloidosis.17–19 Diflunisal, a nonsteroidal anti-inflammatory drug, can stabilize the TTR tetramer; this will in turn reduce tetramer dissociation and subsequent monomer misfolding and aggregation into amyloid.17 Another agent, tafamidis, has slowed the progression of neuropathy in patients with TTR amyloidosis and appeared to be well tolerated in a small trial of patients with wild-type or mutant TTR cardiomyopathy.18 Other approaches with applications in AL or TTR amyloidosis include stabilizing the native structures of other amyloidogenic proteins and preventing and reversing fibrillogenesis, as well as disrupting established deposits with the use of antibodies, synthetic peptides, and small-molecule drugs.19
Varied clinical presentations in cardiac amyloidosis can result in diagnostic delays. When clinical suspicion is high despite negative biopsy results from noncardiac tissues, an endomyocardial biopsy should be performed to establish a diagnosis. Early recognition is crucial. Timely therapy can improve patients' quality of life, and some of the emerging disease-modulating therapies can contribute to prolonged survival times.
Footnotes
Address for reprints: Pradeep K. Bhat, MD, Hamman 330, Case Western Reserve University, 2500 MetroHealth Dr., Cleveland, OH 44109-1998.
E-mail: pradeepkbhat@yahoo.com
References
- 1.Kyle RA. Amyloidosis: a convoluted story. Br J Haematol 2001;114(3):529–38. [DOI] [PubMed]
- 2.Mathew V, Olson LJ, Gertz MA, Hayes DL. Symptomatic conduction system disease in cardiac amyloidosis. Am J Cardiol 1997;80(11):1491–2. [DOI] [PubMed]
- 3.Dubrey SW, Cha K, Anderson J, Chamarthi B, Reisinger J, Skinner M, Falk RH. The clinical features of immunoglobulin light-chain (AL) amyloidosis with heart involvement. QJM 1998;91(2):141–57. [DOI] [PubMed]
- 4.Falk RH, Rubinow A, Cohen AS. Cardiac arrhythmias in systemic amyloidosis: correlation with echocardiographic abnormalities. J Am Coll Cardiol 1984;3(1):107–13. [DOI] [PubMed]
- 5.Chamarthi B, Dubrey SW, Cha K, Skinner M, Falk RH. Features and prognosis of exertional syncope in light-chain associated AL cardiac amyloidosis. Am J Cardiol 1997;80(9):1242–5. [DOI] [PubMed]
- 6.Roberts WC, Waller BF. Cardiac amyloidosis causing cardiac dysfunction: analysis of 54 necropsy patients. Am J Cardiol 1983;52(1):137–46. [DOI] [PubMed]
- 7.Kristen AV, Dengler TJ, Hegenbart U, Schonland SO, Goldschmidt H, Sack FU, et al. Prophylactic implantation of cardioverter-defibrillator in patients with severe cardiac amyloidosis and high risk for sudden cardiac death. Heart Rhythm 2008;5(2):235–40. [DOI] [PubMed]
- 8.Rahman JE, Helou EF, Gelzer-Bell R, Thompson RE, Kuo C, Rodriguez ER, et al. Noninvasive diagnosis of biopsy-proven cardiac amyloidosis. J Am Coll Cardiol 2004;43(3):410–5. [DOI] [PubMed]
- 9.Falk RH, Plehn JF, Deering T, Schick EC Jr, Boinay P, Rubinow A, et al. Sensitivity and specificity of the echocardiographic features of cardiac amyloidosis. Am J Cardiol 1987;59 (5):418–22. [DOI] [PubMed]
- 10.Olson LJ, Gertz MA, Edwards WD, Li CY, Pellikka PA, Holmes DR Jr, et al. Senile cardiac amyloidosis with myocardial dysfunction. Diagnosis by endomyocardial biopsy and immunohistochemistry. N Engl J Med 1987;317(12):738–42. [DOI] [PubMed]
- 11.Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol 1995;32 (1):45–59. [PubMed]
- 12.Skinner M, Sanchorawala V, Seldin DC, Dember LM, Falk RH, Berk JL, et al. High-dose melphalan and autologous stem-cell transplantation in patients with AL amyloidosis: an 8-year study. Ann Intern Med 2004;140(2):85–93. [DOI] [PubMed]
- 13.Palladini G, Perfetti V, Perlini S, Obici L, Lavatelli F, Caccialanza R, et al. The combination of thalidomide and intermediate-dose dexamethasone is an effective but toxic treatment for patients with primary amyloidosis (AL). Blood 2005;105(7):2949–51. [DOI] [PubMed]
- 14.Cohen AD, Comenzo RL. Systemic light-chain amyloidosis: advances in diagnosis, prognosis, and therapy. Hematology Am Soc Hematol Educ Program 2010;2010:287–94. [DOI] [PubMed]
- 15.Rubinow A, Skinner M, Cohen AS. Digoxin sensitivity in amyloid cardiomyopathy. Circulation 1981;63(6):1285–8. [DOI] [PubMed]
- 16.Gertz MA, Falk RH, Skinner M, Cohen AS, Kyle RA. Worsening of congestive heart failure in amyloid heart disease treated by calcium channel-blocking agents. Am J Cardiol 1985;55(13 Pt 1):1645. [DOI] [PubMed]
- 17.Castano A, Helmke S, Alvarez J, Delisle S, Maurer MS. Diflunisal for ATTR cardiac amyloidosis. Congest Heart Fail 2012;18(6):315–9. [DOI] [PMC free article] [PubMed]
- 18.Coelho T, Maia LF, Martins da Silva A, Waddington Cruz M, Plante-Bordeneuve V, Lozeron P, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology 2012;79(8):785–92. [DOI] [PMC free article] [PubMed]
- 19.Sekijima Y, Kelly JW, Ikeda S. Pathogenesis of and therapeutic strategies to ameliorate the transthyretin amyloidoses. Curr Pharm Des 2008;14(30):3219–30. [DOI] [PubMed]